

1 Introduction
The large accumulation of endocrine disruptors, in continental and marine natural waters are the consequence of the worldwide general application of intensive agricultural methods, the large-scale development of the agrochemical and food industry and the high levels found in some specific effluents (textile, pharmaceutical, hospital waste…). For instance, in the Brittany region (France), the Regional Direction of the Environment (DIREN) observes river contamination by phytosanitary products, including pesticides, which can interfere with hormone systems of living beings (endocrine disruptors) [1]. Partly responsible for this pollution, low volumes containing high concentrations of persistent organic pollutants, in the range of concentrations found in some specific industrial and agricultural effluents (unused treatment solution, spray, machine and container washing…) [2], can result in large polluted volumes that are difficult to treat owing to their low pollutant concentrations. One solution would be therefore to treat pollution at its source, as intended in this study.
Pesticides’ impact on the environment is complex and varied according to various factors, such as toxicity and ecotoxicity of the parent molecule or by-product metabolites, synergistic effects with other pollutants, length of the half-life, exposure time and dose, etc. Various acute or chronic poisoning effects on human health have been described [3–6]. There is therefore an urgent need for efficient processes for their removal and, owing to the possible toxicity of the by-product metabolites, total mineralization is mainly targeted.
For this purpose, biological processes, the most cost-effective for wastewater treatment, which are destructive and have been extensively studied [7–11], do not always appear relevant for the removal of recalcitrant compounds, owing to their low biodegradability.
Contrarily, physicochemical techniques have proved their efficiency for such removal. Among them, advanced oxidation processes constitute the most important and widely documented group [2,12,13], owing to the high reactivity of the free •OH radicals produced. However, and due to the lack of selectivity of the free radicals, possible toxic by-products can be generated, which in some cases can appear more toxic than the parent compounds [14,15]. Consequently, the mineralization time is of major importance; a too low processing time can result in toxic by-products, while a long processing time to ensure total mineralization can induce high-energy costs.
It is noteworthy that pesticides biorecalcitrance can be related to the presence of a complex aromatic chain or to the presence of specific bonds, such as nitro- or halogenated bonds. For instance, among the most used pesticides, a three-quarter of them contain halogenated bonds, and the presence of chlorine atoms on phenyl ring is a factor that favors the toxicity of aryl compounds [16]. From this, the development of processes targeting a selective attack of specific functional groups to improve the biodegradability of a given effluent can constitute another approach to treat pesticide-containing effluents.
Indeed, if agricultural effluents are for instance considered, pesticides levels can reach 500 mg·L−1 [2], as it is the case in farm bottom tanks. Hence, a selective attack of specific functional groups can constitute a relevant solution to relieve AOPs drawbacks. The generation of possible toxic by-products could thus be avoided owing to the expected control of the resulting by-products as well as the high-energy costs, since total mineralization is not the objective, due to the expected improvement of biodegradability.
For this purpose and in the case of an electroactive target compound, its electrochemical oxidation or reduction can be carried out for its degradation. Total mineralization can be subsequently completed during biological treatment, since the potential advantages of the strategy of combining physicochemical and biological processes to treat contaminants in wastewater were previously underlined [17–20]. However, the literature dealing with the use of direct electrochemical oxidation/reduction for effluent pretreatment remains scarce. Doan et al. [21] and Ghafari et al. [22] coupled an electrochemical process and a biological treatment for the removal of heavy metals and nitrate, respectively, while regarding organic pollutants, up to now our work seems to be the only one available dealing with the combination of a direct electrochemical process and a biological treatment. It was investigated for the removal of phosmet, an organophosphorous insecticide [23], some antibiotics, tetracycline [24], sulfamethazine [25], and a chlorinated phenoxy herbicide, 2,4-dichlorophenoxyacetic acid [26,27].
