

1 Introduction
Constitutional self-assembly provides evolutional approaches for the generation of functional systems through the implementation of reversible exchanges between different complex architectures of variable functionality [1,2]. Much efforts continue to be undertaken on complex systems, which have been identified as an especially promising means to explore the chemical space with natural selection of their structural and functional behaviours [3]. These new concepts may be connected today with the simple definition of Dynamic Multicomponent Self-assembly in chemical and biological systems [4]. They concern collections of molecular/supramolecular objects reversibly exchanging and continuously organizing at the nano- or macroscopic levels.
A representative example is related to the architecture, the H-bonded supramolecular macrocycle formed by the self-assembly of four guanosines and stabilized by alkali cations. It may embody an important constitutional (spatial and interactional) reorganization with different G-networks, dynamically exchanging between G-ribbons and G-quartets or their stacked tubular G-quadruplexes (Fig. 1) [5a].
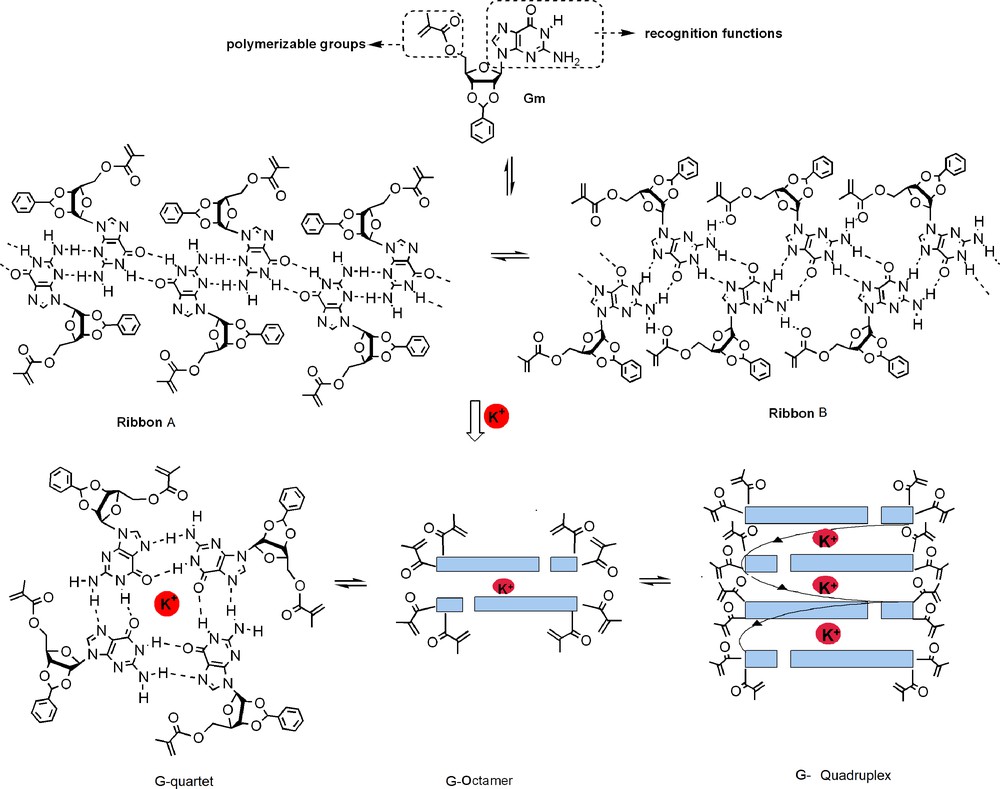
Dynamic exchanges between supramolecular architectures G-ribbons, G-quartets or G-quadruplexes in the presence of ionic chemical effectors (red sphere).
Although discovered in the 1960s [5b], the functionality of the artificial G-quadruplexes as ion-channels and their direct quantification in human cells have been only recently emphasized [6]. Liposomes [7], surfaces [8], dynamers [9], hybrid materials [10,11], mesoporous silica [12] or silicon [13] have been used as scaffolding matrices to stabilize and to orient the anisotropic directional G-mesophases.
