

1 Introduction
EU directive 2010/75/EU [1] requires the drastic reduction of NOx emission in flue gases from the 1st of January 2016. Until now the norms could have been fulfilled in many cases with appropriate primary measures, such as e.g., flue gas recirculation, air staging, low excess air technique, low NOx burners, etc. The BAT methods for coal fired power stations, allowing the fulfilment of the new norms, require now (with the exception of fluidized bed combustion) that either selective non-catalytic reduction SNCR or selective catalytic reduction SCR be introduced in large stationary sources. Selective catalytic reduction (SCR) of NOx with ammonia is the most efficient method of NOx emissions reduction and it is widely used in EU, USA and Japan.
The requirements for in SCR-NH3 are as follows: a catalyst should:
- • be active in reduction of NOx in the oxidizing atmosphere and not sensitive to the changes in oxygen content (flue gases usually contain ca. 6% O2);
- • have a good selectivity to N2 and not lead to a side product N2O, as it is a greenhouse gas ca. 300 times more harmful then CO2;
- • be stable, i.e. not undergo structural or chemical changes under operation conditions;
- • have low activity towards oxidation of ammonia, especially to dinitrogen oxide;
- • have a broad temperature window;
- • not deactivate with H2O, which is always present in flue gases;
- • be resistant towards typical poisons present in flue gases, especially SO2.
Additionally, it would be of advantage if the catalysts could be installed at end-tail configuration where the flue gas has already been cleaned of fly ash and most of sulfur dioxide.
A catalyst most often applied in industry applied catalyst is a monolith with vanadium as active component and titanium as a support with the addition of tungsten oxide or molybdenum oxide promoter to increase the catalyst lifetime by decreasing the harmful influence of sulfur oxides. The temperature window of such catalysts is fairly broad in comparison to e.g., Pt-containing catalysts, but optimum operation temperature is from ca. 573 K to 673 K. This catalyst may be installed either (i) upstream of the electrostatic precipitator (ESP) or (ii) downstream of the ESP and upstream of the desulfurization unit (FGD). In the former case, its life may be shortened by fly ash and sulfur dioxide, and in the latter one by SO2. The operation of SCR downstream of FGD unit requires the reheating of flue gas. This is the main reason why low-temperature catalysts (below 523 K) are searched. Among the best candidates are carbonaceous materials, which, in contrast to V2O5/TiO2 systems, are active at temperature as low as 393 K. Moreover, it was found in many cases that the same active component showed higher NO conversion when supported on active carbon than on Al2O3 or TiO2 [2,3]. There are examples in the literature showing that V2O5/carbonaceous materials are more active than commercial V2O5/WO3/TiO2 [4]. Actually, there exists a DeSOx–DeNOx technology based on activated carbons–BF-Mitsui technology [5,6] which is considered a BAT method under appropriate circumstances. In this method, SO2 is removed from flue gases by adsorption on activated carbon, and NOx are reduced with ammonia, but, due to insufficient activity of carbonaceous materials, the technology has to apply moving beds. It was observed, however, that different types of modification might result in considerable increase in NO conversion.
The main focus of this work is to discuss, based on published literature and the own results of the authors, the possibilities to enhance the catalytic performance of carbonaceous catalysts and to shed light both on the possible preparation procedures, and open questions concerning the design of successful carbonaceous catalysts. Special attention has been paid to carbons containing oxygen and/or nitrogen surface functionalities, used both as catalysts and as supports for active components.
