

1 Introduction
Industrial synthesis of cyclic carbonates from the corresponding alkenes is usually based on a two-step process involving the formation of an epoxide intermediate. Depending on the epoxide nature, the latter is either produced:
- • by the oxidation of alkenes with molecular oxygen in the presence of metallic silver (case of ethylene oxide) [1];
- • by the dehydrochlorination of chlorohydrins with a base;
- • by the direct epoxidation of alkenes with hydrogen peroxide in the presence of titanium silicate catalysts (Enichem process for propylene oxide production) or;
- • by a stoichiometric oxidation reaction involving peroxycarboxylic acids (without catalysts) [2,3].
As highlighted by recent detailed reviews [4–8], cyclic carbonates can be obtained at the laboratory scale by using a wide range of catalytic systems, such as ionic liquids, metal oxides, alkali metal salts, metal complexes, zeolites, organic bases, etc. At the industrial scale, the production of cyclic carbonates from epoxides and CO2 has already been implemented by BASF [9] and Chimei–Asahi Corporation [10], and the current industrial catalysts are halide salts, such as Et4NBr and KI [11].
The two-step conversion of alkenes into cyclic carbonates requires the use of a separation process with substantial energetic costs that could be avoided by the implementation of a “one-pot” synthesis of cyclic carbonates from alkenes. Due to the broad range of catalyst precursors that can be used for both reactions, a variety of combinations is possible. However, coupling alkene epoxidation and CO2 cycloaddition in the same “pot” is not trivial due to strong differences in the optimal working conditions of each step and possible side reactions. Different works dealing with the “one-pot” reaction have been published either using homogeneous [12–15] or heterogeneous [16–21] catalysts.
As far as styrene is concerned, simple catalytic systems using only one type of active phase have been tested but did not yield significant amounts of styrene carbonate. To our knowledge, the best system using a mix of CO2/O2 only afforded a styrene carbonate (SC) yield of 11%. It was proposed by Aresta et al. [17] and involved Nb2O5/NbCl5 as a catalyst. A similar yield was achieved by Sun et al. [12,16] when using tetrabutylammonium bromide (TBAB) as a catalyst and tert-butyl hydroperoxide (TBHP) as an oxidant. SC yields up to 66% were obtained with peroxides and catalytic systems based on the combination of different active phases (one for each reaction), such as Au/SiO2(epoxidation)-ZnBr2/Bu4NBr(cyclocarbonatation) with cumene hydroperoxide [19] or sodium phosphotungstate/n-Bu4NBr/NaHCO3 with H2O2 [20]. A higher styrene carbonate yield (83%) was obtained by Ono et al. [22] by coupling methyltrioxorhenium (MTO) for the epoxidation of styrene with urea–H2O2 and Zn[EMIm]2Br4 (ionic liquid) for the cyclocarbonatation reactions, while using [BMIm]BF4 as a solvent. However, due to the instability of Zn[EMIm]2Br4 in the presence of hydrogen peroxide and the negative effect of CO2 on the epoxidation, the overall reaction had to finally be performed in two steps with the addition of Zn[EMIm]2Br4 and CO2 after epoxide formation.
According to the literature, the direct transformation of alkenes into cyclic carbonates can be achieved. However, the best yields of styrene carbonate is obtained with peroxides. O2, which is the greenest oxidant, is also less reactive. This can explain why the only study dealing with the direct transformation of styrene into styrene carbonate in presence of an O2/CO2 mixture yielded 11% of the desired product. This opens the door to new investigations to find more active and selective catalytic systems. In a previous study [23], it was demonstrated that molecular oxygen can efficiently be used for the epoxidation of styrene in the presence of Mn(Salen) catalyst (59% yield). In a second study [24], we have shown that a new family of quaternary ammonium salts selectively catalyzed the conversion of styrene oxide to cyclic carbonate [best results obtained for n-cetyldimethyl(2-hydroxyethyl)ammonium bromide (HEA16Br): 84% yield]. The aim of the present work was thus to implement Mn(salen)/HEAnX catalytic systems working with O2, CO2, and alkenes. Preliminary studies dealing with our new catalytic system are detailed here under homogeneous conditions. To do so, soluble and commercially available ammonium salts, i.e. choline chloride (HEA1Cl) or HEA16Br and Mn(Salen), have been considered together for the determination of the “one-pot” reaction conditions.
