

1 Introduction
Olefin metathesis, first reported by Banks and Bailey in 1964 [1], is an important method for the production of fine chemicals and polymers [2–4]. Supported W [5–9], Mo [10–12], and Re [13,14] catalysts have been intensively studied for olefin metathesis. Among them, Re2O7/Al2O3 exhibits higher activity and selectivity at low reaction temperatures [15,16], but the high price of Re limits its wide applications. Although WO3/SiO2 is less active than Re2O7/Al2O3 and MoO3/Al2O3, it is the most widely used in industry for the metathesis of butene and ethene due to its excellent stability and resistance to poisoning [17,18]. In a typical process, 2-butene and ethene react to produce propene, and 1-butene in the butene mixture has to first isomerize to 2-butene in order to form propene [19]. Generally, a conversion of 65–70% and a propene selectivity of 90% were obtained on WO3/SiO2 at 250–300 °C, as reported by ABB Lummus [6]. MgO [19] was used together with WO3/SiO2 to isomerize 1-butene in the butene mixture to 2-butene in industrial processes.
The pretreatment conditions of supported WO3 catalysts were found to influence the oxidation state of W, thus affecting its catalytic performance [20–24]. Choung and Weller pretreated WO3/SiO2 with N2 or H2 to form some WO3−x intermediates regarded as catalytically active species [25]. Westhoff and Moulijn found that slightly reduced WO3/SiO2 catalysts showed higher activity in metathesis than calcined samples, and a tungsten oxide composition between WO3 and WO2.95 (formed by H2 reduction of WO3/SiO2 at 875 K) exhibited the highest activity [26]. Basrur et al. inferred the formation of WO2.9 through the appearance of a blue-violet color of the used catalyst, and WO2.9 was regarded as the active species in metathesis [27]. Huang et al. reported that the activity of 10 W/Al2O3–x HY increased with the increase in the zeolite content (HY) [28]. When the zeolite content exceeded 70%, the activity decreased due to the deep reduction in W6+ species. Zaki et al. reported that WO3 could form three intermediate nonstoichiometric oxidation states (WO2.96, WO2.9, and WO2.72) by H2 reduction and the formation process could be adjusted by controlling the reduction conditions [29,30].
Although attempts have been made to explore the active species of WO3/SiO2 catalysts for the metathesis of 1-butene and ethene, there was no systematical investigation of the active species through comprehensive characterization and no systematical research about the effect of pretreatment gases on the catalytic performance. Herein, a systematical research about the change in the physicochemical properties of WO3/SiO2 pretreated by air, N2, N2/H2, and H2 was conducted by comprehensive characterization, and the catalytically active tungsten oxide phase/species were assigned. The correlation between the active tungsten oxide phase/species and the catalytic performance for the metathesis of 1-butene and ethene was concluded.
2 Experimental
2.1 Catalyst preparation
The SiO2 support (surface area 399.1 m2/g, size 3–5 mm) was obtained from Qingdao Haiyang Chemical Co. Ltd., and ammonium metatungstate (AR) was purchased from Sinopharm Chemical Reagent Co. Ltd. MgO tablets (surface area 78.13 m2/g, size 4–5 mm) were prepared by direct compression of MgO powders (99.99%) purchased from Aldrich. WO3/SiO2 (6.0 wt% WO3) was prepared by incipient-wetness impregnation of SiO2 by an aqueous solution of ammonium metatungstate followed by drying at 120 °C for 12 h and calcination in the air at 460 °C for 4 h. The obtained catalyst, denoted as WS, will be further treated by different gases in a fixed-bed catalytic reactor.
2.2 Catalyst pretreatment process and catalytic testing
Nineteen grams of WS (WO3/SiO2 without any gas pretreatment) were blended with 76.0 g MgO tablets, and then loaded into a fixed-bed reactor (internal diameter 3.3 cm). The catalyst was first heated to 400 °C in N2 with a flow rate of 1.0 L/min. At 400 °C, N2, 1/9 H2/N2 (v/v), 1/1 H2/N2 (v/v), or H2 (flow rate 1.0 L/min) were fed into the reactor to treat the catalyst for 30 min. The reactor was further heated to 550 °C and maintained at 550 °C for 2 h under N2 (flow rate 1.0 L/min). The reactor was then cooled down to 280 °C under N2 (flow rate 1.0 L/min), followed by feeding 1-butene and ethene at 280 °C and 3.0 MPa, and at a weight hourly space velocity (WHSV, 1-butene + ethene) of 1.49 h−1 with a 1.6/1.0 molar ratio of ethene/1-butene. The flow rate of ethene was monitored by a mass-flow controller, and 1-butene was pumped into the system and gasified in a preheater. The above catalysts pretreated by N2, 1/9 H2/N2 (v/v), 1/1 H2/N2 (v/v), or H2 were denoted as WS–N2, WS–1/9H2/N2, WS–1/1H2/N2, or WS–H2, respectively.
To obtain air-pretreated WO3/SiO2 (WS–air), WS blended with MgO was first heated to 400 °C in the air (flow rate 1.0 L/min). At 400 °C, air (flow rate 1.0 L/min) was fed into the reactor to treat the catalyst for 30 min. The reactor was further heated to 550 °C and maintained at 550 °C for 2 h under air (flow rate 1.0 L/min) to get rid of residual water. The reactor was then cooled down to 280 °C under air (flow rate 1.0 L/min), which was followed by feeding the reactants. The reaction conditions for WS–air were the same as those described above.
The reaction products were analyzed online using a gas chromatograph equipped with a flame ionization detector (FID), and 1-butene conversion and propene selectivity were calculated according to the literature [28,31].
2.3 Catalyst characterization
Inductively coupled plasma (ICP) analysis was performed on a PerkinElmer OPTIMA 2100 DV optical emission spectroscopy spectrometer to identify the real tungsten oxide contents in the WO3/SiO2 catalysts. Brunauer–Emmett–Teller (BET) surface areas, pore volumes, and pore sizes were obtained using a Micromeritics ASAP 2020 M adsorption apparatus. Prior to measurement, the sample was degassed under vacuum at 200 °C for 3 h. X-ray diffraction (XRD) patterns were recorded with a D8 Advance X-Ray Diffractometer using Cu Kα radiation in the 2θ range from 10° to 70°. Raman spectra were obtained using a Renishaw Raman Spectrometer equipped with a microscope (laser wavelength: 532 nm). UV–vis spectra were recorded using a PE Lambda950 with BaSO4 as a reference.