We have recently showed that sol–gel selection can be used as an irreversible kinetic process in order to stabilize the G-quadruplex and to provide long-range amplification of its supramolecular chirality into hybrid organic-inorganic twisted nanorods. This may be followed by a second inorganic transcription via calcination into inorganic replica materials, when silica microsprings can be obtained [10]. A second used strategy is related to the implementation of dynamic hydrophobic non-covalent [11a] or reversible-covalent iminoboronate [11a] interactions between the supramolecular G-quadruplexes and the siloxane/polymeric constituents. The reversibility of the interactions between components represents a crucial factor and, accordingly, a dynamic reversible hydrophobic interface might render the emergence of organic/inorganic mesophases self-adaptive. They mutually may adapt their 3D spatial distribution based on their own structural constitution, during the simultaneous formation of hybrid self-organized domains. [14] It is known that the multicomponent self-assembly of the G-quadruplexes superstructures can be also achieved by using the reversible metathesis polymerization reaction, as previously demonstrated by Davis et al. [15].
We hypothesized that the stabilization of guanosine supramolecular architectures may be obtained via a classical polymerization process. Herein we describe this strategy by using 5′-methacrylate guanosine, Gm, self-assembling in solution in G-quadruplexes, followed by their irreversible transcription in an organic polymeric network via intermolecular cross-linking of methacrylate end groups. These results provide new insights into the basic features that control the convergence of supramolecular self-organization and polymerization process toward functional materials at the nano(micro)level.
2 Experimental
2.1 Materials and methods
All reagents were obtained from Aldrich and used without further purification. All organic solutions were routinely dried by using sodium sulfate (Na2SO4). Polymerizations were performed under an inert atmosphere of nitrogen, using standard Schlenk techniques. Tetrahydrofuran was dried and distilled prior to use over Na. The initiator, AIBN, was recristallized from ether and stored at −15 °C. 1H and 13C NMR spectra were recorded on an ARX 300 MHz Bruker spectrometer in d6-DMSO or CDCl3 using the residual solvent peaks as references. ESI–MS studies were performed in the positive and negative ion mode using a quadrupole mass spectrometer (Micromass, Platform II). The samples were continuously introduced into the mass spectrometer through a Waters 616 HPLC pump (60 °C; extraction cone voltage: Vc = 30 V). X-ray powder diffraction measurements were performed with Cu Kα radiation at 20 °C using a Philips X’Pert Diffractometer equipped with an Xcelerator detector. The SEM micrographs were obtained with a High-Resolution Transmission Electron Microscopy (HRTEM) JEOL 2010 FEG apparatus, working with an accelerating voltage of 200 kV and a point resolution of 2.0 Å.
2.1.1 2′,3′-O-Benzylidene guanosine
The precursor, 2′,3′-O-benzylidene guanosine was prepared according to the reported procedures [16]. Guanosine (5 g, 17.65 mmol) was suspended in benzaldehyde (50 ml). An excess of ZnCl2 (13.25 g; 97.18 mmol) was added. After stirring at room temperature for one night, the reaction mixture was washed with ether and cold water. The crude solid was recrystallized from an ethanol/water (3:1) mixture to give a yellow product. Yield: 84%. 1H NMR (d6-DMSO, 300 MHz) δ (ppm) 10.65 (s, 1H, NH); 7, 90 et 7.85 (2s, 1H, H7); 7.48-7.31 (m, 5H, aromatics); 6.45 (s, 2H, NH2); 6.11 et 5.85 (2s, 1H, H6); 6.02 (2d, 1H, H1′); 5.25 (m, 1H, H2′); 5.05 (m, 2H, H3′, OH); 4.20 (m, 1H, H4′); 3.55 (m, 2H, H5′).