2 The possibilities of the modification of carbon materials
Carbon materials, especially activated carbons and carbon fibres, are characterized by their textural, as well as chemical and surface properties. They are very important both for adsorption and catalysis but SBET may not always be correlated with catalytic performance. It was recognized that for carbon materials, the decisive role was played by surface chemistry [7,8]. The change in surface chemistry affects adsorption and catalytic properties of the modified carbonaceous materials. Figueiredo and Pereira [7] gave an overview of the reactions catalyzed by carbon materials, together with the type of sites necessary for these processes. Among the reactions important for environmental protection, the following should be mentioned: selective catalytic reduction of NOx with NH3, for which acidic and basic sites are of importance, NO reduction with propene [9], NO or N2O reduction by carbon [10], oxidation of NO, SO2 or H2S [7], which require basic sites, or hydrodehalogenation where pyridinic nitrogen sites were identified as being of primary importance. Apart from those, the influence of surface chemistry of carbonaceous materials was recognized for numerous other catalytic reactions, such as e.g., CO oxidation over carbon-supported Wacker-type catalysts [10], Fischer–Tropsch reaction on Co/carbon nanotubes [11–13], oxidative dehydrogenation [7,8], dehydration of alcohols [7,8], etc. Additionally, the surface chemistry of carbonaceous supports influences the catalytic performance of MeOx/catalysts as it will be discussed later.
In this short review, only carbonaceous materials for the DeNOx process are addressed in more detail. The main focus is on the improvement of their catalytic performance, which arises from chemical surface modification. The specific surface area SBET is believed to be of less importance. Admittedly, Teng et al. [14] found for a series of carbons obtained from the same precursors that there was a correlation between activity and SBET, but some surface in micropores was not totally utilized. Several other authors did not find such correlation, possibly also due to the complicated character of activated carbons porosity and internal area being only partly utilized. It must be mentioned, however, that porosity may play an important role in case bigger pores are present. Such conclusions may be drawn from the works of:
- • Loiland and Lobo [15], who worked on NO oxidation at low temperature over different materials, stated that carbon samples dictated catalytic activity more than the zeolite and metal-organic framework materials, this being the result of irregular pore structure of these materials;
- • García-Bordejé et al. [16], who observed differences in stability for V2O5/AC (micro- versus mesoporous) for a SCR mixture containing SO2. No plugging by accumulated NH4HSO4 was observed for mesoporous carbon in contrast to catalysts based on microporous support;
- • Huang et al. [17] reported that NO conversion for V2O5 supported on carbon nanotubes (CNT) increased with the mean diameter of CNT. The effect was explained by differences in V2O5 dispersion.
On the other hand, Su et al. [18] studied carbon nanotubes with MnOx supported on inner or outer surface of nanotubes. Taking into account that the former showed higher NO conversion than the latter, it seems to suggest that internal diffusion is not the key influencing factor, at least for carbonaceous support with such large porous system.
From the point of view of DeNOx applications, the most interesting modifications are those leading to surface groups containing oxygen or nitrogen surface functionalities. Surface oxygen species present on carbonaceous materials include (i) acidic species, such as carboxylic acids and anhydrites, lactones, lactols and phenols; (ii) neutral species, such as carbonyl or ether species; and (iii) quinone, chromene or pyrone groups whose basic character is still under discussion [7]. A review of types of surface groups on carbonaceous materials, their stability as a function of temperature and the analytical tools used for their characterization was given by Figueiredo and Pereira [7]. To summarize shortly, several types of groups may either form spontaneously on the surface of active carbons or be introduced by different modification techniques [7]. Fig. 1a summarizes the methods resulting in the formation of oxygen or nitrogen surface functionalities. Fig. 1b shows typical oxygen- or nitrogen-containing groups formed on carbonaceous materials.