The images of transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were recorded on a Tecnai F20 Transmission Electron Microscope. The samples were prepared as follows: the catalysts were dispersed in ethanol by sonication, and a few drops of the dispersion were dropped onto a carbon-coated copper grid, which was followed by solvent evaporation in the air at room temperature.
X-ray photoelectron spectroscopy (XPS) data were recorded by a Kratos AXIS ULTRA DLD X-ray photoelectron spectrometer using Al Kα as the exciting source. Data processing was performed using CasaXPS software. The spectra were fit after linear background subtraction.
H2 temperature-programmed reduction (H2-TPR) was carried out in a quartz microreactor. The as-prepared samples (50 mg) were pretreated at 300 °C in Ar for 1 h prior to H2-TPR measurement, which was followed by a temperature-programmed reduction by 1/10 H2/Ar flow (v/v, 50 mL/min) from room temperature to 800 °C with a ramping rate of 5 °C/min. Hydrogen consumption was monitored by a thermal conductivity detector (TCD).
The infrared (IR) spectra of the samples were obtained in a transmission mode in a Bruker Tensor 27 spectrophotometer. A Pyris Diamond thermogravimetric analyzer (TGA) was used to analyze the organic residues in the used catalysts. The experimental run was carried out from 50 to 800 °C with a heating rate of 10 °C/min in flowing air (flow rate 50 mL/min).
3 Results and discussion
The BET surface areas, pore volumes, and mean pore sizes of WO3/SiO2 catalysts with an actual WO3 content of 6.0 wt% determined by ICP are summarized in Table 1. All catalysts after different gas pretreatments showed insignificant changes of BET surface areas, and a slight change in pore volume and pore size was found in 1/1H2/N2 and H2-pretreated catalysts.
Specific surface areas, pore volumes, and mean pore sizes of WO3/SiO2 catalysts pretreated by different gases.
| Catalysts | BET surface area (m2/g) | Pore volume (cm3/g) | Average pore size (nm) |
| WS–air | 332 | 0.83 | 7.5 |
| WS–N2 | 345 | 0.83 | 7.2 |
| WS–1/9H2/N2 | 344 | 0.84 | 7.3 |
| WS–1/1H2/N2 | 339 | 0.89 | 8.3 |
| WS–H2 | 349 | 0.88 | 7.8 |
XRD patterns of WO3/SiO2 catalysts are shown in Fig. 1. As shown in Fig. 1b and 1c, WS-air and WS–N2 exhibited similar XRD patterns assigned to monoclinic WO3. In contrast, WS–1/9H2/N2 (Fig. 1d) and WS–1/1H2/N2 (Fig. 1e) exhibited XRD patterns assigned to tetragonal WO3, suggesting that the thermal treatment of WS in the presence of H2/N2 mixtures induced the phase change of WO3. When pure H2 was used to pretreat WS, nonstoichiometric WO2.92 phase appeared (Fig. 1f), indicating the partial reduction of WO3 [29].

(Color online.) XRD patterns of 6.0 wt% WO3/SiO2 catalysts pretreated with different gases: (a) commercial SiO2, (b) WS–air; (c) WS–N2; (d) WS–1/9H2/N2; (e) WS–1/1H2/N2; (f) WS–H2.
XPS data of WO3/SiO2 after different gas pretreatments are shown in Fig. 2. The XPS curve fitting procedure is that of Doniach and Sunjic [32]. The binding energies of 35.9 and 38.0 eV in Fig. 2a for WS–air were assigned to 4f7/2 and 4f5/2 of W6+ (WO3), respectively [33–35]. As shown in Fig. 2b, WS–N2 exhibited the similar XPS data as WS–air; only W6+ was observed. For WS–1/9H2/N2, WS–1/1H2/N2, and WS–H2, in addition to the binding energies of W6+, the binding energies of 34.6∼35.1 eV and 36.7∼37.2 eV can be assigned to 4f7/2 and 4f5/2 of W5+ [36–39], respectively, confirming the partial reduction of a small portion of WO3 by H2 or H2/N2 pretreatment. The intensities of XPS peaks of W5+ increased with the increase in the H2 content in the gases.

(Color online.) XPS spectra of 6.0 wt% WO3/SiO2 catalysts pretreated with different gases: (a) WS–air; (b) WS–N2; (c) WS–1/9H2/N2; (d) WS–1/1H2/N2; (e) WS–H2.
Table 2 summarizes the molar percentages of W6+ and W5+ obtained by XPS for different gas-pretreated WO3/SiO2. W5+ did not exist in WS–air and WS–N2, while W5+ was observed in the catalysts pretreated by H2 or H2/N2. When the H2/N2 ratio was increased from 1/9 to 1/1, the molar percentage of W5+ increased from 7.1% to 11.3%. For WS–H2, the molar percentage of W5+ approached 17.7%. The relationship between the content of W5+ and the pretreatment gas was first reported here.
Binding energies and molar percentages of W5+ and W6+ species in WO3/SiO2 catalysts pretreated by different gases.
| Catalysts | Binding energies for W4f (eV) | W5+ (%) | W6+ (%) | |||
| W6+ 4f5/2 | W6+ 4f7/2 | W5+ 4f5/2 | W5+ 4f7/2 | |||
| WS–Air | 38.0 | 35.9 | N/A | N/A | 0.0 | 100.0 |
| WS–N2 | 38.1 | 36.0 | N/A | N/A | 0.0 | 100.0 |
| WS–1/9H2/N2 | 38.2 | 36.1 | 36.7 | 34.6 | 7.1 | 92.9 |
| WS–1/1H2/N2 | 38.2 | 36.1 | 37.2 | 35.1 | 11.3 | 88.7 |
| WS–H2 | 38.2 | 36.1 | 36.9 | 34.8 | 17.7 | 82.3 |
The WO3/SiO2 catalysts pretreated by different gases were studied by Raman spectroscopy. Raman peaks at 806, 710, and 268 cm−1 in Fig. 3, were assigned to the symmetric stretching mode of WO, the bending mode of WO, and the deformation mode of WOW in crystalline WO3, respectively [40,41]. Moreover, a band around 979 cm−1 was assigned to the OWO bond of the isolated surface tetrahedral tungsten oxide [42–44]. In this work, WS–air in Fig. 3a and WS–N2 in Fig. 3b exhibited characteristic Raman bands at 806, 710, and 268 cm−1, corresponding to crystalline WO3. For WS–1/9H2/N2 (Fig. 3c) and WS–1/1H2/N2 (Fig. 3d), the Raman peaks at 806 cm−1 became broader, and the peaks at 710 and 268 cm−1 disappeared, suggesting a gradual degradation of WO3 crystallinity [45]. For WS–H2 in Fig. 3e, all the Raman peaks of crystalline WO3 almost disappeared, which is consistent with the reported Raman spectra of WO2.9 [46]. The band at 979 cm−1 indicated the presence of isolated surface tetrahedral tungsten oxide in WS–air, WS–N2, WS–1/9H2/N2, and WS–1/1H2/N2.

