

1 Introduction
Esters, thioesters, and amides of hydroxy carboxylic acids serve as challenging targets to synthetic organic chemists. The activation of a carboxyl group using thionyl chloride or oxalyl chloride cannot be safely used in the presence of a free hydroxy group and initial protection and subsequent deprotection of the hydroxy group is a common requirement in the preparation of these compounds [1,2]. Some amides of substituted salicylic and naphthoic acids have several therapeutic applications and act as antiplaque [3] and anthelmintic agents [4]. Conventional methods for the synthesis of amides involve the treatment of activated derivatives of acids with the corresponding amine. Phenyl esters of salicylic acid have been effectively used for the synthesis of 2-hydroxybenzamides [5]; however, their synthesis from salicylic acids requires harsh conditions [6]. The synthesis of 2-hydroxynaphthamides has been carried out by various methods [7–10], including one starting from 2-hydroxy-3-naphthoic acid and using halogenating agents such as PCl3 [11], and SOCl2 [12]. The indirect routes have severe limitations, and are harsh [8], low yielding [10], or need very low temperatures [7,9]. Thus, preparation of these functionalized carboxamides, which also act as interesting ligands in coordination chemistry, under mild conditions with better yields, would be highly desirable for a range of applications. Unnecessary steps could be avoided if a reaction is designed in such a way that it does not need introduction and later removal of protecting groups and/or sequential separation and purification processes. Functionality tolerance, mild reaction conditions and efficient use of reagents, short reaction times and simple workup leading to the isolation of pure product are among other key factors that must be taken into account.
The carboxamide [–C(O)NH–] group, ubiquitous throughout nature in the primary structure of proteins, is an important ligand construction unit for coordination chemists. In recent years, chemists have paid special attention to the synthesis of cobalt carboxamido complexes of biological importance [13,14].
It is well known from the literature that the formation of metal complexes of weekly acidic organic ligands, in which the acid group is an amide NH, a hydroxide, or a thiol, is difficult [15]. The deprotonation of the ligand is facilitated by the addition of a base such as triethylamine, sodium hydride, or the use of the corresponding metal acetate [16–20]. A suitable alternative approach to the synthesis of these metal complexes is direct electrosynthesis [21–24]. In continuation of our interest in the synthesis of carboxamide ligands and their metal complexes [18–20], we herein report, for the first time, the robust one-pot synthesis of two new carboxamide ligands from the reaction of 4,5-dimethylphenylenediamine with unprotected salicylic acid and 2-hydroxy-3-naphthoic acid and the electrochemical synthesis of their defective dicubane mixed valance tetranuclear cobalt complexes, [Co4(μ3-OH)2(μ2-Me2hybeb)2(CH3CN)6] (1), and [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] (2) (Scheme 1). The spectroscopic properties and X-ray crystal structures of these ligands and their complexes are also presented and discussed.

Synthetic route for the complexes 1 and 2.
2 Experimental
2.1 Material and methods
All solvents and chemicals were purchased from Merck and Aldrich (Pro Analysi grade). Cobalt (Aldrich) was used as a plate (ca. 2.5 × 2.5 cm, 1.0 mm thick). Elemental analyses were performed by using a PerkinElmer 2400II CHNS–O elemental analyzer. Infrared spectra (KBr pellets) were obtained on a FT-IR JASCO 680 plus spectrophotometer. 1H NMR spectra were measured with a Bruker Avance III400 spectrometer (400 MHz). Proton chemical shifts are reported in ppm relative to Me4Si as an internal standard. Electrosynthesis was performed using a potentiostat/galvanostat (SAMA500 Electroanalyzer System).
2.2 Synthesis of the ligands
2.2.1 H4Me2hybeb, L1
A mixture of 3.10 g (10 mmol) of triphenylphosphite (TPP), 1.66 g (5 mmol) of tetrabutylammonium bromide (TBAB), 1.38 g (10 mmol) salicylic acid, and 0.68 g (5 mmol) 4,5-dimethyl-1,2-phenylenediamine in a 25-mL round-bottom flask was placed in an oil bath. The reaction mixture was heated until a homogeneous solution was formed. The solution was stirred for 1 h at 120 °C. The product was precipitated from the final viscous solution by adding 20 mL of methanol, and the resulting white solid was filtered off and washed with cold ethanol. The crude product was recrystallized from a mixture of acetone/dibutyl ether (2:1, v/v). Yield 65%. FT-IR (KBr, cm−1) νmax: 3336 (OH), 3234 (NH), 1636 (CO), 1592 (CC), 1533 (CN). 1H NMR (CDCl3, 400 MHz) δ = 11.81 (s, 2H, OH), 10.33 (s, 2H, NH), 8.01(d, J = 7.9 Hz, 2H), 7.57 (s, 2H), 7.43 (t, J = 7.8 Hz, 2H), 6.95–6.98 (m, 4H), 2.26 (s, 6H). Anal. calcd. for C22H20N2O4 (376.41): C, 70.2; H, 5.36; N, 7.44. Found: C, 70.8; H, 5.60; N, 7.60%.