2 Experimental
2.1 Reagents
Styrene oxide (97%), styrene (≥ 99%), isobutyraldehyde (≥ 99%), n-dodecane (≥ 99%), benzonitrile (99%) and choline chloride (HEA1Cl) (≥ 98%) were purchased from Sigma-Aldrich and used as received. Carbon dioxide (99.9%) and oxygen (99.5%) were supplied by Air Liquide.
2.2 Reaction 1a: Typical epoxidation reaction
In a typical epoxidation reaction, Mn(salen)Cl (16 μmol) (nStyrene/nSalen ≈ 1100) was dispersed in 40 mL of benzonitrile in a double-necked round-bottom flask (100 mL), leading to a brown solution. Then 2 mL of dodecane used as internal standard were added, as well as 2 mL of styrene (St) (17.5 mmol) and 2.28 mL of a sacrificial aldehyde, here isobutyraldehyde (IBA) (50 mmol, nSt/nIBA = 0.35). The flask was fitted under reflux and the solution was stirred and heated to 80 or 120 °C. At the desired temperature, 80 mL/min of O2 were introduced and constantly bubbled in the solution for 3 h. Aliquots (0.5 mL) of the reaction mixture were collected at 20 min, 1 h, 2 h and 3 h to monitor the reaction evolution. The samples were injected in GC and the reactants and products were followed using dodecane as the internal standard.
2.3 Reaction 1b: typical cyclocarbonatation reaction
In a typical cycloaddition reaction, styrene oxide (SO; 17.5 mmol), quaternary ammonium salt (QAS) (0.29 mmol; SO/QAS = 60), the GC internal standard (n-dodecane; 14.4 mmol) and benzonitrile (40 mL) were charged into a 160-mL stainless-steel autoclave equipped with a mechanic stirrer and an electric heater. The autoclave was then purged with N2, the temperature was raised to 120 °C and the system was pressurized with 15 bar of CO2. The reaction mixture was stirred for 23 h (from the time of CO2 introduction) and the CO2 pressure was kept constant at all times of the experiment.
2.4 Reaction 2: “one-pot” reaction
Styrene (8.7 mmol), isobutyraldehyde (25 mmol), 20 mL of benzonitrile (solvent), catalyst 1: Mn(salen) (if present: 5 mg; 7.9 μmol), catalyst 2: HEA1Cl (20.2 mg; 0.145 mmol) and internal standard n-dodecane (4.6 mmol) were introduced in the pressure reactor. Then, the reactor was purged with O2 flow for 5 min followed by introduction of 1 or 5 bar of O2 and 15 bar of CO2. The reactor was then heated to 80 °C and stirred for 3 h at this temperature. The temperature was then raised to 120 °C and maintained at this temperature for 20 h.
2.5 Analysis method
Gas chromatography was performed on a DELSI NERMAG DN200 instrument with a FID detector using a 30-m long, 0.25-mm i.d. and 0.25-μm thick capillary OPTIMA-5-MS column (Macherey-Nagel).
Styrene, styrene oxide, isobutyraldehyde and styrene carbonate amounts were obtained from the integration of their respective GC areas relatively to the one of the internal standard. Conversion, selectivity and yield were calculated as follows:
3 Results and discussion
3.1 Preliminary experiment: reaction of CO2 with an epoxidation crude mixture
The first step of the present work consisted of the study of the cyclocarbonatation reaction starting from an epoxidation crude mixture. This allowed us to look at the feasibility of the QAS-catalyzed cyclocarbonatation reaction within an overall process, i.e. in the presence of secondary oxidation products and Mn(salen).