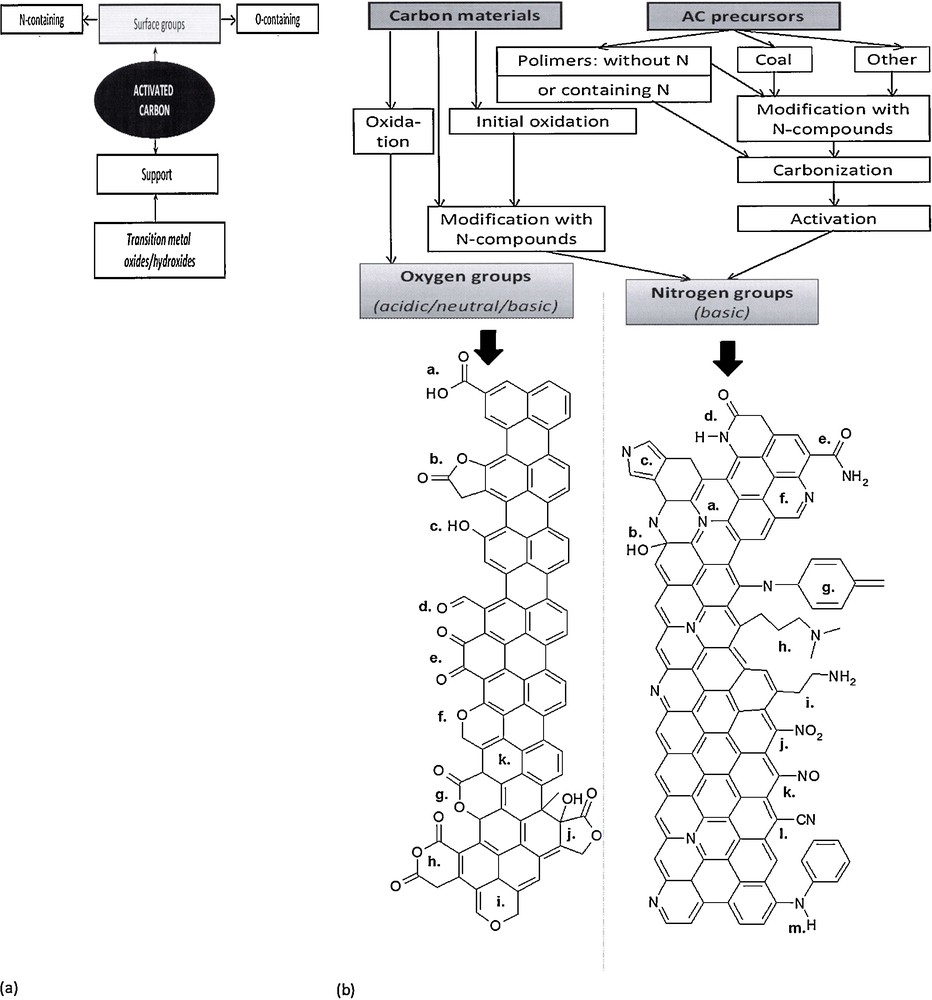
a: the methods of the modification of carbonaceous materials leading to the formation of oxygen and nitrogen functionalities; b: surface oxygen- and nitrogen functionalities on carbonaceous materials. Left: oxygen functionalities: a. carboxyl groups, b. lactone, c. hydroxyl, d. carbonyl, e. quinone, f. ether, g. pyrone, h. carboxylic anhydride, i. chromene, j. lactol, k. π electron density on carbon basal planes. Right: a. quaternary amine, b. pyridine, c. pyrrole, d. lactam, e. amide, f. pyridine, g. imine, h. tertiary amine, i. primary amine, j. nitroso group, k. nitro group, l. nitrile, m. secondary amine.
2.1 Introduction and control of the amount and type of oxygen-containing surface species
Oxygen surface groups may be introduced by different treatments. There is an extensive literature on the subject of the formation of oxygen surface groups and here only examples of typical oxidative procedures with the most important information on the formed surface species are given (Table 1).
Examples of oxidation procedures used for carbonaceous materials.
