

1 Introduction
Nitrogen oxides (NOx) and soot are both present in the exhaust gases of a diesel engine. Thus, the development of the simultaneous removal technology seeks removal of soot and NOx in a single device, which is an ambitious purpose that would provide considerable advantages.
Simultaneous removal was first proposed by Yoshida et al. [1], since then, many different catalytic systems for the simultaneous removal have been investigated [2–18], however most of them were studied under laboratory conditions, using powder catalyst under tight or loose contact. Only few authors have worked with monolithic or foam structured catalysts. In a structured catalyst one can expect loose contact between the soot and the catalyst phase, similar to real contact in a Diesel Particulate Filter [19]. Under such lose contact conditions GHSV becomes an important parameter in terms of assessing catalytic activity, since it can determine the fluid-dynamic patterns and the mass transfer of reactive species in the gas and towards the catalytic layer, and its influence must therefore be carefully analyzed.
In most of the papers published about simultaneous removal, gas flow velocity used is set in a laboratory scale, orders of magnitude smaller than the real one. Wang et al. [20] studied the effect of the gas flow rate in a simultaneous removal system working with a powder catalyst and varying the flow rate from 25 to 150 mL/min. They reported that above 100 mL/min the effect of increasing the gas flow rate is negative, due to the shortened residence time of the reactive species in the catalyst bed, leading to a decrease in the NOx conversion and an incomplete combustion which increases the CO production. However, 100 mL/min is still far away from the gas flow rate found in a real diesel exhaust, which is around 800 L/min, considering a typical catalyst volume, around 1 liter, leading to a gas hourly space velocity (GHSV) around 50.000 h−1 [21,22].
In previous works [23–25] we reported high conversions in the simultaneous removal of soot and NOx for catalyst prepared with a transition metal (Cu or Co) and different alkali or alkaline-earth promoter (K, Ba or Cs), in experiments performed at GHSV of 1300 h−1. In the present work, the activity of these systems when increasing GHSV to a value close to that actually present in a vehicle is studied, in order to see the impact of this parameter on the activity of the different catalysts tested.
2 Experimental
2.1 Catalysts preparation
Supported catalysts were prepared according to the method described elsewhere [23–26]. Cordierite monoliths (2MgO·2Al2O3·5SiO2, Corning, 400 cpsi) were coated employing different alumina suspensions prepared by sol-gel synthesis, using a highly dispersible boehmite (Disperal 20, Salsol GmbH). Alumina load was 5 wt. %. Cu and Co were added to the alumina suspensions as nitrates (Cu(NO3)2·3H2O or Co(NO3)2·6H2O) together with either K or Ba or Cs (KNO3, BaC2O4 or CsNO3) as alkaline/alkaline-earth promoter. The metal to promoter ratio was set at 5:10 and the HNO3 ratio was 0.1, both in wt. % with respect to the alumina load. The mixtures were stirred for 24 h. The coating was then carried out by forced circulation of the suspensions through the channels of the monoliths, with the help of a peristaltic pump. Then, the monoliths were dried in a rotating oven at 60 °C during 24 hours, and subsequently calcined at 450 °C for 4 h.
The different composition catalysts prepared are labelled with the abbreviation of the active metal followed by the abbreviation of the alkali promoter, for example CoBa refers to the catalyst prepared with Co(NO3)2·6H2O and BaC2O4. To refer to the catalysts prepared with an active metal and the different alkali promoters the abbreviation of the metal is followed by Pr, for example, CoPr refers to the catalysts CoK, CoBa and CoCs. On the same way, MeBa refers to the catalysts CoBa and CuBa.
The physicochemical characterization of the prepared catalysts has been previously reported [23–26].
2.2 Catalytic activity
Two different series of experiments were carried out, in the absence of soot and in the presence of soot. To simulate the loading of soot in a Diesel Particulate Filter, a carbon black (Elftex 430, Cabot, SBET: 80 m2/g, primary particle mean size: 27 nm) was selected as a diesel soot surrogate. In order to load the carbon black on the catalyst surface simulating loose contact conditions, each catalyst was introduced for 1 min into a continuously stirred dispersion of 1 g of carbon black in 100 mL of n-pentane, then dried at 65 °C during 1 hour. A similar amount of soot, 300 mg, was tried to load over all the catalyst, however the actual soot load varied from 175 to 400 mg. These variations in soot loading are inherent to the deposition procedure aiming to simulate loose contact conditions but did not affect the final soot oxidation activity registered for each catalyst, i.e. catalytic tests in the presence of the same catalyst under different soot loadings yielded very similar results. Possible loss of soot during a run due to its entrainment was measured for a flow of 15 L/min of N2 during 1 hour, while heating the catalyst at 100 °C. It was confirmed that loss of soot represents maximally 5% of the initial weight loaded (200 mg).