The promising results obtained regarding 2,4-D should be underlined [28,29], since halogenated pesticides are the most widespread pesticides. Indeed, it was shown that mild oxidation of the target compound can be sufficient to improve biodegradability [28], allowing subsequent biological treatment [29]. However in view of the targeted selectivity, the nature of the electrochemical process should be clearly elucidated, namely the involvement or not of free hydroxyl radials in pesticide oxidation; it is discussed in this study.
However, the production of this highly reactive species is closely linked to the electrode material used and to some operating parameters such as oxygen overvoltage. If some electrodes (boron-doped diamond electrodes for instance) are well-known as powerful candidates for •OH generation, it is not at all obvious for some other materials, such as graphite felt, the material used in the laboratory [23,25–27]. Indeed, the use of a graphite felt working electrode with a high specific area in a flow electrochemical cell [30] allows the electrochemical reaction of the electroactive species at macroscale level with low electrolysis times. Even if mechanisms involving •OH have been previously suggested for studies based on the use of graphite felt [31], to our knowledge such production at the surface of such electrode material has never been demonstrated up to now. However, this question appears to be of major importance to understand the electrochemical oxidation phenomena occurring when using such material and to confirm the specificity and selectivity of the considered processes, namely a direct reaction at the electrode surface. For this purpose, three points have to be considered, the nature of the working electrode, the experimental conditions and the indirect determination of the hydroxyl radicals; these points are investigated in this study.
In addition and to complete the mechanism's knowledge, the electrochemical reaction of 2,4-D at the electrode was also examined.
Biological treatment involving activated sludge was then considered to examine the efficiency of the electrochemical pretreatment.
2 Materials and methods
2.1 Chemicals
2,4-Dichlorophenoxyacetic acid (2,4-D) (98%) was purchased from Alfa Aesar (Schiltigheim, France). Chlorohydroquinone (85%) was purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). Acetonitrile (ACN) and formic acid were LC/MS grade from JT Baker (Deventer, Netherlands). All standards were prepared with ultra-pure water (PurelabOptions-Q7/15, Elga, 18.2 MΩ·cm).
2.2 Electrochemical pretreatment
The electrochemical pretreatment was based on a home-made flow cell [28]. Graphite felt used as the working electrode was supplied by Mersen (RVG 4000 – Mersen, Paris La Défense, France) [27]. The dimensions of the graphite felt were 48 mm in diameter and 12 mm in width.
Two interconnected stainless steel plates were used as counter-electrodes and compartments were separated by cationic exchange membranes (Ionac 3470 – Lanxess SAS, Courbevoie, France). The reference electrode (Saturated Calomel Electrode–SCE) was positioned in the middle of the graphite felt. To ensure a good homogeneity of the potential distribution in the three-dimensional working electrode, the felt was located between the two counter-electrodes [30]. The electrolyte solution percolated the porous electrode and the flow rate was monitored by a Gilson minipuls 2 peristaltic pump (Middleton, WI, USA). A VersaSTAT 3 potentiostat (Ametek/Princeton Applied, Élancourt, France) was used to control the potential.
2.3 Biological process
After only one pass through the electrochemical flow cell, the effluent was collected for the subsequent biological treatment, which was carried out in aerobic conditions, using activated sludge purchased from the local wastewater treatment plant (station de Beaurade, Rennes, France). Before use and to avoid any residual carbon or mineral nutrient, it was treated as previously detailed [26].
Experiments were carried out in 250-mL erlenmeyer flasks containing 100 mL of medium, stirred at 250 rpm, kept at 30 °C and triplicated to ensure the reproducibility of the results. The electrolyzed solution (initially 500 mg·L−1 2,4-D) was diluted (five times dilution) to allow a direct comparison with non-treated 2,4-D (100 mg·L−1 – [26]). Minerals were spiked in the medium as highly concentrated solutions to reach the following initial composition (mg·L−1): Na2HPO4, 334; K2HPO4, 208; KH2PO4, 85; CaCl2, 27.4; MgSO4.7H2O, 22.6; NH4Cl, 75; FeCl3.6H2O, 0.26; and the initial pH was adjusted to 7.0 ± 0.2. Activated sludge was added in order to have an initial concentration of 0.5 g L−1 of dry matter. Samples (5 mL) were taken regularly and filtered through 0.45 μm-syringe filters for measurements.