| Type of carbonaceous materials | Oxidant used | Information on surface groups influenced | Characterization method | Reference |
| AC, ACC | HNO3 {ACN}, H2O2 {ACH}, (NH4)2S2O8 in 4 M H2SO4 {ACS} | Oxygen content: ACS > ACN > ACH > AC Total acidity: ACN > ACS > ACH ≈ AC IR: types of groups formed on oxidation depends on oxidizing media | Neutralization with NaOH, Na2CO3 or NaHCO3; IR | [21] |
| AC | 2 M HNO3 (ACN) or H2SO4 (ACS), 363 K | Species decomposing to: CO2: ACN > AC > ACS CO: ACN > ACS > AC | TPD | [80] |
| Nomex rejects ACF | HNO3 (40%) 323 or 363 K 1 or 2 h {ACF-N T/time}, air 573 or 773 K | Sequence of acidity: ACF-N363/2 > ACF-N323/2 > aCF-N323/1 > ACF-air500 > ACF-air300 | NaOH neutralization | [22] |
| AC | H2SO4 (8%) or (NH4)2SO4 and H2SO4, followed by heat treatment (673 K, N2) | Species decomposing to CO2: AC > treated AC CO: treated AC > AC | TPD, NaOH neutralization | [87] |
| AC | 2 M HNO3, H2O2, H2SO4,{AC-N}, AC-H, AC-S, respectively room temp., 48 h | Acidic sites: AC-N > AC-H > AC-S > AC Basic sites: AC > AC-S > AC-H > AC-N | Boehm method, TPD | [89] |
| Viscose-based ACF | Oxygen plasma (10 kV, t = 40 min) HNO3 (40%, 363 K, 2 h) | High increase in the no. of O functionalities (HNO3 > oxygen plasma) | XPS | [25] |
| AC on cordierite monolith | HNO3 (conc. or 2 M) {ACNc, ACN}, H2SO4 (2 M){ACS}, H2O2 {ACH}, all room temp., 24 h | Species decomposing to CO2: ACNc > ACN > ACS > ACH > AC CO: ACH > ACNc > ACN > ACS | TPD, XPS | [23] |
| OCM | HNO3 (5 M, 2 M, 5 M at 80 °C, 1–5 h; 14.55 M at room temp, 1 h); H2O2 (30%, room temp., 4–24 h) | Up to 9% O introduced, mainly as carboxylic, phenol and hydroquinone groups | TPD, XPS | [20] |
| AC | 10 M H2SO4, HNO3, H22, 10 min, boiling temp; air 300 or 400 °C, 5 h | Acidity increase: AC-H2SO4 > AC-HNO3 > AC-air400 > AC-air 300 > AC-H2O2 > AC Basicity: AC-air 400 > AC-air 300 ≈ AC > AC-H2O2 > AC-HNO3 > AC-H2SO4 Different distribution of strength of acidic sites depending on the oxidant | TPD, XPS, Boehm titration, zero point charge | [18] |
The speciation of the surface oxygen species is studied by a variety of techniques, the most popular of them being the temperature-programmed desorption TPD, X-ray photoelectron spectroscopy XPS and infrared spectroscopy, or Boehm method. TPD is able to differentiate between the groups decomposing to CO2 (carboxylic, carboxylic anhydrides) and CO (phenol, ether, hydroquinons, etc.). Using XPS either C 1s or O 1s peak may be analyzed. C 1s peak at ca. 284.6 eV arises from C–C groups. Binding energies higher by ca. 1.6, 3.2 and 4.8 eV are interpreted as, respectively, C–O (alcohol), CO (carbonyl) and COO (carboxyl) groups. Similarly O 1s may be fitted with oxygen in different environments. It must be kept in mind, however, that XPS can register only surface or subsurface groups (2–20 monolayers from the outer surface) and no information on the groups inside, e.g., deeper micropores is obtained [23]. FT–IR is able to register different types of species but for carbonaceous materials the intensity is usually rather low. On the other hand, the Boehm method, which utilizes the neutralization reaction with NaOH, NaHCO3, Na2CO3 and HCl, can differentiate between acidic or basic groups, the former of different strength (strong, medium and weak).
The matter of oxidation is additionally complicated by the fact that both the initial structure of carbonaceous materials and the type of oxidative pretreatment play an important role and only semi-quantitative prediction on the type and number of groups may be generally given.