A reactant gas containing 500 ppmv NO and 5% O2 in N2 was flowed at of 15,000 mL/min through a cylindrical monolithic catalyst (2.5 × 5 cm), corresponding to a GHSV of 36,690 h−1. Temperature programmed experiments were performed at temperatures between 250 and 650 °C, at a heating rate of 5 °C/min. The reactant gas was preheated (250–550 °C, 5 °C/min) before being fed to the catalytic reactor.
The concentrations of the different compounds in the exhaust flow gas were analyzed by the combination of different detectors: a Topaze 32 M chemiluminescence NOx analyzer for the detection of NO and NO2 and MIR2 M IR analyzers for N2O, CO, CO2 and O2. The total amount of NOx reduced is calculated as:
3 Results and discussion
3.1 Experiments in the absence of soot. NO2 formation capacity
Experimental tests in the absence of soot were performed with the aim of verifying the capacity of the catalysts to generate NO2. NO2 is a powerful soot oxidant and its formation step is considered as the key step in the mechanism of soot oxidation in presence of NOx [27].
Fig. 1 shows the evolution of NO2 produced in the presence of the different catalyst, in the absence of soot. The presence of the catalyst causes an increase in the NO2 concentration compared with the blank test. The amount of NO2 formed depends on the metal but mostly on the alkali promoter. For a given alkali promoter, Co is the metal that provides the higher activity for NOx production. Concerning the alkali promoter, we can observe that Ba and K lead to a higher NO2 production. We believe thus that the main mechanism for NO2 formation is not the direct oxidation of NO in the presence of oxygen but rather the formation of nitrates, assisted by the presence of the alkali metal. The presence of the metal can further contribute to the stability of the nitrates formed.
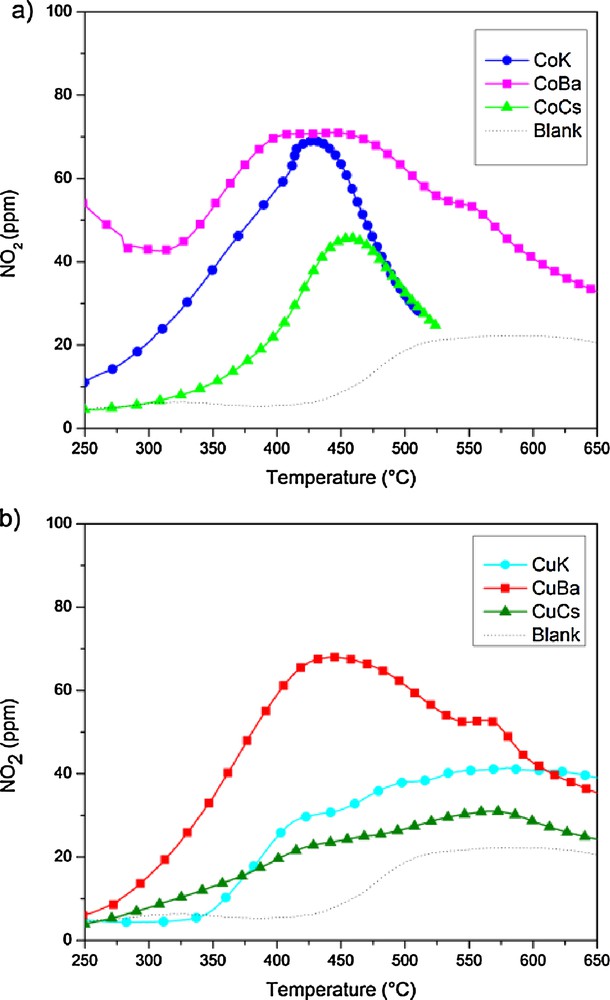
(Colour online.) NO2 production capacity of the different catalysts tested using 500 ppm NO, 5% O2 in N2, 36,690 h–1 GHSV. a: CoPr catalyst; b: CuPr catalyst.