2.4 Analysis
Measurements of the residual 2,4-D and chlorohydroquinone concentration were performed by an HPLC (High Pressure Liquid Chromatography) (Milford, USA) system involving a pump Waters 600, fitted with a Phenomenex Kinetex® C18 2.6 μm column (4.6 mm × 100 mm), along with a Waters 996 Photodiode array detector, a Waters 717 ph Autosampler and controlled through an Empower® 2 program. The mobile phase consisted of acetonitrile and trifluoroacetic acid (TFA) 0.1% in ultra-pure water with a ratio of 30/70 at 1 mL min−1.
2.4.1 Dissolved organic carbon (DOC) measurements
Solutions were filtered on Sartorius Stedim Minisart 0.45-μm GF prefilters (Göttingen, Germany). DOC was measured by means of a Schimadzu TOC-VCPH/CPN Total Organic Analyzer. Reproducible DOC values were always obtained using the standard NPOC (Non-Purgeable Organic Carbon) method.
2.4.2 Chemical oxygen demand (COD) and biological oxygen demand (BOD5)
Chemical oxygen demand (COD) was measured by means of a Test Nanocolor® CSB 160 from Macherey-Nagel (Düren, Germany).
BOD5 measurements were carried out in Oxitop IS6 (WTW, Alès, France). Activated sludge provided by a local wastewater treatment plant (Rennes Beaurade, Bretagne, France) was used to inoculate duplicate flasks and the initial microbial concentration was set to 0.5 g L−1. More details regarding the experimental procedure can be found in previous papers [26,27].
3 Results and discussion
3.1 Electrochemical behavior of 2,4-D
The electrochemical behavior of 2,4-D in acidic medium (pH ≈ pKa = 2.8, naturally obtained when 2,4-D was dissolved in water) showed an electroactivity in reduction and in oxidation [27]. 2,4-D Electrolysis in reduction showed a disappearance of the first reduction signal, which was however not confirmed by HPLC analysis, which indicated no 2,4-D degradation. The disappearance of the reductive signal was found to be closely related to its anionic or molecular form, and thus to the pH of the medium, since the reductive signal disappeared at alkaline final electrolysis pH (11), while it was recovered after acidification of the medium to the initial pH value (2.8) (Fig. 1).

Cyclic voltammetry of a 2,4-D solution (500 mg·L−1) at pH 2.8 (dots), at pH 11 (dashes) and at pH 2.8 after re-acidification (full line). Voltammograms were recorded at 100 mV·s−1 in Na2SO4 0.1 mol L−1 on a glassy carbon electrode (7 mm2).
This electrochemical behavior closely related to the pH and coupled to the analytical results obtained by liquid chromatography led to exclude the initial assumption of a C–Cl bond cleavage and to consider instead a reduction of the carboxylic acid function of 2,4-D. Indeed, the carboxylic acid can undergo a one-electron reaction to yield the corresponding carboxylate anion, which is accompanied by hydrogen release (Reaction 1 – Fig. 2), and the reduction of the formed carboxylate appears difficult in the considered solvent, water. The carboxylic acid can also undergo 2-, 4- or 6-electron reductions to give the corresponding aldehyde, alcohol or alkane, respectively (reactions (2) to (4) in Fig. 2); these reactions can however occur in acidic conditions avoiding the carboxylate reduction [32].

(Color online). Possible reactions for the reduction of the carboxylic function of 2,4-D.
The lack of a reduction wave was expected at alkaline pH, since its anionic form largely predominated owing to the 2,4-D pKa (2.8); this characteristic wave can only be observed in the presence of the protonated form of the compound, namely for pH values below 4.