2.2.2 H4Me2hyneb, L2
H4Me2hyneb was synthesized by a procedure similar to that used for H4Me2hybeb, except that 2-hydroxy-3-naphthoic was used instead of salicylic acid. The crude product was recrystallized from acetone. Yield 80%. FT-IR (KBr, cm−1) νmax: 3304 (OH), 3229 (NH), 1654 (CO), 1582 (CC), 1537 (CN). 1H NMR (CDCl3, 400 MHz) δ = 11.54 (s, 2H, OH), 10.59 (s, 2H, NH), 8.68 (s, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.65 (s, 2H) 7.52 (t, J = 7.2 Hz, 2H), 7.36 (t, J = 7.7 Hz, 2H), 7.31 (s, 2H), 2.29 (s, 6H). Anal. calcd. for C30H24N2O4 (476.52): C, 75.61; H, 5.0; N, 5.88. Found: C, 75.76; H, 4.98; N, 5.92%.
2.3 Electrochemical synthesis
The electrochemical synthesis of cobalt complexes was performed in acetonitrile solution using a platinum wire as the cathode and a cobalt foil as the anode. The ligand was initially dissolved in 70 mL of acetonitrile, and a small amount of tetraethylammonium perchlorate (ca. 10 mg) was then added to the solution as the supporting electrolyte. All syntheses were carried out at room temperature using constant current conditions. In all cases, hydrogen gas was evolved at the cathode during the electrolysis process. Under these conditions, the cell can be summarized as Co(+)/(CH3CN) + H4L/Pt(−), where H4L represents the corresponding 2-hydroxycarboxamide ligand. After completion of the electrolysis reaction, the colored solution was filtered and the filtrate was left undisturbed at room temperature to give crystalline products. The compounds were characterized as solvates of [Co4(μ3-OH)2(μ2-Me2hybeb)2(CH3CN)6] (1), and [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] (2).
2.3.1 Synthesis of [Co4(μ3-OH)2(μ2-Me2hybeb)2(CH3CN)6] (1)
Under these electrochemical conditions, the electrolysis of 70 mL of the acetonitrile solution containing H4Me2hybeb (37.7 mg, 0.1 mmol) and tetraethylammonium perchlorate (ca. 10 mg) at 15 mA for 60 min dissolved 17.0 mg of the Co sacrificial anode (Ef = 0.52 mol·F−1). The solution gradually turned red violet. After completion of the electrolysis reaction, the colored solution was filtered and the filtrate was left undisturbed at room temperature for 5 days to give the dark-red crystals suitable for X-ray crystallography. The compound was characterized as [Co4(μ3-OH)2(μ2-Me2hybeb)2(CH3CN)6].2(H2O).2(CH3CN), 1·2H2O·2CH3CN, with water inherited from solvent and/or air. Anal. calcd. for C60H62Co4N12O12 (1378.94): C, 52.3; H, 4.53; N, 12.2. Found: C, 52.9; H, 4.53; N, 12.7%. FT-IR (KBr, cm−1): νmax: 2311 (w, CN), 1598 (s, CO). It should be noted that in one experiment, crystals closely similar in appearance to 1·2H2O·2CH3CN, but with an approximate composition, 1·4CH3CN, were obtained (vide infra).
2.3.2 Synthesis of [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] (2)
Following a procedure similar to that described for complex 1, using H4Me2hyneb (47.7 mg, 0.1 mmol) and Et4NClO4 (ca. 10 mg) in 70 mL of the acetonitrile solution, 16.3 mg of the sacrificial anode were dissolved (Ef = 0.49 mol·F−1). Dark cuboidal crystals up to 1 mm in size of the product were obtained after 1 week. Satisfactory elemental analysis could not be obtained due to instant desolvation; however, an approximate crystal structure model obtained from weak X-ray single-crystal diffraction data (vide infra) confirmed the presence of the expected [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] complex analogous to 1. The analytical data for a solvate species, similar to that of complex 1, that is [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6].2(H2O).2(CH3CN), 2·2H2O·2CH3CN, are: Anal. calcd. for C76H70Co4N12O12 (1579.18): C, 57.8; H, 4.47; N, 10.64. Found: C, 56.9; H, 5.23; N, 10.6%. FT-IR (KBr, cm−1): νmax: 2302 (w, CN), 1626 (s, CO).