2.1.2 5′-Methacrylate-2′,3′-O-benzylidène-guanosine, Gm
2′,3′-O-Benzylidène-guanosine (2.3 g, 6.73 mmol) was dissolved in anhydrous DMF (60 ml). Three equivalents of triethylamine (2.04 g, 20.20 mmol), DMAP (0.41 g, 3.37 mmol) and methacryloyl chloride (0.98 g, 9.42 mmol) were successively added. The mixture was stirred at room temperature for one night. The solvent was removed in vacuo, and the residue treated with chloroform (30 ml) and water (60 ml). The organic layers were separated, dried (MgSO4) and evaporated in vacuo. The resulting mixture was purified by silica gel column chromatography (10% methanol in chloroform) to give the product as a yellow solid. Yield: 56%. 1H NMR (d6-DMSO, 300 MHz): δ (ppm) 10.80 (bs, 1H, NH); 7.89 et 7.87 (2s, 1H, H7); 7.57–7.42 (m, 5H, aromatics); 6.61 (bs, 2H, NH2); 6.20 (2s, 1H, H6); 6.08 (2d, 1H, H1′); 6.14 et 5.68 (2s, 2H, CH2=); 5.42 (m, 1H, H2′); 5.32 (m, 1H, H3′); 4.53 (m, 1H, H4′); 4.48 (m, 2H, H5′); 1.86 (2s, 3H, CH3). 13C NMR (DMSO-d6, 300 MHz) δ (ppm) 166.30 (C16); 156.75 (C11); 153.78 (C10); 150.54 et 150.45 (C9); 145.97 et 145.39 (C17); 136.01 (C7); 135.50 (C12); 129.80 (C15); 128.40 (C13); 126.97 (C14); 126.31 (C18); 117.04 (C8); 106.73 et 103.09 (C6); 88.19 (C1); 84.78 et 84.27 (C2); 83.11 (C3); 81.87 et 81.59 (C4); 64.75 et 64.50 (C5); 17.95 (C19). ESI–MS (30 V, ESI–MS, CH3CN) m/z = 440.53 [M + H]+.
2.1.3 Polymerization procedures
2.1.3.1 Synthesis in the absence of alkali cations
Gm (50 mg, 0.114 mmol) was dissolved in THF freshly distilled (11 ml, [Gm] = 10−2 M) and the initiator, AIBN (1.86 mg, 0.0114 mmol) was added. Three freeze-pump-thaw cycles were performed. The reaction mixture was stirred at 60 °C for four days. The solvent was removed in vacuo. The polymer was precipitated with excess hexane, and dried under vacuum. Conversion: > 95%; yield: 75%. 1H NMR (DMSO-d6, 300 MHz) δ (ppm) 7.90 (bs, 1H8); 7.10–7.70 (m, 5H, aromatics); 6.61 (bs, 2H, NH2); 5.90–6.30 (bs, 2H, H1′, H6); 5.10–5.60 (m, 2H, H2′, H3′); 4.53 (m, 3H, H4′, H5′).
2.1.3.2 Synthesis in the presence of alkali cations
Gm (50 mg, 0.114 mmol) was dissolved in THF freshly distilled (11 ml, [Gm] = 10−2 M); KCF3SO3 8:1 mol:mmol and then the initiator, AIBN (1.86 mg, 0.0114 mmol), were added. Three freeze–pump–thaw cycles were performed. The reaction mixture was stirred at 60 °C for two days. The solvent was removed in vacuo. The polymer was precipitated with excess hexane, and dried under vacuum. Conversion: > 95%; yield: 75%. 1H NMR (DMSO-d6, 300 MHz): δ (ppm) 7.90 (bs, 1H8); 7.10–7.70 (m, 5H, aromatics); 6.61 (bs, 2H, NH2); 5.90–6.30 (bs, 2H, H1′, H6); 5.10–5.60 (m, 2H, H2′, H3′); 4.53 (m, 3H, H4′, H5′).
3 Results and discussion
3.1 Design of hybrid molecular components
5′-Methacrylate-2′,3′-O-benzylidene-guanosine, Gm, has been prepared in two steps:
- • furanose protection with benzaldehyde leading to acetal G* as an equimolar diastereisomeric mixture [16], followed by;
- • a reaction with methacryloyl chloride to afford the compound Gm as a white powder.