The choice of oxidizing conditions is of primary importance for the surface chemistry of the resulting carbonaceous materials [8,19,20]. Several types of such treatments have been described in the literature: air or different oxygen-nitrogen mixtures at varying temperatures, solutions of HNO3, H2SO4, H2O2, (NH4)2S2O8, etc., at different temperatures and time, in some cases, additionally followed by heat treatment to selectively remove some of the formed surface groups. The most general conclusion is that the type of surface groups, and, as a consequence acidic/basic character of the surface depend predominantly on the type of oxidant [8,19,20]. The number of the groups depends additionally on the concentration of the oxidant, temperature of the treatment and its duration [20]. There is an agreement that higher acidity is obtained by wet treatments rather than through dry ones (air or oxygen-mixtures). HNO3 was most often used to change the type and number of oxygen-containing surface groups. The detailed comparison of the effect of several oxidants on the same types of carbonaceous materials was described by many authors. As examples, articles of Pradhan and Sandle [21], Marbán and Fuertes [22], Boyano et al. [24], Van Pelt et al. [19] and Sanchez-Sanchez et al. [20] are addressed here. Pradhan and Sandle compared the effect of oxidation by the solutions of HNO3, H2O2, and (NH4)2S2O8 in 4 M H2SO4. HNO3 led to the highest increase in acidity while H2O2 application resulted in the number of acidic groups barely higher than the starting carbonaceous material. However, for (NH4)2S2O8, the amount of stronger acidic groups was the highest. IR studies revealed that the groups formed by HNO3 differed from those obtained when using two other oxidants. In both cases, carboxyl and phenol groups were registered, but for (NH4)2S2O8 and H2O2 ketone and ether groups were additionally postulated based on IR spectra. The work of Marbán and Fuertes [22] describes the influence of acidity on the treatment with HNO3aq at different temperatures (323 and 363 K) for 1 or 2 h as compared to the modification by air at 473 and 573 K. Again the primary change was associated with the type of oxidant. The sequence of acidity obtained for activated carbon fiber (ACF) was: HNO3 at 363 K for 2 h > HNO3 at 363 K for1 h > HNO3 at 323 K for 2 h > HNO3 at 323 K for 1 h > air at 573 K for 5 h > 573 K for 3 h >> untreated ACF. The treatment with nitric acid at higher temperatures (363 K) led to ca. 3–4 times higher acidity than air treatment at 573 K.
The statement about the best results obtained when using nitric oxide as oxidation agent was also found to be true for layers of activated carbon introduced onto other supports [24] or ordered mesoporous carbons [20]. Boyano et al. [13] studied the oxidation of such a layer formed by carbonization and activation of polymer blend dip-coated ceramic support. Marked differences in types of the resulting surface complexes were found. The ratio of evolved CO/CO2 formed a sequence depending on the oxidant: concentrated HNO3 < HNO3 < H2SO4 < H2O2. The TPD profiles revealed carboxyl, phenol and ether groups after treatment with HNO3 or H2SO4 and carbonyl, quinone and pyrone species after H2O2 treatment. HNO3 favoured the formation of a high number of carboxylic species decomposing at low temperatures. H2SO4 led to groups decomposing at higher temperatures, tentatively ascribed to lactones. No low-temperature groups decomposing to CO2 were registered after H2O2 treatment.
Sanchez-Sanchez et al. [20] studied the oxidation of ordered mesoporous carbons (OCM) using either H2O2 for different periods (4 or 24 h) or aqueous solutions of HNO3 of different concentrations for 1 to 5 h. The obtained surface groups were characterized by TPD and XPS. When higher concentrations of HNO3 were used, most formed groups were of carboxylic, phenolic and hydroquinone types. H2O2 treatment led to much less oxygen functionalities, mainly phenolic and hydroquinone ones.
Van Pelt et al. [19] compared the types and numbers of groups, as well as their acidity and basicity and strength after the oxidation of commercial activated carbon (AC) with 10 M solutions of H2SO4, HNO3, H2O2 at boiling temperature for 10 minutes and dry air at 300 and 400 °C for 5 h. As found before, acidity of carbons treated by H2SO4, HNO3 and air at 400 °C was significantly increased but the strength of the formed groups was different. AC-H2SO4 exhibited high concentration of strong acidic groups, some weak acidic groups and no functionalities of medium strength. The total number of acidic groups on AC-HNO3 was similar, with a lower number of strong acidic groups (0.55 mmol/g as compared to 0.78 mmol/g for AC-H2SO4), 0.28 mmol/g of medium strong and 0.12 mmol/g weak acidic functionalities. The number of basic groups increased after treatment with air and decreased when treated with acids or H2O2. For AC-H2SO4, no basic groups were found. The strong acidic functionalities for AC-H2SO4 were ascribed to the formation of sulfate and carboxylic species.