2.4 X-ray crystallography
Crystals for X-ray diffraction of L1 were obtained from acetone/dibutyl ether, and of L2 from acetone. Crystals of 1 were obtained from acetonitrile in the form of the solvate 1·4CH3CN and in the presence of more moisture in the form of the solvate 1·2H2O·2CH3CN. In the air, under ambient conditions, both forms decomposed quickly by desolvation. Of these two solvates, only the crystal structure of 1·2H2O·2CH3CN was studied in full detail. X-ray diffraction data were measured at T = 100 K on a Bruker Kappa APEX-2 CCD diffractometer with graphite monochromated Mo Kα (λ = 0.71073 Å) radiation. Unit cell refinement, data reduction, and absorption corrections were performed with the help of programs SAINT and SADABS [25]. The structures were solved with direct methods using program SHELXS97, and structure refinement on F2 was carried out with program SHELXL97 [26]. Anisotropic displacement parameters were used for all non-hydrogen atoms. Hydrogen atoms were inserted in idealized positions and refined riding with the atoms to which they were bonded, permitting OH and CH3 groups to rotate about their CO and CC bond axes. The acetonitrile solvent molecules in the crystal structure of 1·2H2O.2CH3CN were disordered and were therefore removed with procedure SQUEEZE of program PLATON [27], while the ordered water molecules residing in pockets of the Co complex were retained. The cavity occupied by the squeezed acetonitrile solvent molecules is centred at x,y,z = 0,½,0, has a solvent accessible void volume of 204 Å3 (one cavity per unit cell) and an electron count of 48 e compared with an expected value of 44 e for 2CH3CN [27]. The chemical formula of 1·2H2O·2CH3CN is idealized and quantities derived thereof include both kinds of solvent molecules. Molecular graphics (Figs. 1–6) were generated with program Mercury [28]. Crystal data, together with other relevant information on structure determination, are listed in Table 1. The crystal structure of 1·4CH3CN, a CH3CN solvate of 1 without lattice water, was studied provisionally (space group Pī, a = 11.86(2), b = 11.87(2), c = 12.71(2) Å, α = 73.92(2), β = 74.17(2), γ = 87.08(2)°, V = 1654(4) Å3, T = 100 K, Z = 1 for 1·4CH3CN, solvent molecules ordered), but the R-values were too large to justify a report of further details, except that the tetranuclear Co complex was virtually identical to that of 1·2H2O·2CH3CN. It was also attempted to determine the crystal structure of [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] (2). In this case, crystallization from the acetonitrile solution yielded large cuboidal solvent-rich crystals that were extremely prone to instant desolvation when removed from the mother liquor. Only very poor diffraction data could be collected with these crystals (low intensities despite large crystals diffracting only to a resolution of 0.8 Å), which showed a very big tetragonal primitive unit cell of a = 24.30, c = 31.34 Å, V = 18514 Å3, T = 100 K, Z = 8. Only a very approximate structure model could be worked out with these data. It confirmed the presence of the expected [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] complex analogous to 1 in 1·2H2O·2CH3CN, but the complexes formed a very open structure with big pores extending along [001] and large layer-like spaces parallel to (001) that were obviously filled with many disordered acetonitrile solvent molecules, making the solid unstable due to desolvation.
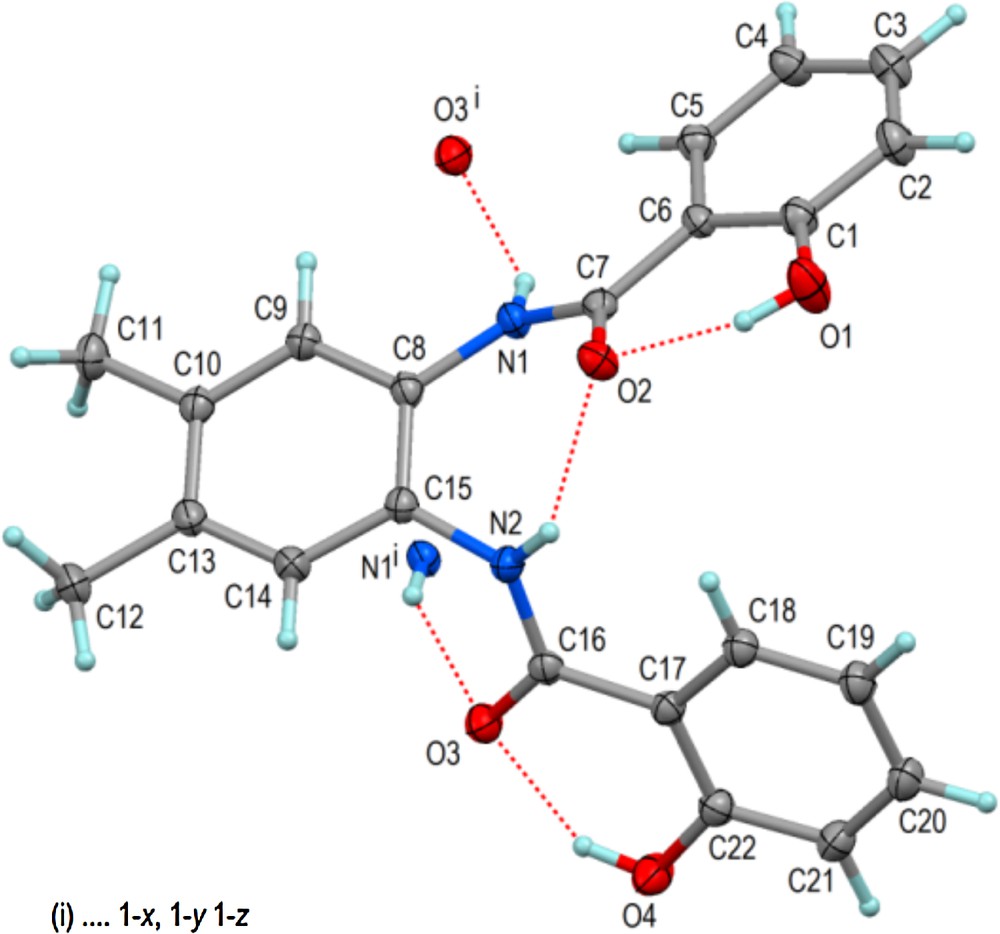
(Color online.) Molecular structure of L1 in solid state showing 60% displacement ellipsoids and hydrogen bonds including those with the centrosymmetric partner molecule. Selected bond distances and angles (Å,°): C1–O1 1.3544(12), C7–C6 1.4815(12), C7–O2 1.2561(11), C7–N1 1.3508(12), C22–O4 1.3504(12), C16–C17 1.4830(12), C16–O3 1.2512(12), C16–N2 1.3495(12); C15–C8–N1–C7 −44.80(14), C8–C15–N2–C16 −118.07(10); O1···O2 2.5658(10), O4···O3 2.5594(10), N1···O3i 3.1702(11), N2···O2 2.6617(11).