The transformation of the carboxylic acid into its ester form should allow one to avoid its carboxylate reduction, which would be helpful to validate the above assumption. Thus, the diethyl ester of 2.4-D was prepared by the reaction of 2.4-D with oxalyl chloride to form the acyl chloride derivative and then with ethanol. However, it was not possible to analyze the synthesized ester in water owing to its low solubility, leading to perform cyclic voltammetry analyses in acetonitrile. As shown in Fig. 3, the synthesized ester was reduced at a lower potential (–1.75 V/ECS) than the initial compound (–1.5 V/ECS). Consequently, the wave observed during 2,4-D reduction was linked to the reduction of the acid to acetate instead of the aromatic ring dechlorination.

Cyclic voltammetry of 2,4-D (20 mmol L−1–dots) and its ester form (20 mmol L−1 – full line) in acetonitrile (KPF6 0.1 mol L−1 – blank: dashes) on a glassy carbon electrode (7 mm2). Voltammograms were recorded at 100 mV·s−1.
The electrochemical behavior of 2,4-D was then examined in oxidation, and hence a similar approach was considered, namely cyclic voltammetry analysis prior to electrolysis to confirm the pretreatment's feasibility. An oxidation wave was observed at 1.6 V/ECS (Fig. 4).
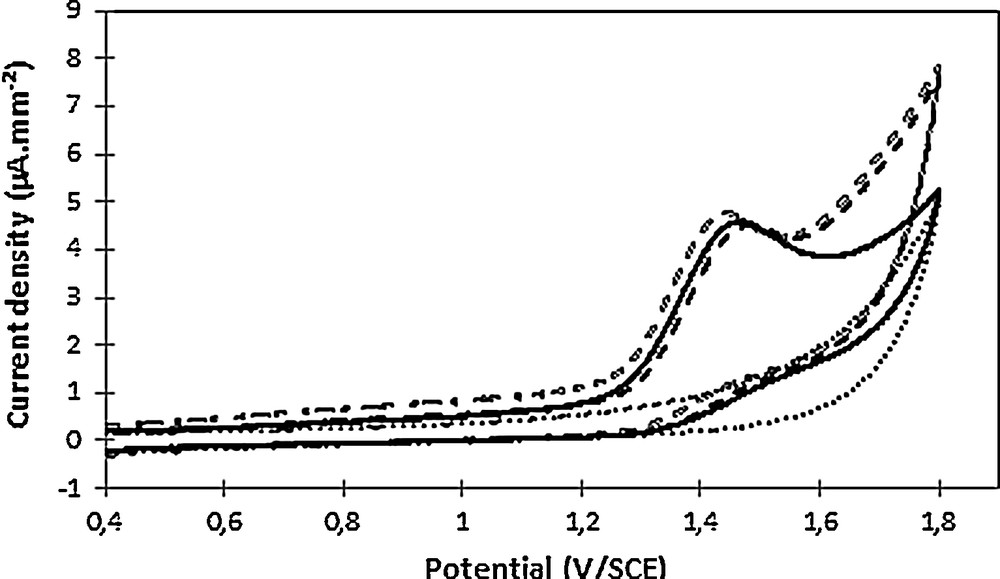
Cyclic voltammetry of Na2SO4 0.1 mol L−1 as supporting electrolyte (dots), in the presence of 100 mg·L−1 2,4-D at pH 3.6 (full line), at pH 11 (empty dashes) and at pH 2.8 after re-acidification (full dashes). Voltammograms were recorded at 100 mV·s−1 on a glassy carbon electrode (7 mm2).
To confirm 2,4-D electroactivity in oxidation, the absence of pH impact on its electrochemical behavior was checked and confirmed, since the oxidation wave was observed at both acidic and alkaline pHs (Fig. 4).