Styrene epoxidation was firstly performed, as described in the experimental part (reaction 1a), in the presence of soluble Mn(salen) under optimal conditions (80 °C for 3 h with dioxygen bubbling at a rate of 80 mL O2/min and a isobutyraldehyde (IBA)/styrene (St) molar ratio of ≈ 3). The styrene conversion was 96% with a styrene oxide selectivity of 60% (Fig. 1A and Table 1, entry 1). The main secondary product was benzaldehyde (BZ; 13% of selectivity). A small amount of isobutyraldehyde was found in the final mixture (0.1 mmol; i.e. 0.4% of the IBA initially introduced), which means that most of isobutyraldehyde was oxidized into isobutyric acid during the epoxidation. The resulting reaction mixture was cooled down, introduced in a pressure reactor and then HEA16Br catalyst was added. In the following, a classical cycloaddition reaction was performed under 15 bar of CO2 at 120 °C, as described in the experimental section (reaction 1b).
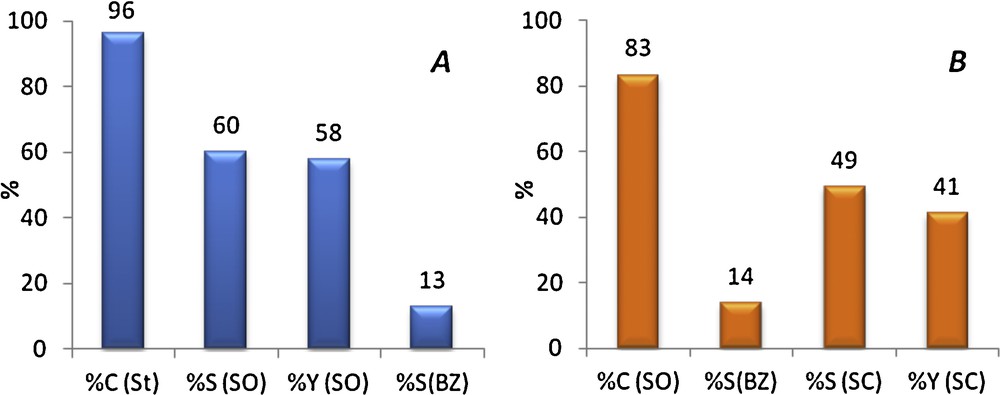
(Color online.) Catalytic results after: (A) 3 h of styrene (St) to styrene oxide (SO) conversion, then (B) 23 h of the crude mixture to styrene carbonate epoxidation (SC). BZ in (B) stands for benzaldehyde.
Comparison of styrene epoxidation results in the round-bottom flask or in the pressure reactor, after 3 h of reaction at 80 °C catalyzed by Mn(salen)a.
| Entry | Vessel | Oxygen source | Styrene conversion (%) | Styrene oxide selectivity (%) | Benzaldehyde selectivity (%) | Isobutyraldehyde conversion (%) |
| 1 | Flask | 80 mL/min | 96 | 60 | 13 | 100 |
| 2 | Pressure reactor | 1 bar O2 (15 bar CO2) | 89 | 8 | 0 | 35 |
| 3 | Pressure reactor | 5 bar O2 (15 bar CO2) | 96 | 35 | 12 | 78 |
a Reaction conditions: styrene (8.7 mmol), isobutyraldehyde (25 mmol), 20 mL of benzonitrile (solvent), catalyst Mn(salen) (5 mg; 7.9 μmol) and internal standard n-dodecane (4.6 mmol) at 80 °C for 3 h.
In a typical CO2 cycloaddition reaction to styrene oxide using HEA16Br (reaction 1b in the experimental part), the styrene oxide conversion is 98%, with 86% of carbonate selectivity. In the present case, 83% of the styrene oxide produced during the first part was converted, but with a lower styrene carbonate selectivity (49% instead of 86%) (Fig. 1B), affording a total yield of 24% of styrene carbonate (calculated as nSC,23h/nSt,0h × 100). Given the performance of the two reactions considered separately, the theoretical overall yield of styrene carbonate should have been around 49%. Then, even if the total styrene carbonate is lower than expected, this preliminary experiment clearly shows that the chosen catalytic system is able to directly transform styrene into styrene carbonate in the presence of molecular oxygen and carbon dioxide.
The decrease in selectivity is attributed to the formation of a main secondary product (noted as by-product A). GC–MS analysis using electron ionization (EI) did not allow a complete identification but partial data are compatible with a 1,2-diol monoester produced from the reaction of styrene oxide with isobutyric acid issued from isobutyraldehyde oxidation [25].