The 1H, 13C NMR, ESI-MS spectra are in agreement with the proposed formula. The generation of G-quartet architectures can be achieved by using mixtures of Gm, potassium triflate, and KTf in different molar ratios (Fig. 1).
3.2 Generation of G-quartet architectures in solution
The formation of H-bonded self-assembled architectures of Gm has been studied in solution by 1H NMR spectroscopy and ESI-MS spectrometry. The 1H NMR spectrum of Gm in DMSO-d6 shows well-defined sharp signals, mostly indicative of the presence of monomeric species in solution. In CDCl3, the peaks broaden, indicating a dynamic equilibrium. These fast exchanges at the NMR time scale are considerably slowed down by lowering the temperature (up to –50 °C), when three sets of signals at δ 12.25, 12.38, 12.48 ppm, showing N–H and NH2 cross-peaks (Fig. 2a), reminiscent of a slow exchange between H-bonded interdigitated ribbons A and B and G-quartets (Fig. 1), as previously observed [17].
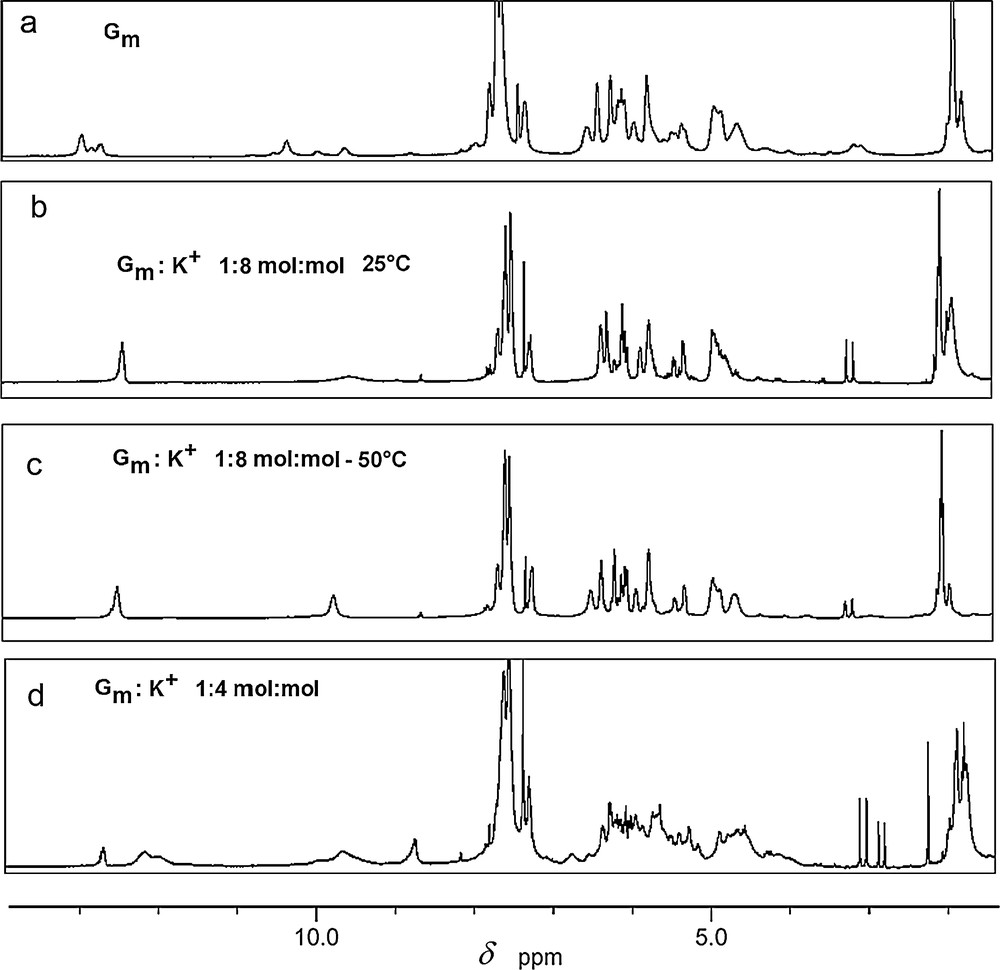
1H NMR titration (300 MHz, CDCl3) of Gm with KTf: a: Gm at 25 °C followed by the addition of; b: 1/8 equiv KTf at 25 °C; c: 1/8 equiv KTf at –50 °C; d: 1/4 equiv KTf at 25 °C.