3.2 Nature of the electrochemical process
Since no 2,4-D degradation was detected during cathodic reduction experiments in aqueous medium, electrolyses were therefore performed in oxidation. However, one major question remained, which was the nature of the electrochemical process involved in 2,4-D oxidation: are free radicals involved in 2,4-D degradation?
Electrodes can be classified as “active” and “non-active” according to their reactivity vis-à-vis the reaction of oxygen formation [33]. In a first step, a plane graphite electrode was used to allow a fine observation of oxygen formation on the material (graphite). The obtained results were then compared to well-known electrode materials, such as platinum or vitreous carbon. At pH 2.7, namely close to the experimental conditions in the presence of 2,4-D, it can be observed an oxygen release from about 1.5–1.6 V/SCE on graphite and platinum and from 1.6–1.7 V/SCE for vitreous carbon (it should be remembered that it is usually observed near 2.5 V/SCE on a BDD) [34]. The shape of the curve recorded on graphite suggested a resistive character of the material before oxygen release, which may be related to the nature of the electrode. This phenomenon was systematically observed, irrespective of meticulous electrode polishing, long degassing time or electrochemical surface cleaning, namely a chronoamperometry at 0 V/SCE before cyclic voltammetry analyses. This relatively early oxygen release on graphite suggested an “active” electrode nature, which was not in favor of •OH production in the considered experimental conditions.
3.2.1 The nature of the working electrode
Current densities can vary according to the considered surface as highlighted in Fig. 5. A cylindrical graphite felt of 10 mm in diameter and 12 mm in height was considered. The total geometric surface of the cylinder was 5.34 cm2. From BET analysis and the felt's density (given by the manufacturer), 0.7 m2·g−1 and 0.088 g·cm−3, respectively, the BET surface was 580 cm2 (the cylinder volume was 0.942 cm3), namely a ratio close to 109 between both surfaces. However, it can be observed that the surface given by BET measurements is most likely overestimated, since it is deduced from the adsorbed nitrogen amount, while, owing to its low size, nitrogen can penetrate pores that are not accessible to water. Another probably more realistic approach is to use cyclic voltammetry and especially the oxidation (or reduction) current of potassium ferricyanide (K3[Fe(CN)6], 4.9 mM) to estimate the electrochemical electrode surface. Indeed, the ferricyanide peak current is linked to the electrode surface through the following relation [35]:
| (1) |
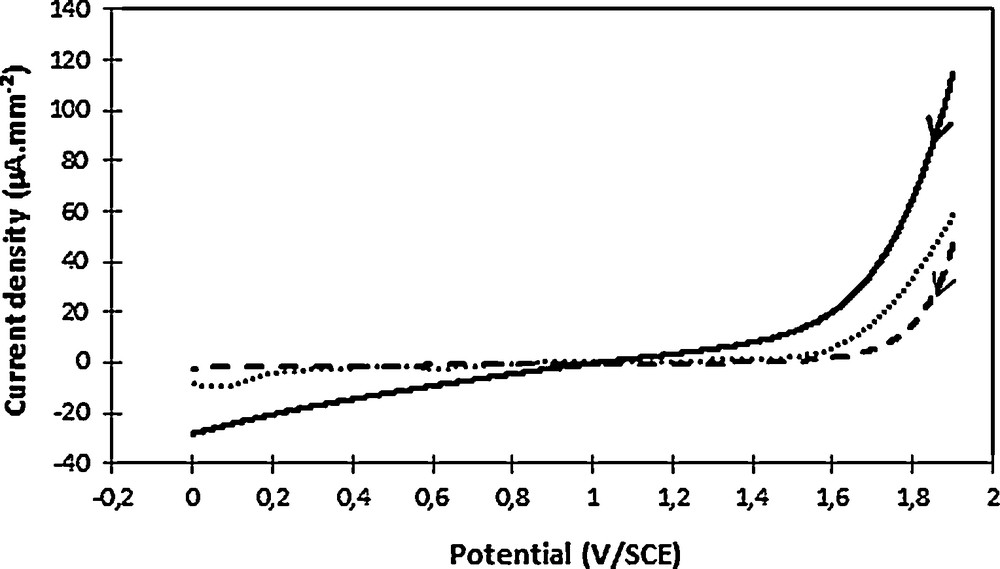
Oxygen evolution observed in phosphate buffer (pH 2.7) on various electrode materials: graphite (full line, 12.6 mm2), vitreous carbon (dashes, 7 mm2) and platinum (dots, 4.5 mm2). The scan direction was indicated by the arrow. Voltammograms were recorded at 100 mV·s−1.