Packing diagram of L1 showing OH···O and NH···O hydrogen bonds by dashed red lines. C-bonded H atoms are omitted for clarity. The yellow field emphasizes the pairwise cyclical link of two inversion-related molecules by the hydrogen bond N1H1n···O3i (i1–x,1–y,1–z). The brown lines indicate π–π stacking between inversion-related phenyl rings C1 through C6 (plane to plane distance 3.3442(4) Å, ring slippage 2.004 Å). Green dashed lines indicate the CH···O interactions C2H2···O4(x,y,z–1) with C···O = 3.2808(16) Å and C5H5···O3(1–x,1–y,1–z) with C···O = 3.3791(14) Å. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
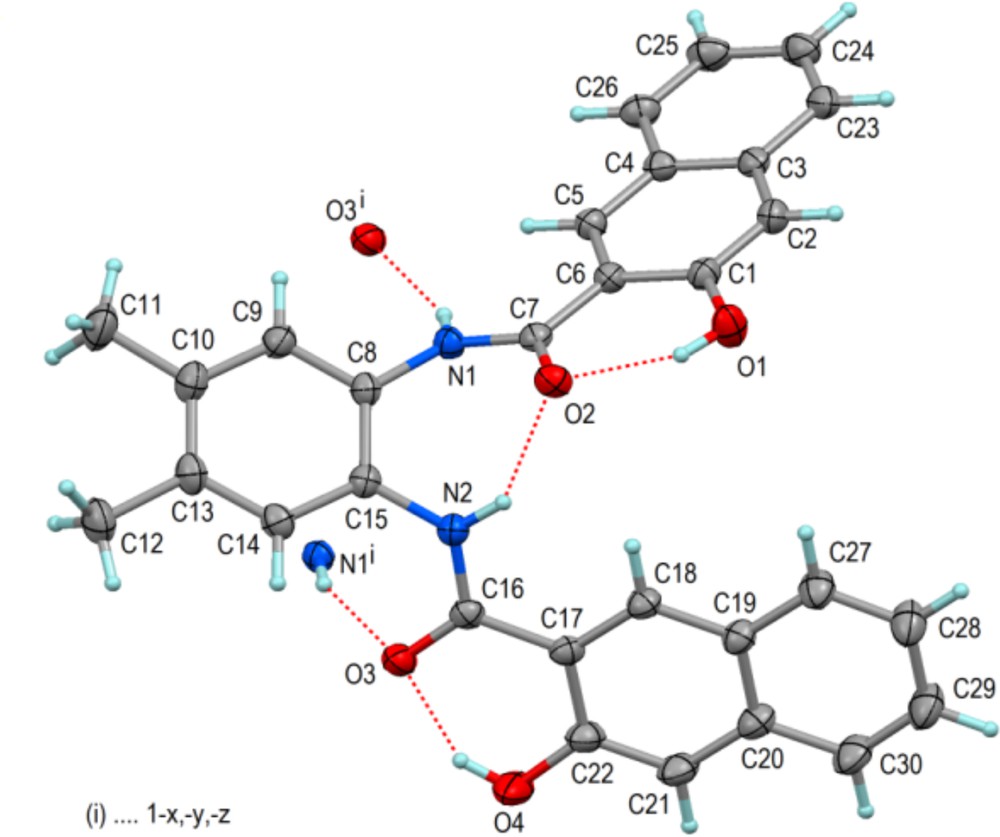
(Color online.) Molecular structure of L2 in the solid state showing 60% displacement ellipsoids and hydrogen bonds including those with the centrosymmetric partner molecule. Selected bond distances and angles (Å,°): C1–O1 1.3563(13), C7–C6 1.4861(14), C7–O2 1.2563(13), C7–N1 1.3449(13), C22–O4 1.3586(14), C16–C17 1.4849(15), C16–O3 1.2561(13), C16–N2 1.3423(14); C15–C8–N1–C7–54.01(16), C8–C15–N2–C16–135.83(11); O1···O2 2.5751(11), O4···O3 2.5461(12), N1···O3i 2.9499(12), N2···O2 2.7067(12).