(Color online.) Raman spectra of 6.0 wt% WO3/SiO2 pretreated with different gases: (a) WS–air; (b) WS–N2; (c) WS–1/9H2/N2; (d) WS–1/1H2/N2; (e) WS–H2.
UV–vis DRS spectra of WO3/SiO2 pretreated by different gases are shown in Fig. 4. All catalysts showed two absorption bands at 215 and 250 nm, assigned to isolated [WO4]2−tetrahedral species and octahedral polytungstate species, respectively [47–49]. Huang et al. and Zhao et al. ascribed the absorption between 400 and 800 nm to reduced W species such as W4+ and W5+ [28,31]. In our present work, the absence of absorption between 450 and 800 nm in Fig. 4a and 4b indicated that W species in WS–air and WS–N2 were in the W6+state, which is consistent with the XRD and XPS results. For the H2/N2 or H2-pretreated catalysts, the intensity of the band between 450 and 800 nm increased with the increase in H2 content, suggesting the increase of the content of partially reduced W species, which is consistent with the XPS results. The formation of partially reduced W species by H2 reduction of WO3/SiO2 allows the intervalence charge transfer from W5+ to W6+ in the substoichiometric WO3−x [50], which explains the observation that the color of the catalyst gradually changed from yellow (WO3) to deep blue (WO2.92).
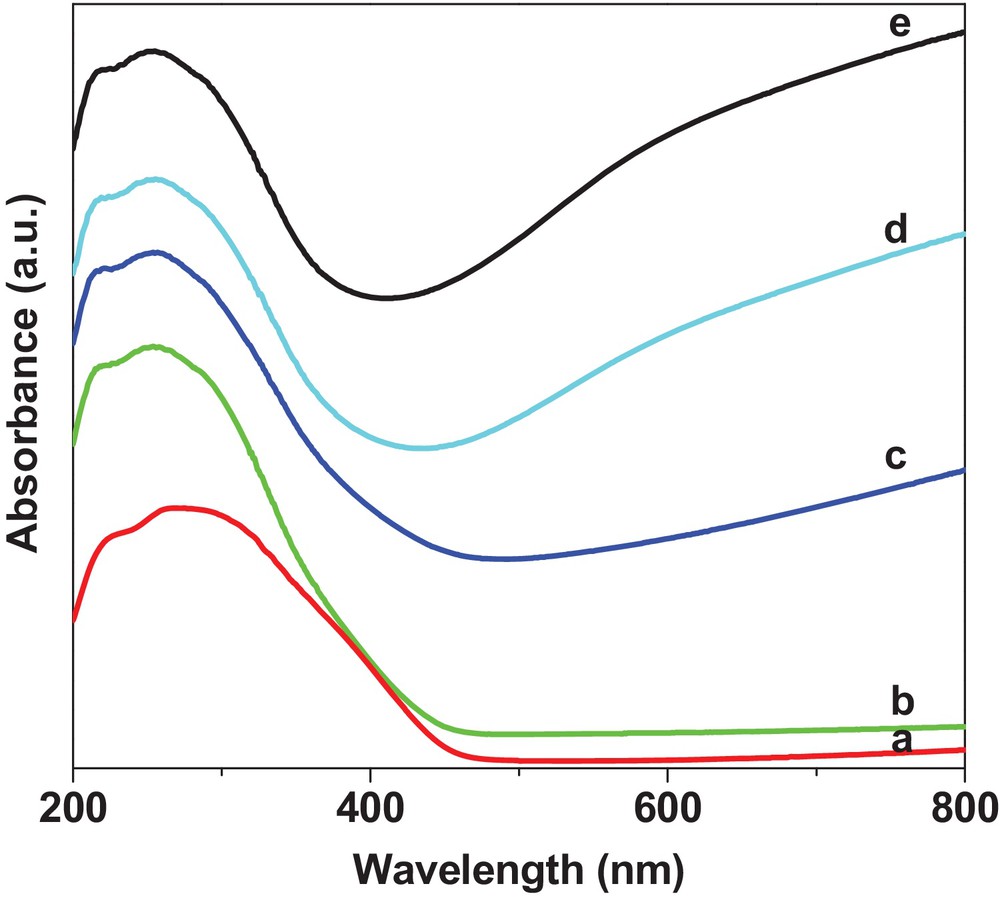
(Color online.) UV–vis spectra of 6.0 wt% WO3/SiO2 catalysts pretreated with different gases: (a) WS–air; (b) WS–N2; (c) WS–1/9H2/N2; (d) WS–1/1H2/N2; (e) WS–H2.
H2-TPR was used to monitor the reduction process of WS–air, WS–N2, and WS–1/1H2/N2. As shown in Fig. 5a, the main H2 consumption peak for WS–air was observed at 602–711 °C, with a very slight hump of H2 consumption centered at 450 °C, while the first obvious H2 consumption peak for WS–N2 in Fig. 5b was at 417–520 °C, suggesting that WS–N2 was more easily partially reduced by H2 than WS–air. For WS–1/1H2/N2 in Fig. 5c, the first H2 consumption peak was further shifted to lower temperatures starting at ca. 400 °C, indicating the easiest reducibility of WS–1/1H2/N2.