3.2 Towards a “one-pot” reaction
The next step implied the use of epoxidation and cyclocarbonatation catalysts mixtures from the beginning. Ideally, one set of reaction conditions should be used throughout the overall process, i.e. unique solvent, temperature and pressure. Commercially available, choline chloride was used as a representative soluble quaternary ammonium salt.
The separate reactions have individually been optimized in the same solvent (benzonitrile) and these conditions are described in the experimental part (Sections 2.2 and 2.3). However, the cycloaddition of CO2 was carried out under CO2 pressure and, up to now, the epoxidation of styrene was performed under an O2 flow, which implies the optimization of styrene epoxidation under static conditions in the presence of a CO2/O2 gas mixture. First experiments were then dedicated to epoxidation tests with soluble Mn(salen) catalyst under O2/CO2 static mixtures. The procedure involved O2 and 15 bar of CO2 as required by the cyclocarbonatation part of the whole reaction.
3.2.1 Preliminary study: optimization of the dioxygen pressure
Firstly, epoxidation tests were carried out under atmospheric pressure of O2 and 15 bar of CO2 (Table 1, entry 2), all other reaction parameters being equal to the bubbling condition. Styrene conversion was important (89%) but styrene oxide selectivity was very low (8%) compared to the one obtained with O2 bubbling (60%, Table 1, entry 1). So, it appears that 1 bar of O2 is not sufficient to perform the reaction at the expected extent, as also suggested by the low conversion of isobutyraldehyde (only 35%) within the time interval. Even more, although ∼ 90% of styrene was consumed, no major secondary products were detected by GC. Possible formation of styrene oligomers or/and polymers is possible but has not been proved. The fact that no styrene carbonate is formed in these conditions shows that Mn(salen) does not catalyze the cycloaddition reaction. The lower selectivity obtained in the pressure reactor can also be due to the presence of CO2 that could induce the formation of polymeric side products in the presence of Mn(salen). The oxygen pressure was thus raised to 5 bar, affording a gaseous mixture containing 20% of O2 and 80% of CO2. In this case, Table 1 (entry 3) shows that styrene oxide selectivity rose to 35%, benzaldehyde was formed with the same selectivity as in the round-bottom flask (12%) and styrene conversion was almost total. Although the obtained styrene oxide selectivity was still lower than under O2 bubbling conditions, the oxygen pressure was not incremented due to the risks of manipulating oxygen at high temperatures, namely the flammability limit of the compounds inside the reactor.
3.2.2 Single-temperature pathway
In the single-temperature pathway, the reaction mixture containing the two catalysts [HEA1Cl and Mn(salen)], isobutyraldehyde, styrene and benzonitrile was pressurized under 5 bar of O2 and 15 bar of CO2 and then heated either to 80 or 120 °C (Fig. 2 – pathway A). The results after 23 h are presented in Table 2 (entry 1 and 2) and show that at both temperatures styrene was totally consumed but no styrene carbonate was detected.
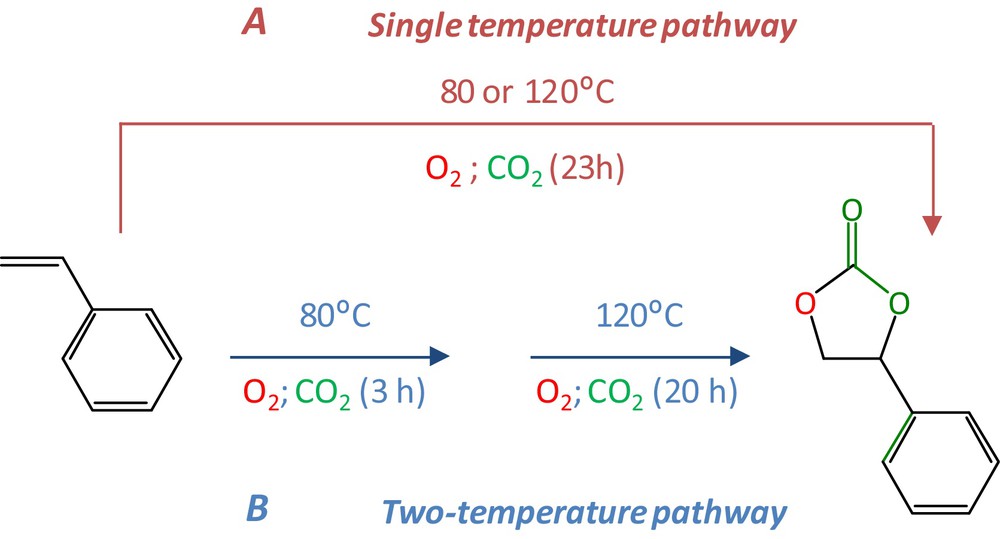
(Color online.) Single versus two-temperature pathway for the “one-pot” reaction.