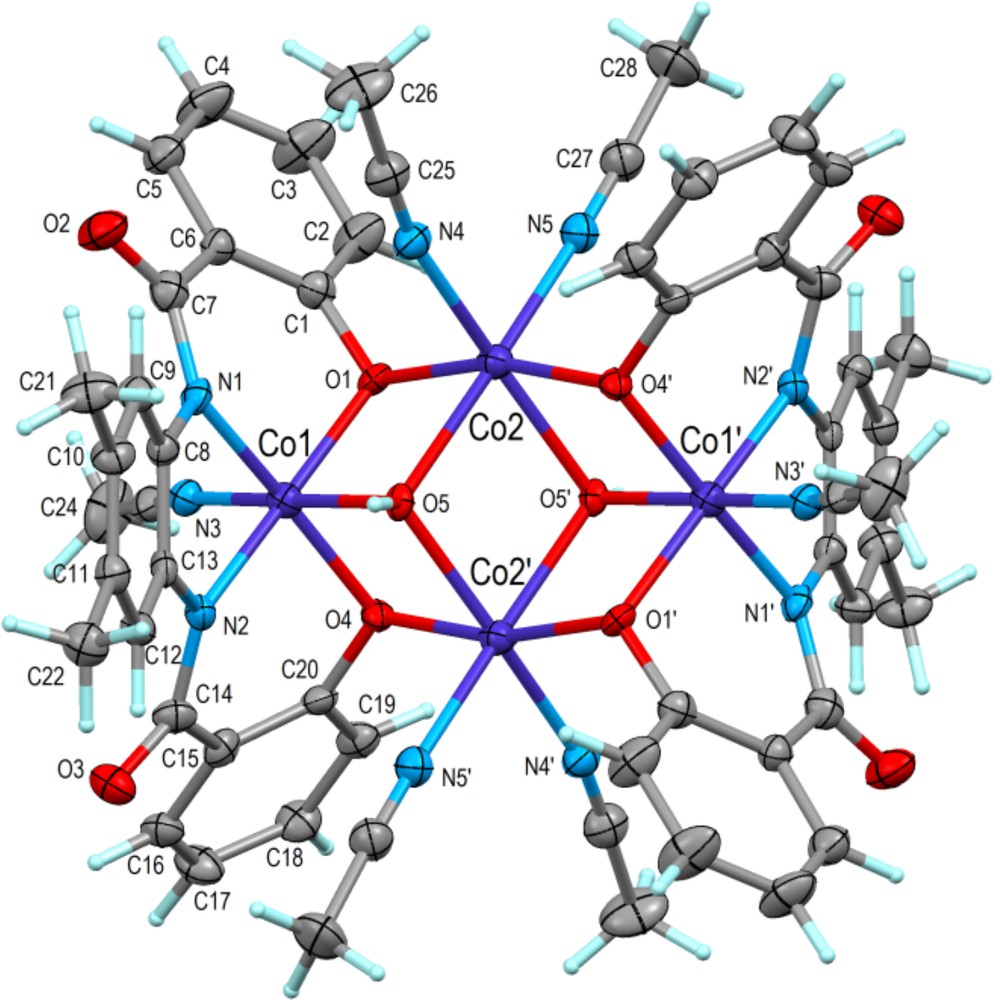
(Color online.) Molecular structure of [Co4(μ3-OH)2(μ2-L1)2(CH3CN)6] in 1·2H2O·2CH3CN showing 50% displacement ellipsoids. The complex is centrosymmetric and atoms related by inversion (1–x, 1–y, 1–z) are indicated with primes. The defective dicubane core (see text) is represented by Co1, Co2, O1, O4, O5 and their centrosymmetric equivalents.

(Color online.) Space filling diagram of the tetranuclear complex [Co4(μ3-OH)2(μ2-L1)2(CH3CN)6] in 1·2H2O·2CH3CN showing also the triclinic unit cell. View direction as in the previous figure with one OH group on the left-hand side in the central cavity of the complex right above the three oxygen atoms. The disordered CH3CN solvent molecules (not shown) are near this cavity in the crystal lattice, and the two water molecules (not shown) are between the two pairs of CH3CN ligands coordinating Co2 and Co2′.
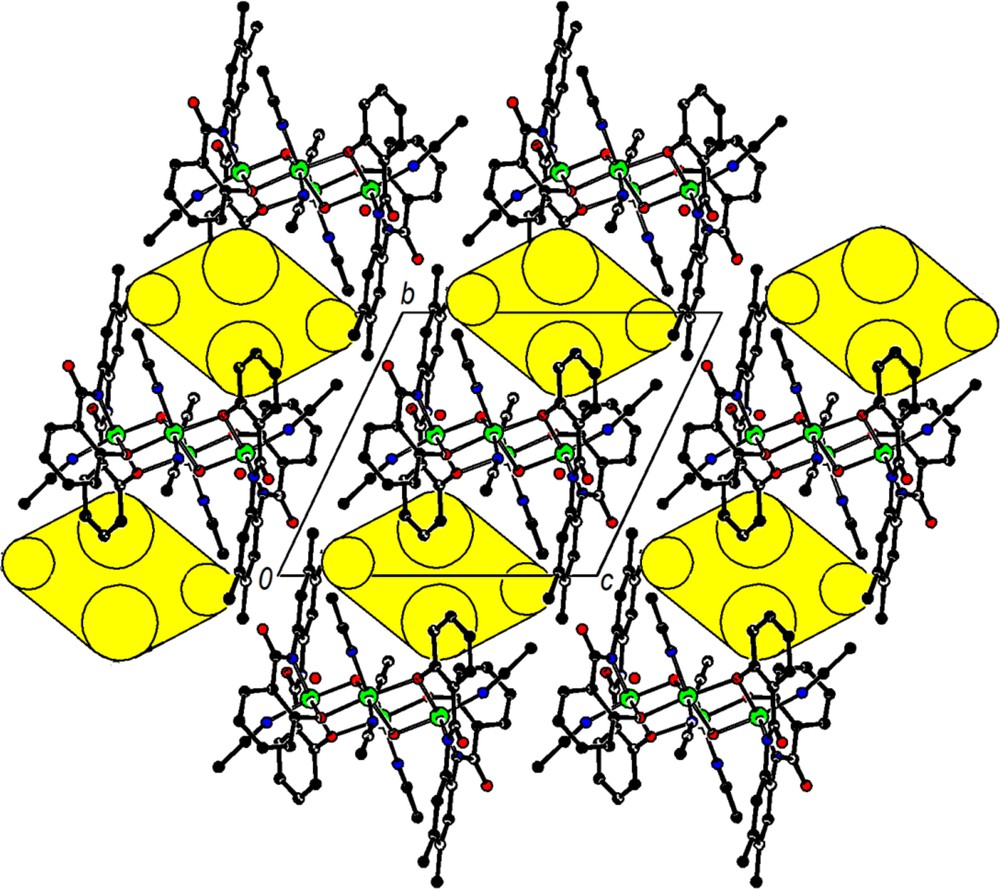
Packing diagram of [Co4(μ3-OH)2(μ3-L1)2(CH3CN)6] in 1·2H2O.2CH3CN in a view down the a-axis with ball-and-stick representation and hydrogen atoms omitted. The yellow ellipsoids centered at 0, ½, 1 and 0, 1½, 1 symbolize the outlines of the cavities housing the disordered CH3CN solvate molecules. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
Crystal data and structure refinement for L1, L2, and 1·2H2O·2CH3CN.