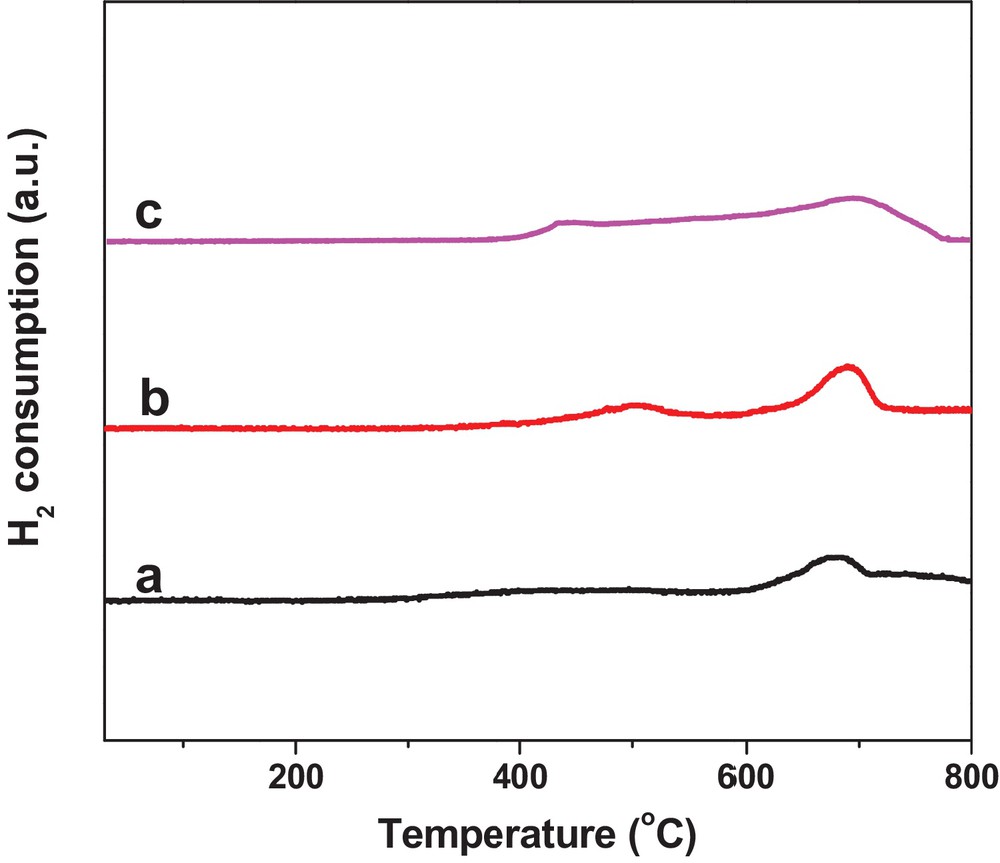
(Color online.) H2–TPR profiles of the pretreated 6.0 wt% WO3/SiO2 catalysts: (a) WS–air; (b) WS–N2; (c) WS–1/1 H2/N2.
In the literature, the deep reduction of WO3/SiO2 catalysts was reported to be unfavorable to butene and ethene metathesis [28,51]. From the H2-TPR data (Fig. 5), we can see that the deep reduction of catalysts occurred above 600 °C. That is the reason why we pretreated the catalysts (in air, N2, 1/1H2/N2, 1/9H2/N2, or H2) consistently at 400 °C. XPS data in Table 2 also confirmed that W6+ can be partially reduced to W5+ at 400 °C by H2/N2 or H2, and the ratio of W5+/W6+ can be adjusted by the ratio of H2/N2, thus providing opportunities for studying the effect of these parameters on catalytic performance.
TEM images of WS–N2 and WS–1/1H2/N2 are shown in Fig. 6a and 6b, respectively. WO3 particles were clearly observed on SiO2. The HRTEM images of WS–N2 and WS–1/1H2/N2 are shown in Fig. 6e and 6f, respectively. The lattice spacing (0.363 nm) of WS–N2 in Fig. 6e is consistent with that of (200) planes of monoclinic WO3 detected by XRD in Fig. 1c. Moreover, the lattice spacing with 0.392 nm of WS–1/1H2/N2 in Fig. 6f is consistent with that of (001) planes of tetragonal WO3 detected by XRD in Fig. 1e.

TEM (top) and HRTEM (bottom) images of WS–N2 (a, e); WS–1/1H2/N2 (b, f); the used WS–N2 (c, g); the used WS–1/1H2/N2 (d, h). Scale bars of (a), (b), (c), and (d) are 100 nm; scale bars of (e), (f), (g), and (h) are 5 nm.
The catalytic performance of WO3/SiO2 catalysts with different gas pretreatments was tested for metathesis of 1-butene and ethene in a fixed-bed reactor. The catalysts first converted 1-butene to 2-butene, and further catalyzed the metathesis of 2-butene and ethene to propene. In industrial processes, the reactant butene contains 1-butene and 2-butene, and MgO is used to convert the 1-butene fraction to 2-butene in order to obtain high product yield. In this study, WO3/SiO2 catalysts blended with MgO tablets were used for reaction testing. Below, the real catalyst systems were the WO3/SiO2 catalysts with MgO, unless otherwise specified.
To begin with, MgO was tested in the reaction to investigate the role of MgO. As shown in Fig. 7a, 34.2% conversion of 1-butene was achieved on MgO, and no propene was detected, suggesting that the only role of MgO was to convert 1-butene to 2-butene.
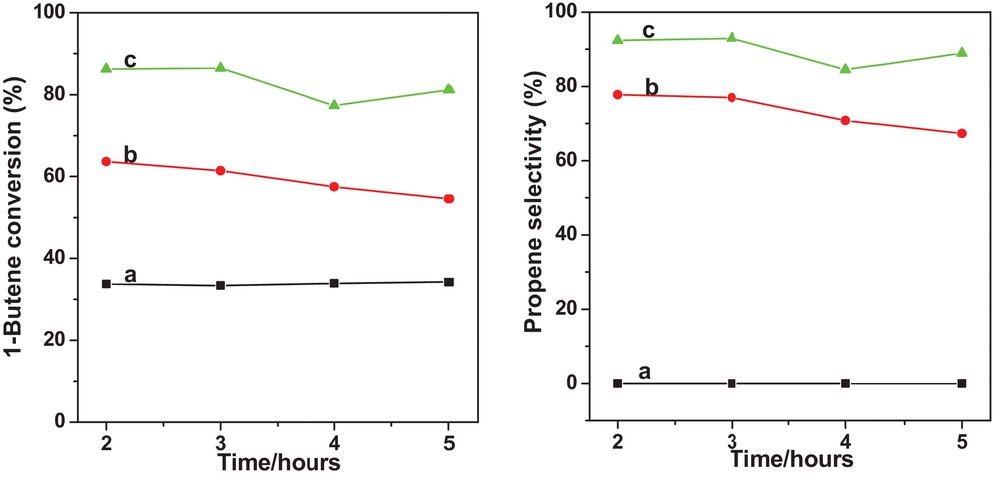
(Color online.) 1-Butene conversion (left panel) and propene selectivity (right panel) over different catalysts: (a) MgO; (b) WS–1/1H2/N2; (c) WS–1/1H2/N2 + MgO. Reaction conditions: temperature: 280 °C; pressure: 3.0 MPa; C2H4 = 0.83 L/min and 1-C4H8 = 0.52 L/min; WO3/SiO2: 19.0 g; MgO: 76.0 g.