Apart from dry or wet physicochemical oxidation, low temperature oxygen plasma was also proposed to increase acidic properties. Huang et al. [23] treated viscose-based activated carbon fibers (ACF) by oxygen plasma and compared the results with ACF oxidized by 40% HNO3 at 363 K for 2 h. XPS studies showed that both treatments led to a high increase in C–O, CO or COO− groups. Under the used conditions of plasma treatment (10 kV, 40 minutes), the amount of O surface functionalities was however lower for oxygen plasma than for HNO3.
The concentration of different types of oxygen groups may be additionally controlled by heating under vacuum at appropriate temperatures, as they decompose to CO2, CO and/or H2O in determined temperature regions [23]: carboxylic from 373 to 673 K, lactone from 463 to 923 K, phenol from 873 to 973 K, carbonyl from 973 to 1253 K, anhydride from 623–673 K to ca. 900 K, ether at 973 K and quinine from 973 to 1253 K.
Vivo-Vilches et al. [25] showed that there was a reorganization of oxygen functionalities during thermal treatment in nitrogen. XPS indicated that the content of oxygen on the external surface decreased with the increase in temperature. Three-quarters of the CO2 species originating from the decomposition of carboxylic groups inside the pores from 300 to 500 °C were again fixed as semiquinone (evolving CO) while the treatment at 700 °C prevented re-oxidation of the surface and decreased oxygen content. Additionally, open porosity favoured the formation of CO bonds.
2.2 Introduction and control of the amount and type of nitrogen-containing surface species
Nitrogen functionalities may be introduced in two ways (Fig. 1a): either by (i) treatment of activated carbons, carbon fibers or chars with nitrogen-containing molecules e.g., ammonia [26–33] urea [34,35], melamine or melamine + urea [36], pyridine [37,38] or mixtures, such as e.g., ammonia-air [27,39], ammonia and nitrogen [40], etc., and/or (ii) carbonization and activation of carbon material precursors untreated or treated with N-compounds: model compounds, such as acridine, carbozole [41,42], 9-aminoacridine, 9-cyanoanthracene, 2-aminoanthracene [42], sucrose [43], nitrogen-containing organic salts (e.g., tetrasodium EDTA) [44], N-containing (or modified) polymer precursors [45], polymers or resins [42,46–51], copolymers [52,53] or coal [54,55]. Plasma treatments should also be mentioned [e.g., 57–59].
The types of the N-functionalities that may be present on, or introduced onto, the surface of carbonaceous materials are summarized in Fig. 1b.
An extensive overview of different details of the preparation of N-modified carbonaceous materials is given in [8]. Here, only selected examples are given.
The interpretation of N-functionalities is more difficult than that of O-containing groups. The methods most often used to identify them are X-ray photoelectron spectroscopy (XPS) and infrared spectroscopy FT–IR. In case of XPS, there is no total agreement in the literature concerning the assignment of binding energy of N 1s. Especially the assignment of amine and amide groups seems problematic. Some authors assigned the peak at binding energy (BE) of ca. 398 eV to amine and amide groups [59], while most others [41,60–64] assign one of the peaks at BE between 399.5–400.8 eV to these groups. On the other hand, the assignment of other types of groups is less controversial. The peaks at binding energies of 398.3–398.8 eV [e.g., 62,65,66], 399.5–400.9 eV [e.g., 62,65], 401.1–402.5 [62,67], 402–405 eV [59,62,66,68] are interpreted as pirydinic, pyrrolic/pyridone, N-Q (quaternary nitrogen) and N-Ox (N-oxides) groups, respectively.
Similarly, as in the case of oxygen surface functionalities, the type of N-surface groups depends on preparation conditions: type of modifier, temperature of modification and the history of the carbonaceous sample. Additionally, thermal treatment following the introduction (or formation) of these groups results in either their reconstruction to other types of species or, in certain cases, decomposition (desorption).