| Compound | L1 | L2 | 1·2H2O·2CH3CN |
| Empirical formula | C22H20N2O4 | C30H24N2O4 | C56H52Co4N10O10·2(H2O)·2(CH3CN) |
| Formula weight | 376.40 | 476.51 | 1378.94 |
| Temperature (K) | 100(2) | 100(2) | 100(2) |
| Radiation, wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | Pī | C2/c | Pī |
| a (Å) | 9.6854(2) | 27.3312(9) | 12.0549(6) |
| b (Å) | 9.9691(2) | 12.3272(4) | 12.4484(6) |
| c (Å) | 10.4536(2) | 17.8035(6) | 12.7005(6) |
| α (°) | 87.793(2) | 90.0 | 65.574(3) |
| β (°) | 63.452(2) | 125.968(2) | 63.112(3) |
| γ (°) | 85.496(2) | 90.0 | 85.976(3) |
| V (Å3) | 900.12(3) | 4854.7(3) | 1531.60(13) |
| Z | 2 | 8 | 1 |
| Dcalc (Mg/m3) | 1.389 | 1.304 | 1.495 |
| Crystal size (mm) | 0.44 × 0.35 × 0.30 | 0.40 × 0.30 × 0.20 | 0.34 × 0.20 × 0.16 |
| μ (mm−1) | 0.097 | 0.087 | 1.136 |
| F(000) | 396 | 2000 | 710 |
| θ Range (°) | 2.0–30.0 | 1.9–30.0 | 2.0–25.0 |
| Reflections collected | 19,534 | 46,455 | 16,757 |
| Independent reflections | 5208 [Rint = 0.025] | 7071 [Rint = 0.032] | 5127 [Rint = 0.047] |
| Completeness | 99.2% | 99.7% | 95.0% |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan |
| Max. and min. transmission | 0.96 and 0.89 | 0.96 and 0.85 | 0.84 and 0.66 |
| Data/restraints/parameters | 5208/0/257 | 7071/0/329 | 5127/150/375 |
| Goodness-of-fit on F2 | 1.02 | 1.04 | 1.09 |
| Final R indices [I > 2σ (I)]a | R1 = 0.0406, wR2 = 0.1116 | R1 = 0.0427, wR2 = 0.1104 | R1 = 0.0542, wR2 = 0.1372 |
| R indices (all data) | R1 = 0.0458, wR2 = 0.1179 | R1 = 0.0567, wR2 = 0.1237 | R1 = 0.0937, wR2 = 0.1575 |
| Largest difference in peak and hole (e Å−3) | 0.49 and −0.45 | 0.44 and −0.26 | 0.96 and −0.65 |
a R1 = ∑||Fo|–|Fc||/∑|Fo|, wR2 = {∑[w(Fo2–Fc2)2]/∑[w(Fo2)2]}1/2.
3 Results and discussion
3.1 Synthesis
Two new tetranuclear neutral cobalt complexes were obtained by electrochemical oxidation of cobalt metal in a cell containing an acetonitrile solution of tetradentateamido-phenolato ligands H4L. Elemental analyses indicate that cobalt ions react with the ligands in a 4:2 M ration to afford the neutral complexes [Co4(μ3-OH)2(μ2-L)2(CH3CN)6], where L represents the deprotonated tetraanionic (N2O2) ligand. These complexes were obtained, as crystalline solids, directly from the filtrate of the final electrolysis solution giving in case of 1 (incorporating L1) the solvates 1·4CH3CN or 1·2H2O·2CH3CN depending likely on the amount of water inherited from solvent and/or air. The electrochemical efficiency Ef of the cell, defined as the number of moles of metal dissolved per Faraday of charge, was close to 0.5 mol·F−1 in all cases, which is compatible with the following mechanism:
- • cathode: H4L + 4 e− → 2 H2 + L4−
- • anode: 2 Co → 2 Co2+ + 4e−
- • solution: 2 Co2+ + ½ O2 + H2O → 2 Co3+ + 2 OH−
- • solution: 2 Co2+ + 2 Co3+ + 2 L4− + 2 OH− + 10 CH3CN → [Co4(μ3-OH)2(μ2-L)2(CH3CN)6]
The oxidation of Co(II) to Co(III) ions is apparently due to the presence of atmospheric O2, and the H2O molecules from traces of water provide the bridging OH− groups. The ligands L1 and L2 and the resulting complexes were characterized by microanalysis, IR spectroscopy, and X-ray diffraction.