Catalytic results after 23 h of the reaction of styrene carbonate formation from styrene or styrene oxide using a single-temperature pathway.
| Entry | Catalyst | T (°C) | Pressure (bar) | Substrate | Substrate conversion (%) | SO selectivity (%) | SC selectivity (%) | nIBA (mmol) –IBA %C – |
| 1 | Mn(salen) + choline | 80 | 5 O2 (15 CO2) | Styrene | 100 (St) | 25 | 0 | 0 –100 – |
| 2 | Mn(salen) + choline | 120 | 5 O2 (15 CO2) | Styrene | 100 (St) | 3 | 1 | 0– 99 – |
| 3 | Cholinea | 80 | (15 CO2) | Styrene oxide | 6 (SO) | – | 45 | – |
| 4 | Cholinea | 120 | (15 CO2) | Styrene oxide | 83 (SO) | – | 82 | – |
| 5 | Cholinea | 80 | (30 CO2) | Styrene oxide | 8 (SO) | – | 14 | – |
a Performed without isobutyraldehyde nor Mn(salen).
The quasi-absence of styrene oxide and styrene carbonate at 120 °C was expected (Table 2, entry 2). Indeed, a typical epoxidation reaction at 120 °C (reaction 1a) led to total styrene conversion, but only 6% of styrene oxide selectivity. As mentioned in previous studies, it seems that for temperatures higher than 90 °C, the epoxidation path is by-passed by the possible styrene polymerization reaction [26].
When the reaction was carried out at 80 °C (Table 2, entry 1), the styrene oxide selectivity reached 25%, but no styrene carbonate was detected. Actually, this temperature was optimized for the epoxidation reaction, but it is insufficient for the cycloaddition of CO2 to epoxides in the tested conditions. Indeed, Table 2 shows that when the cyclocarbonatation of styrene oxide was performed at 80 °C under 15 bar of CO2, using HEA1Cl as the catalyst, a styrene oxide conversion rate of only 6% was obtained (entry 3), as opposed to 83% at 120 °C (entry 4). Unfortunately, raising the CO2 pressure to 30 bar did not help to promote styrene carbonate formation at 80 °C, as seen in Table 2 (entry 5).
As expected from the separated cyclocarbonatation and epoxidation experiments, a single-temperature pathway has been proven to be inadequate.
3.2.3 Two-temperature pathway
Differences in optimal temperatures for the epoxidation (80 °C) and cyclocarbonatation steps (120 °C) are important drawbacks. However, styrene epoxidation at 80 °C is fast (total styrene conversion in 3 h) and styrene oxide cyclocarbonatation is inexistent at this temperature, which should allow the accumulation of styrene oxide for the second part of the transformation of styrene to styrene carbonate at 120 °C. This is the reason why it was decided to vary the operating temperature throughout the reaction (Fig. 2).
In this case, styrene and isobutyraldehyde (IBA) were contacted with both catalysts under 5 bar of O2 and 15 bar of CO2 in the pressurized reactor and the reaction was carried out at 80 °C for 3 h, then at 120 °C for 20 h without any separation step (Fig. 2 – pathway B). For more details, see experimental part (reaction 2).
The reaction evolution was monitored by sample collection at t ∼ 3 h, just before [3 h (80 °C)] and after [3.25 h (120 °C)] raising the temperature at 120 °C, then at t = 23 h [23 h (120 °C)]. All the corresponding results are presented in Fig. 3.