WS–1/1H2/N2 (without MgO) showed an average conversion of about 60% and an average propene selectivity of 74% during 5 h on stream (Fig. 7b). WS–1/1H2/N2 blended with MgO exhibited an average conversion of ca. 80% and an average propene selectivity of 89% (Fig. 7c). The enhancement in 1-butene conversion for the MgO-blended WO3/SiO2 is due to the extra 1-butene conversion provided by MgO. The increase in propene selectivity with the addition of MgO is due to the fact that MgO decreases the concentration of 1-butene, thus minimizing the formation of the high-molecular-weight compound via 1-butene oligomerization [52].
The catalytic performances of WO3/SiO2 with different gas pretreatments are summarized in Table 3, in which the product was sampled for online analysis at 2 h on stream. WS–air showed 33.5% of 1-butene conversion and 0.0% of propene selectivity, nearly the same as that of MgO, suggesting that WS–air was inactive. In contrast, WS–N2 showed a conversion of 75.0% and a propene selectivity of 84.1%. All the H2- and H2/N2-pretreated catalysts exhibited enhanced 1-butene conversion and propene selectivity. Among them, WS–1/1H2/N2 exhibited the highest 1-butene conversion and propene selectivity.
Catalytic performances of MgO and WO3/SiO2 catalysts with different gas pretreatments for metathesis of 1-butene and ethene to propene.
| Catalystsa | 1-Butene conversion (%) | Selectivity (%) | |
| Propene | 2-Butene | ||
| MgO | 34.2 | 0.0 | 100.0 |
| WS–air + MgO | 33.5 | 0.0 | 100.0 |
| WS–N2 + MgO | 75.0 | 84.1 | 15.9 |
| WS–1/9H2/N2 + MgO | 77.8 | 85.8 | 14.2 |
| WS–1/1H2/N2 + MgO | 86.3 | 92.3 | 7.7 |
| WS–H2 + MgO | 83.4 | 90.5 | 9.5 |
a Reaction conditions: temperature: 280 °C; WHSV (1-butene + ethene) of 1.49 h−1; pressure: 3.0 MPa; C2H4 = 0.83 L/min and 1-C4H8 = 0.52 L/min; WO3/SiO2: 19.0 g and MgO: 76.0 g; reaction time: 2 h.
The propene yield (conversion × selectivity) of WO3/SiO2 catalysts decreased in the following order: WS–1/1H2/N2 (79.7%) > WS–H2 (75.5%) > WS–1/9H2/N2 (66.8%) > WS–N2 (63.1%) > WS–air (0.0%). Although WS–N2 and WS–air exhibited similar XRD, XPS, Raman, and UV–vis data, WS–N2 exhibited excellent 1-butene conversion and propene selectivity, whereas WS–air was inactive, indicating the formation of some active WO3 phase and W species during metathesis on WS–N2.
Postmortem analysis of the used WS–air, WS–N2, and WS–1/1H2/N2 was carried out to investigate the change in the physicochemical properties of the catalysts during the reaction. XRD patterns of the used catalysts are shown in Fig. 8. As shown in Fig. 8a, the used WS–air exhibited the same monoclinic WO3 phase as WS–air before the reaction. In contrast, the WO3 phase of WS–N2 was gradually transformed from the monoclinic phase before the reaction (Fig. 1c) to the tetragonal phase after the reaction (Fig. 8b, 8c and 8d), suggesting the phase change during the reaction. Moreover, the WO3 phase of WS–1/1H2/N2 transformed from the tetragonal phase before the reaction (Fig. 1e) to the WO2.92 phase after the reaction (Fig. 8e), suggesting that the reactants or products could reduce the WO3 phase under certain conditions. From the XRD data, it is inferred that the tetragonal WO3 or partially reduced WO3−x phase were active phases. Furthermore, air-pretreated monoclinic WO3 phase was stable and inactive, whereas the N2-pretreated monoclinic WO3 phase could be transformed into an active tetragonal WO3 phase during the reaction.
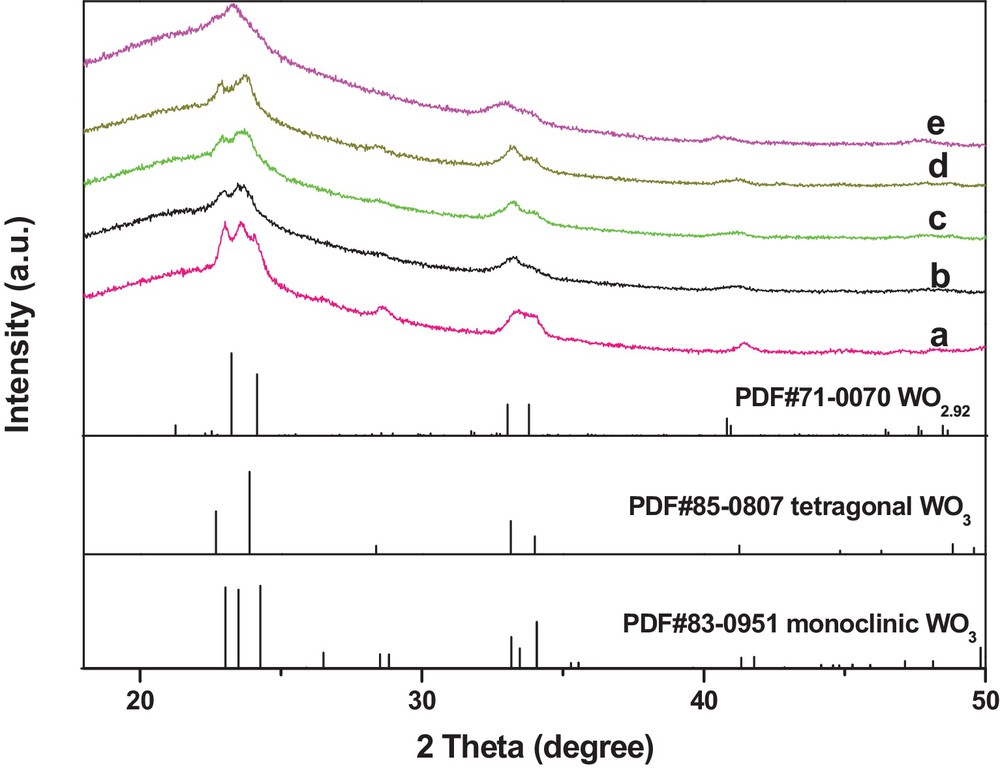
(Color online.) XRD patterns of 6.0 wt% WO3/SiO2 catalysts pretreated with different gases and collected after reaction testing: (a) the used WS–air, 10 h (after a 10-h reaction); (b) the used WS–N2, 20 min; (c) the used WS–N2, 6 h; (d) the used WS–N2, 10 h; (e) the used WS–1/1H2/N2, 10 h.