A 0.5 M phosphate buffer at pH 2.7 was used and an approximate diffusion coefficient value of 0.76 × 10−5 cm2/s was considered for ferricyanide (in KCl 0.1 M at 25 °C). From this, the electrochemical surface was 55.05 cm2, namely between the geometric surface and that deduced from the BET measure.
If compared to the plane graphite electrode case, the more linear oxygen release observed on the working electrode highlighted a lower conductivity (Fig. 6). The geometric surface seems to minimize the active surface area of the electrode owing to the high current density obtained (Fig. 6). In addition to the external volume, the internal felt volume seems therefore involved in the electrochemical reaction. Contrarily, the BET surface seems to overestimate the active surface area owing to the negligible current density obtained. The electrochemical surface appears consistent if the beginning of oxygen release, occurring at 1.5–1.6 V/SCE, namely close to that observed on a plane graphite felt, is considered.
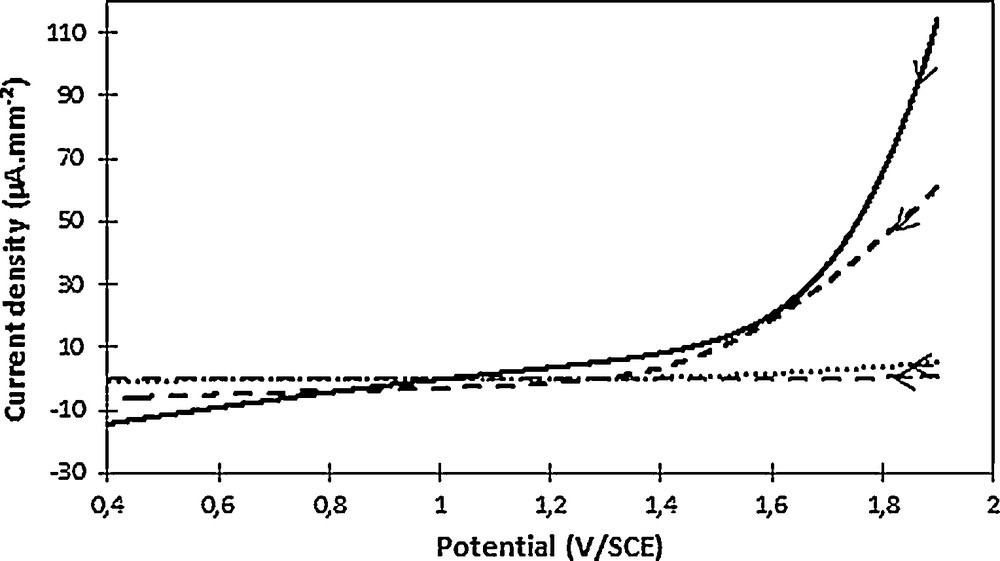
Oxygen evolution observed in phosphate buffer (pH 2.7) on a plane graphite electrode (full line, 257 mm2) and on a graphite felt for various current densities: considering the geometric surface (full dashes), the BET surface (empty dashes) and the electrochemical surface (dots). The scan direction was indicated by the arrow. Voltammogram recorded at 100 mV·s−1.
From this, the oxidation potential of water on graphite felt remains low (1.5–1.6 V/SCE), showing that it can be considered as an “active” electrode and hence the nature of the working electrode was also in favor of a direct 2,4-D oxidation rather than •OH generation.