The IR spectra of the free ligands exhibit characteristic vibrations of OH and amide NH at 3336 and 3234 cm−1 for L1 and 3304 and 3229 cm−1 for L2, respectively. The absence of υ(O–H) and υ(N–H) vibrations in the IR spectra of the two cobalt complexes confirms that the ligands are coordinated in their deprotonated form [29]. The CO stretching vibration band of the free carboxamide ligands, appearing at 1636 cm−1 for L1 and 1654 cm−1 for L2, shifts to the lower frequencies upon coordination of the deprotonated amide, and appears at 1598 cm−1 for 1 and 1626 cm−1 for 2, which is in agreement with the data reported for the related complexes [30]. These results conform to the structural data obtained by X-ray crystallography (vide infra).
3.2 Description of the structures
The molecular structures of the ligands L1 and L2 in the solid state are presented in Figs. 1 and 3 with selected geometric data in the captions. Despite distinctly different crystal structures (Table 1), both molecules show similar features and conformations. The phenolic OH groups donate short intramolecular hydrogen bonds to the adjacent carbonyl O atoms, whereas the NH groups donate one intra- and one intermolecular hydrogen bond to carbonyl O atoms. The intermolecular NH···O hydrogen bonds link centrosymmetric molecule pairs cyclically. Apart from van der Waals interactions, further coherence of the crystal structures is provided by some CH···O and π–π-stacking interactions, as shown exemplarily for L1 in Fig. 2.
The structural diagram of 1 appears in Fig. 4 and selected bond distances and angles are listed in Table 2. It is apparent from Fig. 4 that the title tetranuclear complex consists of two centrosymmetrically related dinuclear asymmetric units, and its structure can be described as a defective dicubane with one missing corner per cube [31,32]. The common face is formed by Co(2), O(5) and related atoms Co(2′) and O(5′) [(′) = 1 − x, 1 − y, 1 − z]. The tetranuclear Co complex in this crystal structure has point symmetry–1 (Ci) with a centre of inversion at x,y,z = ½,½,½. The complex exhibits in addition a high degree of non-crystallographic local symmetry according to point group 2/m (C2h) where the 2-fold axis passes through the two Co2 atoms of the complex and the mirror plane is perpendicular to it and passing through the two Co1 atoms and the acetonitrile molecule N3–C23–C24 (Fig. 4).
Bond lengths [Å] and angles [deg] for the two independent Co atoms of [Co4(μ3-OH)2(μ2-L1)2(CH3CN)6] in 1·2H2O·2CH3CN.
| Bond distances | |||
| Co(1)–N(1) | 1.892(3) | Co(2)–O(1) | 2.050(2) |
| Co(1)–N(2) | 1.895(3) | Co(2)–O(4)#1 | 2.051(2) |
| Co(1)–N(3) | 1.925(3) | Co(2)–N(4) | 2.100(3) |
| Co(1)–O(5) | 1.911(3) | Co(2)–N(5) | 2.103(3) |
| Co(1)–O(1) | 1.929(2) | Co(2)–O(5)# | 12.106(2) |
| Co(1)–O(4) | 1.933(2) | Co(2)–O(5) | 2.109(2) |
| Mean value | 1.914 | Mean value | 2.087 |
| Bond angles | |||
| N(1)–Co(1)–N(2) | 84.82(16) | O(1)–Co(2)–O(4)#1 | 166.72(12) |
| N(1)–Co(1)–N(3) | 94.23(16) | O(1)–Co(2)–N(4) | 98.22(13) |
| N(1)–Co(1)–O(5) | 91.65(13) | O(1)–Co(2)–N(5) | 92.45(13) |
| N(1)–Co(1)–O(1) | 91.67(14) | O(1)–Co(2)–O(5)# | 193.53(11) |
| N(1)–Co(1)–O(4) | 173.63(13) | O(1)–Co(2)–O(5) | 76.22(10) |
| N(2)–Co(1)–N(3) | 94.39(16) | O(4)#1–Co(2)–N(4) | 90.73(13) |
| N(2)–Co(1)–O(5) | 91.43(13) | O(4)#1–Co(2)–N(5) | 97.25(13) |
| N(2)–Co(1)–O(1) | 174.08(13) | O(4)#1–Co(2)–O(5)# | 176.37(10) |
| N(2)–Co(1)–O(4) | 90.71(14) | O(4)#1–Co(2)–O(5) | 93.13(11) |
| N(3)–Co(1)–O(5) | 172.07(14) | N(4)–Co(2)–N(5) | 90.72(15) |
| N(3)–Co(1)–O(1) | 90.62(13) | N(4)–Co(2)–O(5)# | 1165.74(13) |
| N(3)–Co(1)–O(4) | 90.64(13) | N(4)–Co(2)–O(5) | 96.35(13) |
| O(5)–Co(1)–O(4) | 83.91(10) | N(5)–Co(2)–O(5)# | 196.83(13) |
| O(5)–Co(1)–O(1) | 83.89(10) | N(5)–Co(2)–O(5) | 167.37(13) |
| O(1)–Co(1)–O(4) | 92.39(12) | O(5)#1–Co(2)–O(5) | 78.60(12) |
The two different Co atoms of the complex show octahedral coordination figures, Co1(N3O3) and Co2(N2O4). The mean bond lengths are Co1(N,O) = 1.91(2)Å and Co2(N,O) = 2.09(3) Å, and are in good agreement with the values reported for analogous Co(II)–Co(III) complexes [31–33].