The addition of the KTf (K+: Gm 1:8, mol: mol) to a CDCl3 solution of Gm causes significant changes in the 1H NMR spectrum (Fig. 2b–d). At 25 °C, the latter shows two peaks for the N–H proton: one sharp peak at 12.7 ppm and one broad peak at 9.7 ppm, reminiscent of the dominating presence of a symmetrical D4 octamer in a fast dynamic exchange with the corresponding G-quadruplex (Fig. 2b). Lowering the temperature, these signals are sharpening and slowly shifting (Fig. 2c). They correspond to two different inner and outer quartets of the G-quadruplex, exchanging slowly with G-octamers in solution at the NMR time scale. [18] The (Gm)8K+ octamer is stable over a temperature range of approximately 60 °C. The 1H NMR spectra in CDCl3, recorded at different temperatures, show small chemical shifts for the protons NH(1) and NH(2), indicating strong hydrogen bonding (Fig. 2e). Upon further addition of the potassium triflate (K+: Gm 1:4, mol: mol), a new set of broad signals appears, consistent with the formation of unsymmetrical exchanging species in solution (Fig. 2d). Further valuable insights are obtained from the ESI mass spectra of compound Gm and of the related K+:Gm 1:8 mol:mol mixture. The positive ESI–MS spectrum of Gm clearly shows the presence of protonated monomers, dimers, trimers, and tetramers as follows: m/z = 440 [Gm + H+], 495 [Gm + 3H2O + H+], 879 [2Gm + H+], 935 [2Gm + 3H2O + H+], 1318 [3Gm + H+], 1372 [3*Gm + 3H2O + H+], 1757 [4Gm + H+]. When eight or more equivalents of KTf are added to the solution of Gm, the ion pattern changes significantly; a new peak at m/z = 2382 is mainly present in the spectrum. This peak correspond to a species with a molar ratio Gm:K+ of 16:3 mol:mol. High-resolution measurements gave a 0.33 amu spacing in the isotopic distribution, indicating the presence of a triple-charged ion and a formula of [Gm]16[3K]3+.
3.3 Polymerization processes of Gm
Having shown that mixtures of Gm equilibrated with K+ cations form G-octamer and G-quadruplex superstructures in equilibrium in solution, we next evaluated their ability to form higher-order superstructures in the polymeric state, by using a kinetically irreversible polymerization step. The generation polymethacrylate G-quadruplex material, {GmK}n, can be achieved by using a pre-equilibrated guanosine derivative, Gm, and KTf in THF, followed by a polymerization process performed in the presence of AIBN at 60 °C. We also carried out for reference the polymerization process in the absence of K+ cations, resulting in the formation of a polymethacrylate G-ribbon material, {Gm}n. Then, these polymers were precipitated with hexane, and dried under vacuum.
FTIR and NMR spectroscopic analyses of these materials demonstrate the formation of polymerized methacrylate networks. The polymerization reactions have been monitored by 1H NMR after the disappearance of the signals at 5.7 ppm corresponding to the methacrylate groups spectra. These reactions may be considered to be complete after four days for the G-ribbon material, {Gm}n, while the polymeric G-quadruplex material, {GmK}n, can be achieved in two days. This indicates that the polymerization processes are favoured in the presence or in the absence of K+ templating ions, which may self-assemble the methacrylate groups on the periphery of the tubular G-quadruplex superstructures in a more favourable spatial distribution for the polymerization process. The FTIR spectrum of polymeric materials shows the disappearance of the broad νMeth vibrations at 1631 cm−1, initially observed for the molecular precursor Gm. Evidence for H-bonding and K+ complexation was obtained from the vibration shifts of the –C=O bonds, detected before and after the polymerization reaction, only in the presence of K+ cations, at νC=O = 1686 to 1654 cm−1.