The phase change of WS–N2 and WS–1/1H2/N2 after the reaction was further confirmed by TEM studies. TEM images of the used WS–N2 and WS–1/1H2/N2 are shown in Fig. 6c and 6d, respectively, and HRTEM images of the used WS–N2 and WS–1/1H2/N2 are shown in Fig. 6 g and 6 h, respectively. The lattice spacing with 0.196 nm in Fig. 6 g was consistent with that of (002) planes of the tetragonal WO3, confirming the phase change from monoclinic WO3 before the reaction to tetragonal WO3 phase after the reaction for WS–N2. Moreover, for the used WS–1/1H2/N2, the lattice spacing of 0.380 nm in Fig. 6 h was ascribed to that of (011) planes of WO2.92, which is consistent with the phase change evidenced by the XRD pattern in Fig. 8e.
To investigate the oxidation state change of W species after the reaction, XPS studies of the used WS–air, WS–N2, and WS–1/1 H2/N2 were conducted. As shown in Fig. 9a and Table 4, the used WS–air exhibited similar XPS data with WS–air before the reaction, showing the absence of reduced W5+. However, XPS data of the used WS–N2 collected at 20 min (Fig. 9b), 6 h (Fig. 9c), and 10 h (Fig. 9d) on stream illustrated the presence of a small portion of W5+, and the molar percentage of W5+ increased from 5.0% at 20 min to 14.3% at 10 h, indicating the formation of some W5+ during the reaction. The appearance of W5+ for WS–N2 during the reaction is consistent with the H2-TPR data in Fig. 5, where WS–N2 was easier to be reduced than WS–air. Moreover, the molar percentage of W5+ of the used WS–1/1H2/N2 increased from 11.3% before the reaction in Table 2 to 17.8% after the reaction, as can be seen in Table 4, further confirming that the reactants or products in the metathesis of 1-butene and ethene can partially reduce WO3.
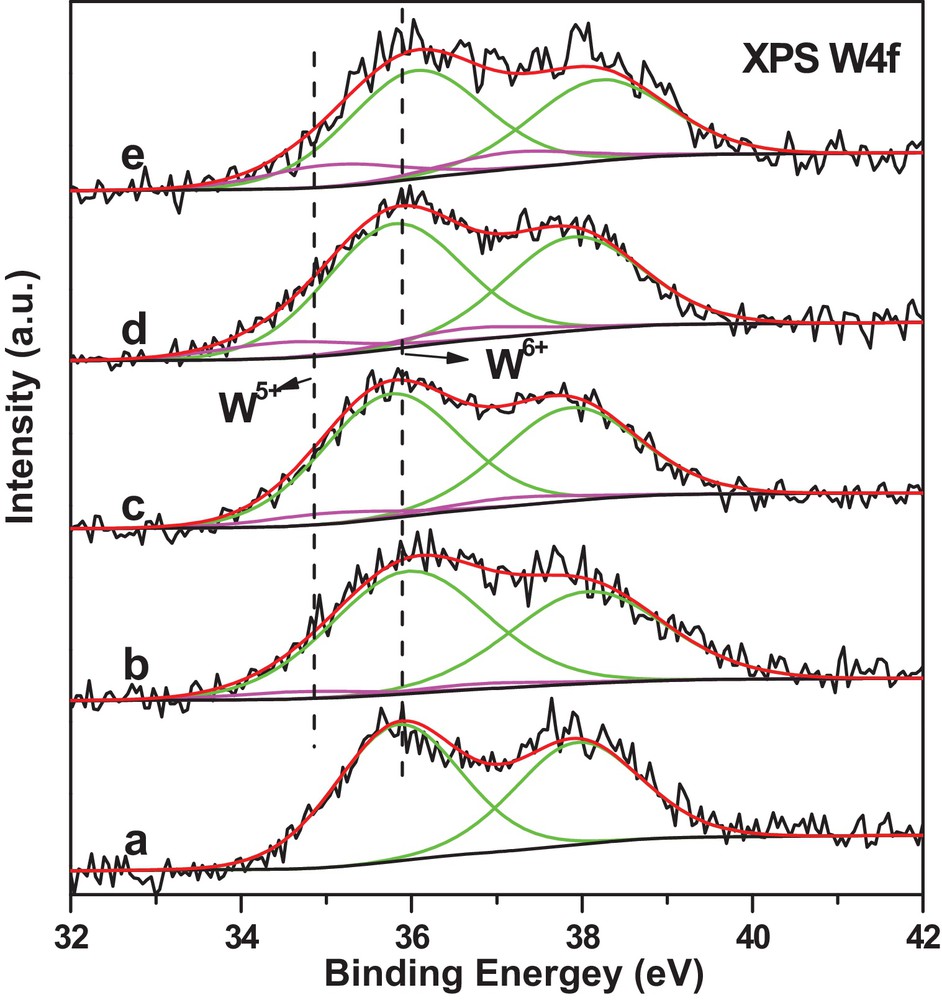
(Color online.) XPS spectra of 6.0 wt% the used WO3/SiO2 catalysts pretreated with different gases and collected after reaction testing. (a) WS–air, 10 h (after a 10-h reaction); (b) WS–N2, 20 min; (c) WS–N2, 6 h; (d) WS–N2, 10 h; (e) WS–1/1H2/N2, 10 h.