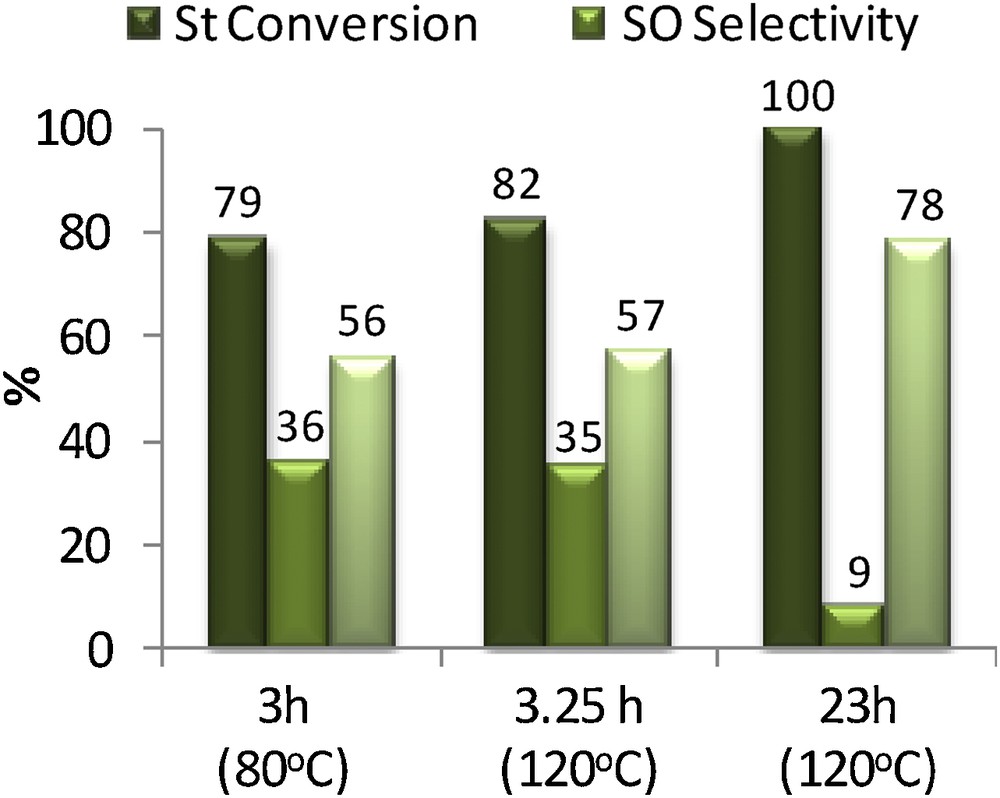
(Color online.) Catalytic results of styrene carbonate formation from styrene through the two-temperature pathway (B).
After 3 h, styrene oxide yield was 28% (St conversion 79%), isobutyraldehyde conversion was of about 56% and no carbon-dioxide-derived products were detected. The temperature was then raised to 120 °C to favor the cyclocarbonatation of styrene oxide. This procedure took about 15 min, during which the system composition did not change, as shown in Fig. 3 [3 h (120 °C)]. After 23 h, about 70% of the styrene oxide formed in the first stage was converted as well as the remaining styrene. Unfortunately, no styrene carbonate was detected at the end. Gas chromatography analyses evidenced many side products, including previously detected by-product A and a major one, noted B, formed at 120 °C. These compounds were not detected in the tests of styrene oxide cyclocarbonatation catalyzed by HEA1Cl at 120 °C for 23 h (Table 2, entry 4). EI–MS data for did not allow a careful characterization of the molecule. The mass spectrum of B is characterized by fragments with m/z = 77 and 105, which are compatible with benzoyl derivatives that could be formed through the over-oxidation of styrene oxide or styrene carbonate itself. It has to be noted that compound A itself did not hamper the production of cyclic carbonate, which means that other reasons have to be found to explain the absence here of styrene carbonate after 23 h.
Thus, a series of tests was conducted in order to understand the possible influence of the constituents of the epoxidation reaction at 120 °C on styrene oxide conversion to styrene carbonate.