3.2 Interaction soot/NO2
Fig. 2a shows the NOx removal and soot conversions using a CoK catalyst, at 1300 h−1 and 36,690 h−1 GHSV. When increasing the GHSV, NOx removal is dramatically affected due to the decrease of the contact time. Soot conversions also decrease but to a lesser extent. The reason is that NOx removal is calculated on the basis of the total amount of NOx or soot entering the system. Increasing GHSV from 1300 to 36,690 h−1, the amount of NOx increases 300 times. In contrast, due to the higher volume of the catalyst, the amount of soot loaded on the surface is 15 times higher for the experiments performed at 36,690 h−1 GHSV. Due to this different variation of the amount of NOx and soot, it is more appropriate to compare the results in terms of total amounts converted per gram of catalyst (Table 1). Note that, in the experiment performed with the catalyst CoBa, the CO2 production was under the detection limit of the analyzer. In these cases the values of soot oxidized given in the table is due to the CO produced, however, it is right to assume it as negligible.
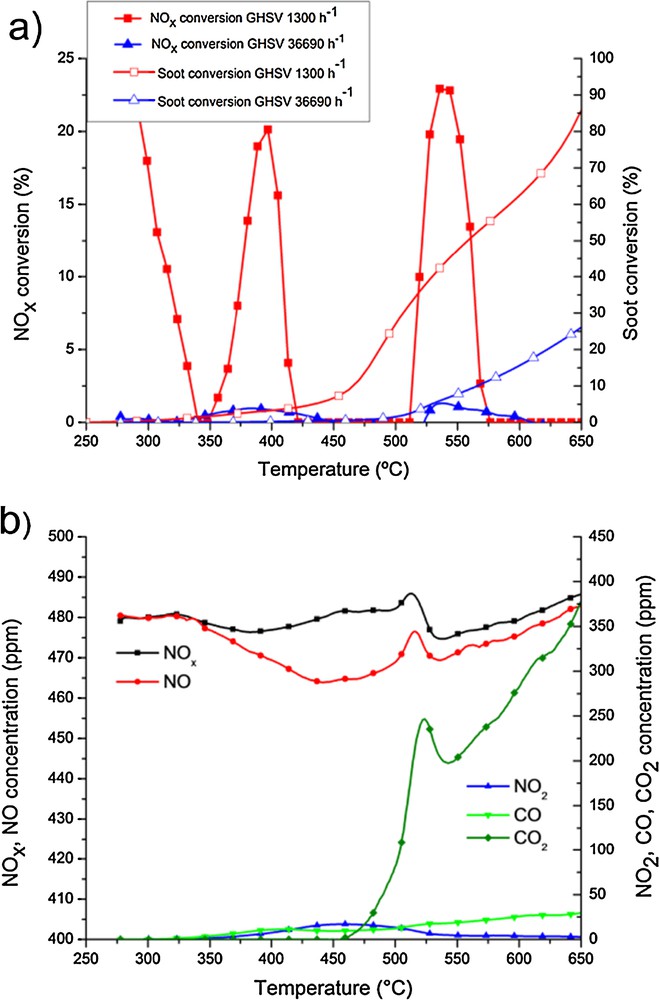
(Colour online.) CoK-containing catalyst. a: comparison of experiments performed using 36,690 h–1 and 1300 h–1 GHSV; b: evolution of the concentration of the different species when using 36,690 h–1 GHSV.
Comparisons between the experiments performed using 36,690 h–1 and 1300 h–1 GHSV.
| Catalyst | 36,690 h–1 | 1300 h–1 | ||||
| NOx reduced (mol)/catalyst | Soot oxidized (mg)/catalyst | NOx reduced (mol)/catalyst | Soot oxidized (mg)/catalyst | |||
| CoK | 0.0151 | 102.8 | 0.89 | 0.0141 | 160 | 0.98 |
| CoBa | 0.0219 | 4* | – | 0.018 | 140 | 0.97 |
| CoCs | 0.0521 | 55.5 | 0.91 | 0.0266 | 84 | 0.98 |
| CuK | 0.0164 | 25.63 | 0.73 | 0.0017 | 176 | 0.97 |
| CuBa | 0.0116 | 17.12 | 0.6 | 0.0089 | 130 | 0.98 |
| CuCs | 0.0133 | 31.78 | 0.86 | 0.0071 | 65 | 0.96 |
When increasing GHSV, similar tendencies in the reaction pathway are observed in spite of decreased soot conversion and NOx removal. As previously reported at 1300 h−1 [23], NOx concentration decreases first at temperatures between 260 and 420 °C. No N2 evolution was recorded during this period, therefore the diminution of NOx concentration within this range of temperatures is assigned to the adsorption of N-species on the catalyst surface. We presume that such species are most probably nitrates, formed through an adsorption mechanism favored by the presence of the alkali promoter.