There is a general agreement, that after high temperature treatments with N-modifiers, pyrrolic and/or pyridinic functionalities are formed [26,29,41,57,58,40], while N-groups formed at lower temperatures are still under discussion.
Yang et al. [40] studied the formation of N-functionalities for wood-based activated carbon pretreated with HCl and subsequently treated with a mixture of ammonia and nitrogen at 250, 450, 650 and 850 °C. XPS of N 1s revealed the formation of pyridinic (binding energy [BE] 398.7 eV), imine/amine/amide (BE 399.5 eV), pyrrolic (BE 400.1 eV), quaternary N (BE 401.3 eV) and N-oxide (BE 403.1 eV). The surface nitrogen content increased with temperature, but the speciation was different for low and high temperature treatments. Over 450 °C, more pyridinic and pyrrolic groups were registered, while low temperature (250 °C) was beneficial for the formation of imine/amide/amine species. The maximum content of the latter was registered after treatment at 450 °C. Yang et al. postulated that carboxylic groups reacted with ammonia at low temperature to form amide species while imine/amide/amine resulted from the reaction of carboxylic anhydrides with NH3.
The decrease of the number of amine groups with the increase of treatment temperature was also confirmed by Zhai et al. [69] although these authors actually studied the high-temperature (650–850 °C) modification with ammonia.
On the other hand, it must be mentioned that not all authors agree that only amide/imine/amine groups result from low temperature treatment. Although in most cases, such groups were registered [27], additionally also nitriles, protonated amides or lactams were postulated.
Jansen and Bekkum [27] found evidence that a modification method was a decisive factor for the formation of certain N-species: ammoxidation leading to amides, amination to lactams and imides and the following heat treatment to pyridine and pyrrole groups. These findings are in partial disagreement with the article of Wachowski et al. [59] who studied activated carbons containing N-functionalities prepared by two procedures: (i) carbonization of activated carbon precursor, followed by ammoxidation and activation, or (ii) ammoxidation of the precursor, followed by carbonization and activation or only activation. Ammoxidation was carried out at 523 or 573 K. According to Wachowski et al. [59], the dominant nitrogen species found by XPS in precursor ammoxidized at 523 K were pyrrolic, pyridinium and quaternary N-functionalities while imine, amine and imide species were additionally registered in the sample ammoxidized at 573 K. Yang and Yang [57] suggested that pyrrolic nitrogen groups provided active sites for the formation of the conjugated structure. As expected, imine, amine and imide groups disappeared after activation or carbonization. Sousa et al. [70] compared different N-compounds modifications: nitric (V) acid, urea and melamine. They suggested that AC chemically modified presented pyridine (N6) and pyrrole (N5) groups on their surface. The N-Q groups present in pure AC were converted to N6 by subsequent treatments. Modification with HNO3 gave nitro complexes on activated carbons surface, with urea–possibly amide-like species. These species underwent dehydration (e.g., pyridone and pyridine) or decarbonylation (e.g., pyrrole) reactions to form more stable heterocyclic aromatic moieties.
Treatment of carbonaceous materials with N-containing molecules results in the increase of their basic properties, at the expense of acidic properties [26,71]. This was confirmed either by direct determination of the acid–base character e.g., by the method of Boehm [26], by enhanced sorption of acidic molecules, such as HCl [71], SO2 [72–76], H2S [36,72,77] or CO2 [32,78] or increased catalytic activity in certain reactions requiring acidic and/or basic sites, such as conversion of isopropanol or acetonylacetone [33]. Conversion of isopropanol via dehydration to propene is ascribed to the presence of acidic sites, while the formation of acetone depends on the presence of both acidic and basic Lewis sites. The cyclization of acetonylactone leads to the formation of 2,5-dimethylfuran or 3-methylcyclopent-2-en-1-one, the former on Brønsted and the latter on basic sites. Krzyżyńska et al. [33] showed that the treatment with ammonia at 973 or 1173 K increased the basic character, the effect being higher for higher treatment temperature.