3.2.2 The operating conditions
The electrochemical oxidation of 2,4-D was performed at 1.6 V/SCE. Only few studies involving chronoamperometry are available in the literature. However, analytical studies show that the potentials corresponding to the currents applied to BDD electrodes in view of herbicides degradation can easily exceed 3.2 V/SCE [36], namely well above the oxidation potential of water on a graphite electrode.
The applied current densities varied widely according to the electrode materials used, the operating conditions (electrolyte and pollutant concentrations, temperature…), but usually remain in the range of some tens to a hundred mA·cm−2 [37–39]. The difficulty to determine the current density applied on the graphite felt has been previously discussed. Indeed, for the graphite felt implemented in the percolation flow cell (48 mm × 12 mm), the geometric surface was 54.3 cm2 and those given by the BET and electrochemically were 13,300 and 1270 cm2. From this and for a usual current of 200 mA, the current density values were 3.7, 0.015, and 0.157 mA cm−2 for the geometric, the BET, and the electrochemical surfaces, respectively. Since the active surface should be considered with caution, the current densities appeared low if compared to those generally reported in the literature regarding AOPs, namely involving hydroxyl radicals formation (in the magnitude of some tens of mA·cm−2).
3.2.3 Dosage of hydroxyl radicals
The above arguments are based on analytical observations and assumptions; while the best proof of the formation of radicals remains their dosage. •OH are known for their strong reactivity with benzene (kinetic rate constant k = 8 × 109 L·mol−1·s−1), showing the relevance of this compound as a radical scavenger [40,41]. Since phenol is the sole product of benzene hydroxylation, the amount of •OH produced can be deduced from the HPLC monitoring of benzene degradation (λ = 200 nm) linked to phenol production (λ = 270 nm). The absence of benzene oxidation at the electrode at 1.6 V/SCE was first confirmed. Electrolysis was then performed in the flow cell with recycling of the electrolyzed solution. Neither noticeable benzene degradation nor phenol formation was observed, confirming the absence of •OH formation in the considered experimental conditions, despite the recycling of the electrolysis solution through the cell to favor phenol accumulation.
In the light of the above results, a possible involvement of •OH can be rejected in the mechanism of 2,4-D oxidation. Consequently, the proposed pretreatment can be considered as a “direct” electrochemical process instead of an advanced electrochemical oxidation process.
3.3 Feasibility of the combined process
The BOD5/COD ratio increased from 0.04 initially to 0.25 for the oxidized solution. Owing to this biodegradability improvement, and even if the limit of biodegradability (0.4 [19,42]) was not reached, a biological treatment of the pretreated solution was performed.
Prior to the biological treatment step, possible biosorption on an activated sludge of 2,4-D, on the one hand, and the major by-product, chlorohydroquinone [27], for the electrolyzed effluent, on the other hand, was assessed and found to be negligible [26].
The evolution of the mineralization, as well as of 2,4-D and of chlorohydroquinone during activated sludge culture is displayed in Fig. 7; the low 2,4-D amount (Fig. 7) should be related to its almost total removal at the end of the electrochemical pretreatment (96% removal), while the mineralization level remained limited, 34% [26]. Contrary to the behavior found for non-pretreated 2,4-D solutions, for which an acclimation period of seven days was needed before noticeable 2,4-D degradation can be observed [26], no significant lag phase can be noticed in the case of the electrolyzed effluent. Indeed, a 66% decrease of the initial DOC amount in the pretreated effluent was measured after only two days of culture (Fig. 7), without significant involvement of biosorption, illustrating a readily assimilation of some degradation products. Mineralization continued until day 5 of culture, since a 77% mineralization yield was measured. Beyond this time, no really significant decrease of the initial dissolved organic carbon was measured, since only less than 10% further mineralization was obtained after 21 days of culture, leading to a final mineralization yield during the biological treatment of 85%, namely 93% overall mineralization yield by means of the combined process. It should be observed that a biological treatment carried out in similar conditions but lacking any carbon source, especially 2,4-D, was used as a “blank” test; its DOC value, namely 17 mg·L−1, was considered as the reference (100% mineralization) [26]. Residual 2,4-D and the main degradation product, chlorohydroquinone, were also monitored, showing their total removal was completed within the first two days of biological treatment for the former and after 14 days for the latter, in agreement with the total 2,4-D removal also observed after 14 days of culture for the non-pretreated effluent [26]. Owing to the presence of a non-negligible residual DOC amount, the presence of refractory degradation products in the oxidized effluent can be assumed, which was not assimilated even after 21 days of activated sludge culture. Activated sludge acclimation to the by-products should be therefore subsequently considered to improve the biological mineralization of the electrolyzed solution.