The bis-amidate-phenolate ligand (ONNO) is tetradentate and tetraanionic, because the phenolic oxygen atoms O1 and O4 and the amide nitrogen atoms N1 and N2 are clearly deprotonated. The oxygen atom O5, which links one Co1 and two Co2, is part of an OH group and not a water molecule: irrespective of whether Co1 and Co2 are di- or trivalent, the three CoO5 bonds add up to a total of about one valence bond unit [33,34], and then necessarily O5 is an OH group. The ligands of the four Co atoms of the tetranuclear complex, 2(ONNO)4− and 2OH1−, bear a total charge of 10−. Taking the Co(O,N) bond lengths and bond valence analysis as provided by program PLATON [27,34,35] into account, Co1 is trivalent (CoIII), while Co2 is divalent (CoII), and the entire complex is electroneutral.
A space filling representation of complex 1 (Fig. 5) shows that its surface has many protruding parts and deep cavities as well. This together with electroneutrality should result in a good solubility, even in relatively apolar solvents. For crystal packing (Fig. 6), the surface structure means that packing efficiency is low and solvents are needed to fill cavities in the crystal lattice. Under such conditions, any crystals are expected to form comparatively unstable solvates, which conforms to the observations. Hydrogen bonding of the two OH groups of the tetranuclear complex would be possible in principle, e.g., to CH3CN molecules, but it is not observed in the crystal structures of [Co4(μ3-OH)2(μ2-L1)2(CH3CN)6]. In cationic tetranuclear complexes of this type, it has been observed, however [31,32]. The water molecule in 1·2H2O·2CH3CN shows a short contact O6w···O2(x–1,y,z) of 2.603 Å, suggesting a hydrogen bond though hydrogen atoms of the water molecule (O6w) were not located. Three further neighbours of O6w, namely the acetonitrile CN carbon atoms C25(1–x,1–y,1–z) and C27(1–x,1–y,1–z) and the CH3 carbon C26(x–1, y, z),are at distances of 3.13 to 3.18 Å suitable for weak OH···π and CH···O interactions that may help to stabilize the crystal structure (contacts to C25 and C27 are side-on to the respective CN groups).
As an alternative to the interpretation of the tetranuclear core of the complex as a defective dicubane structure [vide supra and refs 31,32], the assembly of the four Co(N,O)6 octahedra in 1 may be considered as a lacunar fragment of the brucite layer, Mg(OH)2, in which four coordination octahedra with a diamond arrangement are joined via five shared edges with two OH groups as triple junctions. Such assemblies represent hexagonal closest packing and are comparatively common in solid-state chemistry [36]. They have been found for instance in [(Ti,Zr)4(O-alkyl)16] complexes [37], in a variety of salts of [(SbIII,BiIII)4(Cl,Br,I)16]4− anions like [Mg(CH3CN)6]2[Bi4Cl16] [38], and in a considerable variety of transition metal complexes with polyfunctional organic ligands [30,31,39].
4 Conclusion
Here, we have presented a benign synthesis of two new carboxamide ligands L1 = 1,2-bis(2-hydroxybenzamido)-4,5-dimethylbenzene (H4Me2hybeb) and L2 = 1,2-bis(2-hydroxy-3-naphthamido)-4,5-dimethylbenzene (H4Me2hyneb) from unprotected hydroxyl aromatic carboxylic acids and the first electrochemical synthesis of their hydroxo-bridged mixed valance tetranuclear cobalt complexes, [Co4(μ3-OH)2(μ2-Me2hybeb)2(CH3CN)6] (1), and [Co4(μ3-OH)2(μ2-Me2hyneb)2(CH3CN)6] (2). The X-ray crystal structures of L1, L2, and the solvate complex 1·2H2O·2CH3CN were determined. The coordination geometry around both CoII and CoIII centers in 1 and 2 are distorted octahedra. X-ray data indicates a discrete mixed valence (Co2II–Co2III) defect dicubane molecular unit where each cobalt nucleus attains a distorted octahedral geometry. The Co4 molecular unit is held together via strong bridges provided by two tetraanionic Me2hybeb4− ligand moieties forming two CoII and two CoIII cores. Lattice solvent molecules play a significant role in supramolecular architecture.
Acknowledgements
Partial support of this work by the Research Council of the University of Isfahan and Isfahan University of Technology is gratefully acknowledged. The X-ray center of the Vienna University of Technology is acknowledged for providing access to the single crystal diffractometer.