Further insights into the self-organization and morphology of the polymeric materials were obtained by X-ray powder diffraction (XRPD). The XRPD patterns of the G-ribbon, {Gm}n, and G-quadruplex, {GmK}n, materials present well-resolved peaks. It is noted that the crystallinity of the resulting G-quadruplex, {GmK}n, polymer is higher when compared with the Gm precursor and the non-templated G-ribbon, {Gm}n, analog, in view of the more sharper signal peaks, indicative of long-range structured material. For the Gm precursor, the Bragg diffraction peak at 2θ = 3.4° corresponds to a characteristic distance d = 25.6 Ǻ, compatible with the length of (Gm)2, whereas the peak at 2θ = 6.8° corresponds to a distance d = 13.1 Ǻ, which is the monomeric length. After polymerization, the large Bragg diffraction peak at 2θ = 4.5° corresponds to a characteristic distance d = 22.4 Å, compatible with the more compact G-ribbon, {Gm}n, polymeric analogue. This contraction of the unpolymerized system, resulting in the formation of a more compact polymeric structure, has been as previously observed for similar systems. [19] The XRPD pattern of the G-quadruplex, {GmK}, polymer presents a Bragg diffraction peak at 2θ = 3.2°, corresponding to a distance d = 27.5 Ǻ, compatible with the diameter of the G-quartet superstructure. In the wide-angle region, an additional peak appears at 2 θ = 26.5°, corresponding to d = 3.6 Ǻ and indicative of the π-π stacking distance between two planar G-quartets.
Scanning electron microscopy (SEM) micrographs reveal that the Gm precursor, the G-ribbon, {Gm}n, and the G-quadruplex, {GmK}, materials present superior long-range organisation at micrometric level, as previously observed for similar materils [10,11]. Moreover, while the initial precursor Gm is a compact dense solid with a microfiber morphology, the G-ribbon, {Gm}n, and the G-quadruplex, {GmK}, polymeric analogues consist of crystalline microrods and microsprings, respectively (Fig. 3). This morphology is certainly dependent on the superior emergence of collective domains adapting along the G-quadruplex supramolecular polymer interfaces. Interestingly, the latter has a slightly twisted rod-like morphology, related to the transfer of the supramolecular chirality of G-quadruplex at the micrometric scale in the resulted polymeric system (see a similar example in ref. [10]).

SEM images of a: Gm microfibers and polymeric, b: {Gm}n microfibers, or c: {GmK}n microsprings.
4 Conclusions
In conclusion, the results presented here reveal a new strategy for transcribing and fixing the supramolecular guanosine architectures in self-organized constitutional polymeric networks. In particular, the use of a classical polymerization process represents a useful strategy for improving the compatibility of supramolecular G-guanosine and polymethacrylate networks. A dynamic self-assembly of supramolecular systems prepared under thermodynamic control may in principle be connected to a kinetically controled polymerization process in order to extract and select the H-bonded self-organized networks under a specific set of experimental conditions. Such “dynamic marriage” between supramolecular self-assembly and polymerization process that synergistically communicate leads to higher self-organized hybrid materials with increased micrometric scales. More generally, applying such consideration to polymeric materials leads to constitutional materials, in which supramolecular and polymeric domains are connected. This might provide new insights into the basic features that control the design of functional constitutional architectures. Considering the simplicity of this strategy, possible applications on the synthesis of more complex architectures might be very effective, reaching close to novel expressions of complex matter.
Acknowledgement
This work was financially supported by DYNANO, PITN-GA-2011-289033 www.dynano.eu; ANR Blanc International DYNAMULTIREC, No. 13-IS07-0002-01 and UEFISCDI.