3.3 Influence of epoxidation constituents on the cyclocarbonatation of styrene oxide
Blank styrene oxide cyclocarbonatation tests were performed under 15 bar of CO2 at 120 °C using HEA1Cl as a catalyst in the presence of O2, Mn(salen) as well as isobutyraldehyde (IBA) considered alone or in association.
A first series of experiments was carried out in the presence of O2 or Mn(salen) or isobutyraldehyde (Table 3, entries 2 to 5). The corresponding results are compared to those of the reference styrene oxide cyclocarbonatation test catalyzed by HEA1Cl under 15 bar of CO2 at 120 °C (test 1).
Catalytic results after 23 h of styrene oxide cyclocarbonatation blank tests performed in the presence of epoxidation constituents.
| Test | Catalyst | Atmosphere (bar) | IBA | SO conversion (%) | SC selectivity (%) | IBA conversion (%) | Secondary products | |
| CO2 | O2 | |||||||
| 1 | Choline | 15 | – | No | 83 | 82 | – | NO |
| 2 | Choline | 15 | 5 | No | 82 | 87 | – | NO |
| 3 | Choline + Mn(salen) | 15 | – | No | 94 | 100 | – | – |
| 4 | Mn(salen) | 15 | – | No | 20 | 0 | – | A and B |
| 5 | Choline | 15 | – | Yes | 96 | 74 | 9 | A and B |
| 6 | Choline + Mn(salen) | 15 | 5 | No | 78 | 94 | – | NO |
| 7 | Choline + Mn(salen) | 15 | – | Yes | 57 | 46 | 26 | A and B |
| 8 | Choline | 15 | 5 | Yes | 79 | 0 | 100 | A and B |
Test 2 in Table 3 shows that the introduction of molecular oxygen inside the reactor has no influence on the epoxide conversion or carbonate selectivity. On the other hand, the addition of Mn(salen) (Table 3, test 3) seems to have a beneficial effect with styrene oxide conversion and styrene carbonate selectivity values of almost 100%. Mn(salen) complexes are usually reported to be inactive for CO2 cycloaddition to epoxides [27,28], except for a paper published by Jutz et al. [29] dealing with the synthesis of propylene carbonate in supercritical CO2 under harsh experimental conditions, i.e. 140 °C. In fact, when the cycloaddition reaction was performed with Mn(salen) alone (test 4), 20% of styrene oxide was converted, but no carbonate was detected. Therefore, it seems that the benefits observed in test 3 are probably the result of a synergetic effect due to the presence of both HEA1Cl and Mn(salen).
Finally, test 5 indicates that the addition of isobutyraldehyde to the reference styrene oxide cyclocarbonatation reaction mixture seemed to favor epoxide conversion, while the styrene carbonate selectivity decreased. Indeed, two secondary products (A and B) were formed at the expense of the desired styrene carbonate, lowering its selectivity from 82 to 74% with a 9% IBA conversion. As a matter of fact, it appears that the presence of isobutyraldehyde is detrimental to the cyclocarbonatation reaction but IBA alone cannot explain the total absence of styrene carbonate formation in the “one-pot” conditions.
To conclude, according to the results of tests 2 to 5 (Table 3), it seems that alone, the epoxidation constituents are not responsible for hampering the styrene carbonate formation in the overall reaction following the two-temperature pathway.
In a second series of experiments, the epoxidation constituents have been introduced two at a time into the reaction mixture of the reference cyclocarbonatation test using choline chloride (HEA1Cl) as a catalyst. Both results are presented in Table 3 (entries 6–8). In the figure, overlapping areas correspond to the simultaneous presence of the components associated with each round.
The presence of both Mn(salen) and O2 (see test 6, Table 3) raises the styrene carbonate selectivity again, this time from 82% in their absence, to 94%. Indeed, whether in the absence (entry 3, 100% selectivity) or presence of O2 (entry 6, 94% selectivity), Mn(salen) always favors the selectivity for the desired product. This increase is only present with choline since, as previously shown (test 4, Table 3), Mn(salen) itself does not afford the cyclic carbonate.