Nitrate species are decomposed at higher temperatures in the form of NO and NO2. NOx concentration decreases again at temperatures higher than 500 °C. This low NOx concentration peak can be ascribed to the chemical reduction of this species, hence N2 evolution was measured in this same temperature interval (experiments performed using Ar as carrier gas). This maximal NOx removal is preceded by a marked evolution of NO2 that sets off the soot oxidation reaction. The peak of maximal NO2 concentration corresponds to the evolution of CO and CO2, i.e. a considerable increase in their concentration in the gas exiting the catalyst. NO evolution corresponds to the oxidation of soot in the presence of NO2. In addition to this, soot is oxidized by oxygen at high temperatures. Note that CO is formed as a by-product as a consequence of incomplete oxidation of soot. When working at 36,690 h−1 GHSV, similar evolution of the different gaseous species involved in the reaction can be observed in Fig. 2b.
Fig. 3a shows the concentration profiles of the different species present in an experiment performed using a CuK catalyst under 36,690 h−1 GHSV. NOx profile follows the same trend than in the case of CoK catalyst, less NOx are adsorbed, however similar amount of NOx are converted to N2. NO2 production capacity of the CuPr catalyst is in all the cases lower than for the CoPr catalyst. Thus, soot oxidation capacity is also lower. Fig. 3b compares the NO2 profiles obtained in experiments performed using CuK catalyst in the presence and in the absence of soot. In the presence of soot there is a decrease in the evolution of NO2 that corresponds to the beginning of soot oxidation, confirming that NO2 is directly involved in this reaction. The increase in soot oxidation due to the presence of NO2 was previously reported [27]. Systems containing Pt are very active in the oxidation of NO to NO2 and subsequently in the oxidation of soot [27,28]. However, when catalysts are based on Pt, only partial reduction of NO2 to NO was claimed to occur. Whereas in the presence of other systems [1–20,23–25], successful NOx conversion to N2 is possible. The reaction pathway for this reaction is still not clear. In the presence of a perovskite catalyst, a reaction pathway for the combined removal of NOx and soot was proposed by Fino et al. [4]. Two mechanisms take place: soot combustion causing oxygen vacancies in the crystalline catalyst structure (two surfaces adjacent oxygen vacancies serve as active sites for the chemisorption of two NO molecules, this allows us to fill up the oxygen vacancies to facilitate the generation of a molecule of N2), accompanied by the adsorption of NO on the surface (of both the catalyst and the carbon material), and subsequent reduction to N2. However, we cannot provide here further results confirming this reaction pathway.
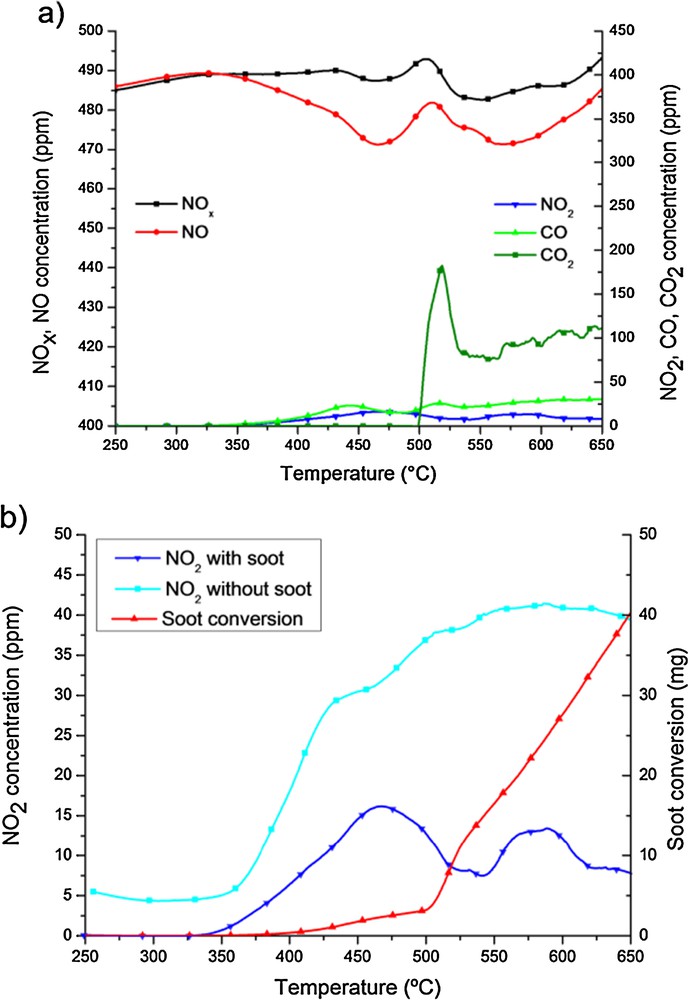
(Colour online.) CuK-containing catalyst. a: evolution of the concentration of the different species in the experiment performed using 36,690 h–1 GHSV; b: comparison of NO2 evolution in the experiments performed in the presence and in the absence of soot.