Binding energies and molar percentages of W5+ and W6+ species in the used WO3/SiO2 catalysts pretreated by different gases.
| Catalysts | Binding energies for W4f (eV) | W5+ (%) | W6+ (%) | |||
| W6+ 4f5/2 | W6+ 4f7/2 | W5+ 4f5/2 | W5+ 4f7/2 | |||
| WS–Air–10 h | 37.9 | 35.8 | N/A | N/A | 0.0 | 100.0 |
| WS–N2–20 min | 38.0 | 35.9 | 36.7 | 34.6 | 5.0 | 95.0 |
| WS–N2–6 h | 37.8 | 35.7 | 37.1 | 35.0 | 7.9 | 92.1 |
| WS–N2–10 h | 37.9 | 35.8 | 36.8 | 34.7 | 14.3 | 85.7 |
| WS–1/1H2/N2–10 h | 38.2 | 36.1 | 37.1 | 35.0 | 17.8 | 82.2 |
The W5+ content of WS–N2 was significantly increased from 0.0% to 14.3% during the 10-h reaction, while the W5+ content of WS–1/1H2/N2 was slowly increased from 11.3% to 17.8% during 10 h on stream. The significant increase in W5+ for WS–N2 may significantly influence the catalytic performance of WS–N2 during the first several hours on stream if the W5+ species were catalytically active. The catalytic studies mentioned above were carried out at 3.0 MPa and 280 °C in a 200-mL fixed-bed reactor with the 3–5-mm catalysts lumps. Under the above reaction conditions, 1-butene liquid at 3.0 MPa had to be pumped into a preheater to gasify. Because 1-butene contacted the catalyst layer later than ethene, the early stage of reaction cannot be correctly sampled and analyzed. To correctly investigate the initial stage of reaction, the reaction was carried out at 0.1 MPa and 430 °C in a 5-mL fixed-bed reactor with the 20–40 mesh catalysts. The 1-butene and ethene at 0.1 MPa were fed into the reactor in a gas state so that the early stage of the reaction can be studied.
The initial catalytic performances at 0.1 MPa of WS–N2 and WS–1/1H2/N2 in a microreactor are shown in Fig. 10. The reaction was carried out at 430 °C to clearly observe the catalytic performance change with the reaction time. As shown in Fig. 10b, 1-butene conversion and propene selectivity over WS–1/1H2/N2 increased slowly during the first 80 min. However, WS–N2 exhibited a significant growth of catalytic activity and selectivity (Fig. 10a). The phase change from monoclinic to tetragonal by postmortem XRD study and the appearance of W5+ during the reaction by postmortem XPS study can explain the significant activity growth during the early stage of reaction for WS–N2, and the tetragonal WO3 phase and W5+ were ascribed to the active phase and species. The appearance of W5+ was necessary for the formation of sufficiently active and stable W carbene species responsible for metathesis [53,54]. Under these reaction conditions, WS–N2 illustrated a better catalytic performance than WS–1/1H2/N2, suggesting that WS–N2 was more suitable for high-temperature metathesis at 0.1 MPa.
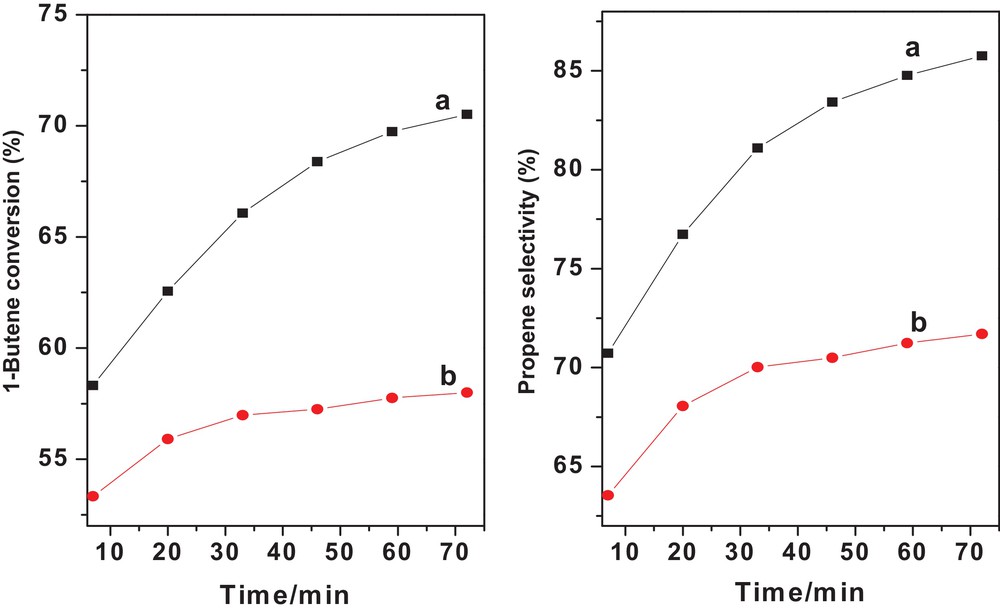
(Color online.) 1-butene conversion (left panel) and propene selectivity (right panel) over different catalysts: (a) 1 g WS–N2 + 1.5 g MgO; (b) 1 g WS–1/1H2/N2 + 1.5 g MgO. T = 430 °C; P = 0.1 MPa; ethene, 10 mL/min; 1-butene, 5 mL/min.
The reaction profiles of 1-butene conversion and propene selectivity over WS–N2 and WS–1/1H2/N2 are shown in Fig. 11. The reaction was again carried out at 3.0 MPa and 280 °C in a 200-mL fixed-bed reactor with the 3–5-mm catalysts lumps. The 1-butene conversion/propene selectivity of WS–1/1H2/N2 decreased from 86.3%/92.3% to 74.0%/83.3% after 10 h on stream. For comparison, the 1-butene conversion/propene selectivity of WS–N2 decreased from 75%/83.5% to 72.8%/82.7% after 10 h on stream.