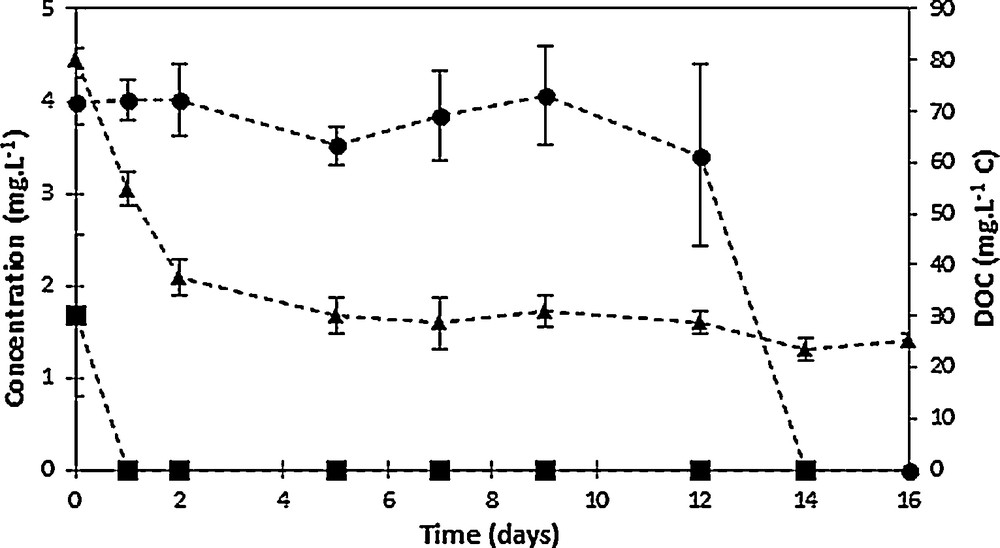
Time-courses of 2,4-D (circles), chlorohydroquinone (squares), and dissolved organic carbon (triangles, right axis) concentrations during activated sludge culture on oxidized 2,4-D solutions (five-fold dilution of a 500 mg·L−1 2,4-D solution electrolyzed at 1.6 V/SCE in 0.1 M Na2SO4).
4 Conclusion
The feasibility of an electrochemical process for pesticide pretreatment was shown, since it improved significantly the mineralization rate and thus significantly shortened the length of the biological treatment if compared to non-pretreated 2,4-D solutions. The absence of hydroxyl radical formation was demonstrated, showing that the pretreatment can be considered as a “direct” electrochemical process instead of an advanced electrochemical oxidation process. However, its selectivity was not as high as expected owing to the various degradation products identified [27] and to the biorefractory degradation products remaining at the end of activated sludge culture. Improving selectivity constitutes the main objective in the continuation of this work. For this purpose and since most of the persistent organic pollutants are halogenated compounds, the specific removal of halogen groups from the substrate could be considered. Works are in progress in the laboratory regarding the electrocatalytic reduction of carbon–halogen bonds of some halogenated target compounds, as well as the influence of the dehalogenation on the biodegradability of these compounds.
Acknowledgments
We thank Dr Elisabet Dunach-Clinet for helpful discussions.