At 1300 h−1 GHSV MeBa catalysts showed lower soot oxidation activity than MeK catalyst [24], however at 650 °C, 70% soot oxidation and 60% NOx removal were reached. It was concluded that the presence of Ba enhanced the adsorption of NOx with higher thermal stability, resulting in its desorption at higher temperatures. A study performed by Materrese et al. [29] working with Pt–K and Pt–Ba catalysts found different thermal stability for nitrates formed over Ba; as a result, the activity for soot combustion was lower. Pisarello et al. [30] also reported the formation of more stable nitrate species over Ba/CeO2 than over K/CeO2 catalyst, resulting in a lower activity for the simultaneous removal of soot and NOx.
Fig. 4 shows the experiments carried out in the absence (a), and in the presence of soot (b), using a CoBa catalyst at 36,690 h−1 GHSV. The evolution of NOx shows a desorption peak at temperatures higher than 550 °C. It is important to notice for the MeBa catalysts that NO2 is produced in the same range of temperatures than for the MeK catalyst. However, the NO2 concentration profile (Fig. 4) is nearly the same in the absence and in the presence of soot which means that the catalyst hardly promotes soot oxidation by NO2.
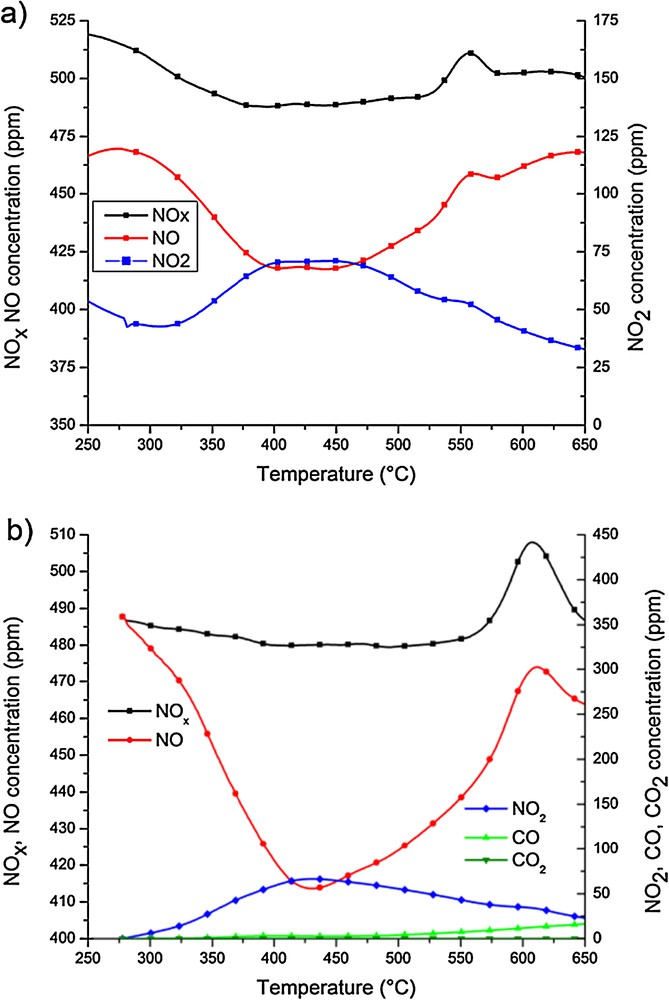
(Colour online.) CoBa-containing catalyst. Evolution of the concentration of the different species in the experiment performed using 500 ppm NO, 5% O2 in N2 36,690 h–1 GHSV (a) without soot, (b) with soot.