When mixing both Mn(salen) and isobutyraldehyde (test 7, no dioxygen), the styrene oxide conversion dropped to 57% and the styrene carbonate selectivity to 46%. In the presence of both O2 and isobutyraldehyde [test 8, no Mn(salen)], styrene carbonate was hampered, whereas 79% of styrene oxide was converted. This decrease was not verified when the cyclocarbonatation reaction was performed with the epoxidation crude mixture (see Section 3.1) where most of the aldehyde was consumed before performing the cyclocarbonatation step (consumption of 99.6% of the IBA initially introduced). As a matter of fact, the hampering of styrene carbonate formation in the two-temperature pathway experiment must come from remaining IBA before raising the temperature to 120 °C after the epoxidation step. Isobutyraldehyde might react with oxygen during this second step until its total consumption (test 8). In fact, gas chromatography measurements showed that secondary products A and B are detected every time isobutyraldehyde is present in the reaction medium.
According to the more recently proposed mechanisms of aerobic olefin epoxidation in the presence of a metal complex and a sacrificial aldehyde, the role of the catalyst is both to initiate the radical mechanism by hydrogen abstraction from the aldehyde and to facilitate the oxygen transfer from the corresponding peroxycarboxylic acid (RCO3H) to the olefin leading to the epoxide [30]. Isobutyraldehyde is also known to autoxidize at rather high temperature in the presence of oxygen alone and without the need of a catalyst, forming an acylperoxy radical by hydrogen abstraction [31]. Both mechanisms thus state the formation of radical species that may react with the epoxide, explaining its consumption with no carbonate formation in the presence of both O2 and aldehyde [32,33].
4 Conclusions
In this work, styrene carbonate has been obtained from styrene with a 24% yield in a consecutive two-step process involving O2 bubbling and Mn(salen) to afford styrene oxide, followed by its cyclocarbonatation catalyzed by HEA16Br. To get styrene carbonate directly from styrene in “one-pot” conditions using epoxidation and cyclocarbonatation catalysts mixtures from the beginning, styrene epoxidation under O2 bubbling had to be transposed to static conditions under 5 bar of O2. The styrene epoxidation under these conditions led, however, to a decrease in styrene oxide selectivity (from 60 to 35%) and a drop of IBA conversion (from 100 to 78%). Such decrease in styrene oxide selectivity was attributed to the incomplete conversion of isobutyraldehyde due to insufficient O2 pressure.
The direct transformation of styrene to styrene carbonate at either 80 or 120 °C with both catalysts has been proven to be inadequate since no styrene carbonate was detected. In fact, at 120 °C, the epoxidation path is by-passed by the possible styrene polymerization reaction (100% of styrene conversion) and, at 80 °C, the cycloaddition of CO2 to styrene oxide was not efficient since only 6% of styrene oxide was converted. When the two steps were performed in “one-pot” conditions by increasing successively the temperature from 80 °C (3 h) to 120 °C (23 h) in the presence of both catalyst and the O2 + CO2 mixture, no styrene carbonate was formed, but many by-products with benzoyl fragments were obtained.
The simultaneous presence of isobutyraldehyde and O2 seems to be the main factor hampering styrene carbonate formation at 120 °C. Indeed, it was confirmed by several cyclocarbonatation blank tests that alone, the epoxidation constituents [O2, IBA or Mn(salen)] are not responsible for the absence of styrene carbonate in the overall transformation. On the contrary, whenever IBA is introduced, the styrene carbonate selectivity decreased and secondary products always appeared. Even more, when the reaction was performed with both IBA and O2, no styrene carbonate was formed at all, even with a styrene oxide conversion of 79%. The effect of these two constituents is probably related to a radical mechanism that is initiated at high temperatures (> 80 °C) upon the autoxidation of isobutyraldehyde.
Dioxygen/carbon dioxide mixtures are cheap and widely available, which is the reason why the direct oxidative carboxylation of alkenes in the presence of these two reactants deserves further studies. In the short term, the next step will consist in optimizing aldehyde consumption at the beginning of the whole “one-pot” process, i.e. before rising the temperature. This could be accomplished by a fine-tuning of the amount of O2 introduced, since it has been verified that a pressure of 5 bar probably limits aldehyde consumption.
Acknowledgements
The authors would like to thank Pr A. Roucoux and Dr A. Denicourt for their gift of HEA16Br. X. Zhou (UPMC student) is acknowledged for her work.