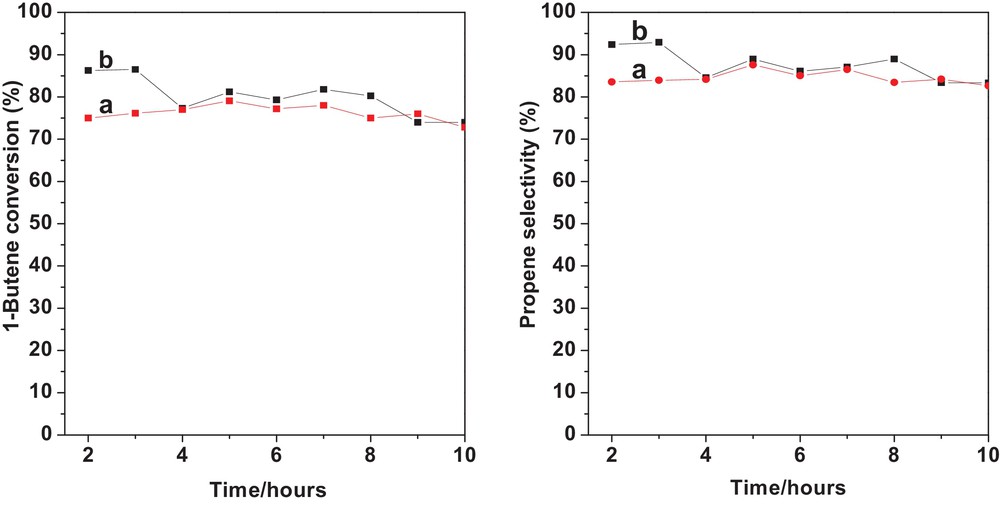
(Color online.) The reaction profiles of 1-butene conversion (left panel) and propene selectivity (right panel) of gas-pretreated catalysts: (a) WS–N2 + MgO; (b) WS–1/1H2/N2 + MgO. Reaction conditions: temperature, 280 °C; WHSV-(1-butene + ethene), 1.49 h−1; C2H4 = 0.83 L/min and 1-C4H8 = 0.52 L/min; pressure, 3.0 MPa; WO3/SiO2, 19.0 g, and MgO, 76.0 g.
It should be emphasized here that the reaction conditions in this study were more critical than those in industrial processes. Pure 1-butene was used in this work, while industrial processes used mixtures of 1-butene and 2-butene, and 2-butene was more favorable to the metathesis of butene and ethene to propene [55–58].
In the literature, the deep reduction of WOx was found to be unfavorable to metathesis [28,51]. In this study, the XRD patterns of the used WS–1/1H2/N2 revealed the formation of a WO2.92 phase after the reaction. Therefore, no deep reduction occurred.
FT-IR spectra and TGA profiles of the fresh and used WS–1/1H2/N2 are illustrated in Fig. 12. Some organic residues on the catalyst were indicated by the appearance of the antisymmetric stretching vibration of the methyl group at 2925 cm−1 in FT-IR spectra of the used catalyst in Fig. 12a [59]. Moreover, TGA analysis in Fig. 12b provides another evidence of organic residues on the used catalyst. Compared with the fresh catalyst, an additional 5.5 wt% of weight loss of the used catalyst was observed. The catalyst's deactivation is possibly due to the deposition of heavy organic compounds onto catalysts during the reaction.
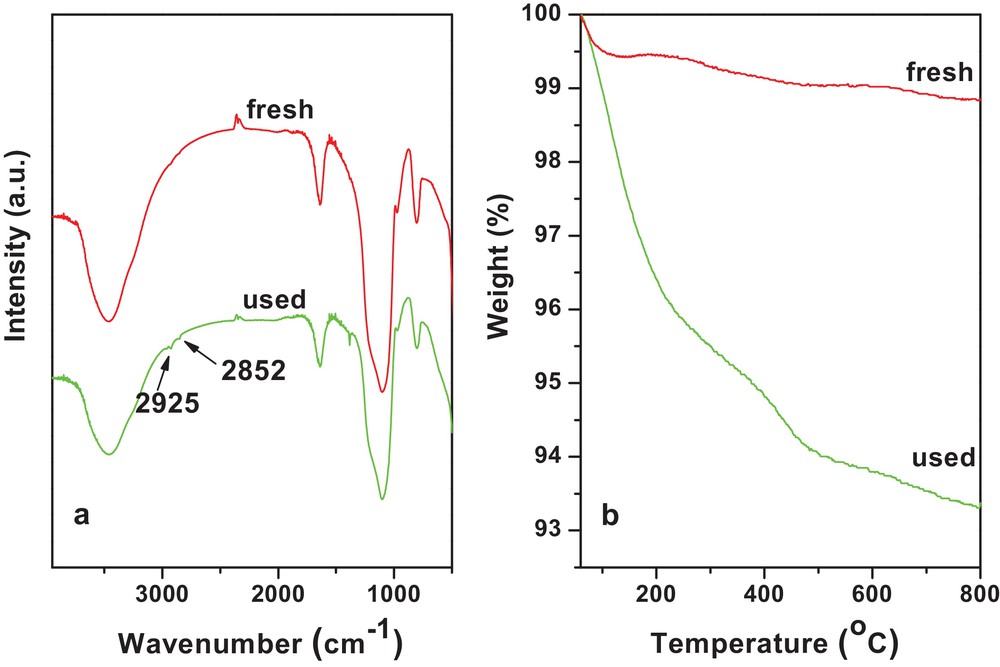
(Color online.) (a) FT-IR spectra of the fresh and used WS–1/1H2/N2; (b) TGA profiles of the fresh and used WS–1/1H2/N2.
4 Conclusions
WO3/SiO2 catalysts pretreated by air, N2, 1/9H2/N2, 1/1H2/N2 and pure H2 were characterized by XRD, N2 adsorption–desorption, Raman, UV–vis, TGA, FT-IR, TEM, XPS and H2-TPR, and tested in the metathesis of 1-butene and ethene to propene. WS–air with monoclinic WO3 phase was inactive, and the monoclinic WO3 phase cannot be transformed into an active phase during the reaction. WS–N2, WS–H2/N2, and WS–H2 showed enhanced catalytic performance. The tetragonal WO3 and partially reduced WO2.92 were active phases for the reaction. The tetragonal WO3 phase can be formed by pretreatment in H2/N2 mixtures or from the transformation of monoclinic WO3 of WS–N2 during the reaction. All active WO3/SiO2 catalysts after the reaction contained W5+ species, which are assigned to active W species.
It should be cautioned that the conclusion about the active W species and crystallographic phases may be dependent on the SiO2 nature of the support, which may not be the case with other supported catalysts. In addition, the characterization in our study was conducted ex situ, but not in situ. The interaction between the reactants/intermediates and the active sites may be dynamic during the reaction. Additional work is still needed to better understand the reaction mechanism.
As for the implication of the current work to industrial catalysis, we would like to propose that perhaps the pretreatment conditions (i.e., pretreatment gas, temperature, duration of pretreatment) should be strictly controlled to get the best performance in olefin metathesis. In addition, the option of using in situ activation of catalysts (i.e., for WS–N2 during the reaction) should be avoided, just in order to get stable activity and selectivity in industrial reactors.
Acknowledgements
The authors are grateful for the financial support from the Ministry of Science and Technology of China under Grant 2012DFA40550, and Ningbo Municipal Natural Science Foundation under Grant 2013A610038.