NO2 production for MeCs catalysts is lower than the ones observed using MeK or MeBa catalyst (Fig. 1). In the case of CuCs catalyst, the NO2 production is the lowest and adsorption of NOx is low; in spite of that, oxidation of soot is higher than the one obtained when using CuK catalyst and the amount of NOx reduced is similar. When Cs is added to a Co-containing catalyst, NOx adsorption takes place and reduction of NOx increases, being higher than for CoK catalyst, but soot conversion occurs to a lower extent, this points to a different reaction mechanism taking place predominantly, in which NO adsorbed species oxidize soot. Krishna and Makkee [31] demonstrate, for Ce- and Pt-Ce catalysts, that NO species adsorbed are implied in the soot oxidation process. Ito et al. [32] reported the same effect when Cs was added to MnOx–CeO2 catalysts.
Table 1 shows a comparison of the results obtained for all the catalysts tested at 1300 h−1 and 36,690 h−1; the total amount of NOx reduced (mol), mg of soot oxidized and CO2 selectivity during each experiment is displayed.
In this study, the soot oxidation capacity decreases when increasing the GHSV. This could be due to the higher dilution of the NO2 produced, resulting in a low NO2 concentration in the gas phase which leads to a lower reaction rate. Also the selectivity to CO2 is affected, which is caused by the shortened residence time of the reaction gas in the catalyst bed that leads to the incomplete combustion of CO.
For CoK and CoCs catalyst there is a moderate decrease in the amount of soot oxidized and in the selectivity to CO2. However, when using CuK and CuCs catalyst there is a more pronounced decrease in both values, in particular for CuK. Due to the low capacity of the CuK catalyst to produce NO2, the dilution of NO2 leads to an even higher decrease in the amount of soot oxidized.
The most drastic decrease in soot oxidation when increasing GHSV occurs for MeBa catalyst. In this case, the reason cannot be attributed to a lack in the NO2 production, but to the failure of Ba to improve the contact between the catalyst and the soot. When increasing the GHSV this contact becomes even more crucial, resulting in an almost non-existent capacity to oxidize soot. According to those results, Castoldi et al. [33] demonstrated the poor mobility of the active surfaces species for Ba catalyst, which results in a low reactivity for soot oxidation under loose contact conditions.
In spite of the low activity of Ba-containing catalyst for soot oxidation, the amount of NOx reduced by this catalyst increases when increasing the GHSV; the reason could be that some of the NOx adsorbed remain in the surface of the catalyst. The formation of stable Ba-nitrate species of high thermal stability has been previously reported [29,30].
For CoK, the amount of NOx reduced per gram of catalyst remains unchanged when increasing GHSV, pointing out that these catalysts, when working at 1300 h−1, were at the limit of its capacity. Rising the amount of molecules of reagent that pass through the catalysts do not lead to an increase in the activity.
In contrast, for CoCs and CuCs the amount of NOx reduced per gram of catalyst increases when increasing the GHSV. However, the increase in the amount of NOx reduced is not significant compared with the amount of NOx that enters the system, leading to low conversions.
4 Conclusions
Cordierite-supported structured catalyst for the simultaneous removal of soot and nitrogen oxides were prepared with several alumina-based suspensions containing K, Cs or Ba as the alkali promoter, and Co or Cu as active metals. These catalysts were tested in soot oxidation by NOx by varying the GHSV. The increase of the GHSV leads, for all the catalysts tested, to a considerable decrease in the amount of soot oxidized and in the selectivity to CO2.
The evolution of the different NOx species and the catalytic behavior towards the oxidation of soot depended strongly on the type of alkali promoted present in the catalyst formulation. Potassium seems to offer the highest ability towards the formation of adsorbed nitrate species. Barium results in the formation of thermally stable nitrates whereas cesium possesses the lowest ability to form nitrates. This was clearly reflected in the evolution of NO2 registered for the different catalysts. NO2 evolution, i.e. formation, can be directly related to soot oxidation catalytic activity, and, in turn, to the overall activity of the catalyst to activate the simultaneous soot oxidation-NOx reduction mechanism. Increased mobility of K species with respect to Ba ones is also playing a role towards decreased activity in the presence of the Ba-containing catalyst. However, at such high GHSV activity is strongly influenced by mass transfer and we believe that hindered diffusion phenomena and too short contact time between the gas phase and the active components of the catalyst are the main facts leading to the observed decrease in catalytic activity.
Acknowledgements
Authors thank Government of Aragón–La Caixa for financial support (GA-LC-030/2008) and CSIC (201280E123). S. Ascaso thanks European Social Fund and CSIC for her JAE doctoral grant.