The confirmation that the treatment temperature influences the basicity was also given by Yang et al. [40] who modified activated carbon with an ammonia-nitrogen mixture at different temperatures (250 to 850 °C) and compared the resulting groups to non-modified carbonaceous material. The increase in the number of groups did not present a linear dependence on temperature. The number of basic species was 0, 0.156, 0.295, 0.328 and 0.343 mequiv/g, respectively, for non-modified active carbon and the samples modified at 250, 450, 650 and 850 °C.
3 The influence of oxygen surface groups on the catalytic performance in DeNOx
Several authors paid attention to the role of oxygen-containing surface species in SCR-NH3 [22,24,79–83]. The general conclusion was that the presence of such species led to the increase in activity. The observed gain in conversion was in most cases rather small, generally of the order of 10% or lower, depending on the used type of oxidative pretreatment [e.g., 20,84,85]. The dependence of NO conversion on temperature showed in most cases a minimum both for non-oxidized and oxidatively treated activated carbons at around 423–473 K (or 523 K) [83,84]. Thus, the positive effect of oxygen functionalities usually decreased over 453 K. Additionally, a deterioration of catalytic performance with time on stream was observed [83], most probably resulting from the removal of acidic surface sites.
Table 2 summarizes most important experimental information on catalytic activity of carbonaceous materials with the increased number of oxygen functionalities. The increase in SCR activity was found by the following.
Modified carbonaceous materials as catalysts for NO reduction with NH3 if not stated otherwise.
| Carbonaceous material | Type of modification | Reaction conditions | Information on catalytic activity | Reference |
| AC | H2SO4, heated at 373 K, 473 K, 573 K in N2 | 700 ppmv NO, 700 ppmv NH3, 4 vol% air, rest He | NO conversion increased by H2SO4 treatment. Higher increase for higher following heat treatment | [85] |
| AC | Wet impregnation: glucosamine, uracil, picolylamine, 2-hydroxy-methyl-pyridine, followed by treatment in N2, and in some case activation in CO2 | 500 ppm NO, NH3, 2% O2, He; T 385–550 K; mcat = 0.3 g; GHSV 500 h−1 | NO conversion highest for modification with glucosamine | [94] |
| Polyacrylonitrile-based AC | H2SO4, heat treatment in N2 at 623–873 K | RC: NO p = 2.7–3.3 kPa, T = 423–623 K | N2 formation rate: ACF > ACK ACF oxidized > ACF Optimal heat treatment following modification by acid 673–773 K | [86] |
| Phenol–formaldehyde resin-based AC | m-phenylenediamene added to resin, gasification in N2 from room temp. to 1073, followed by gasification in oxygen at 673 K | Flow rate 300 cm3/min; T = 383–503 K, NO, NH3 conc. 350–950 ppm | Increase in NO conversion for N-doped carbons, with a maximum for 7.8 wt% N A correlation between NO adsorption capacity and activity in SCR | [96] |
| Viscose-based ACF | HNO3 {ACF-N} or oxygen plasma {ACF-op} | SCR: 1000 ppm NO, 81,000 ppm NH3, 53% O2, He; flow: 3365 cm3/min; GHSV: 9686.8 h−1; mcat = 0.2 g; T = 393–513 K (every 30 K) | A small increase in NO conversion: ca 15–10% (depending on T) for ACF-N ca 5% for ACF-op | [24] |
| Polyarylamide-based ACF | SCR: 700 ppm NO, 800 ppm NH3, 3% O2, Ar; flow: 300 cm3/min; mcat = 2 g; T = 273–673 K | Strong inhibitory effect of water on NO conversion NO reduction higher for ACF with higher basicity | [103] | |
| Commercial ACF: polyacrilonitrile-based, prepared from coal pitch, or phenolic resin; laboratory petroleum-based | Heat treatment at 1073 K {HT}, NH3 at 1073 K {AM}, oxidation by HNO3 followed by NH3 at 1073 K {ONA}, ammoxidation {AO} | SCR: 700 ppm NO, 800 ppm NH3, 3% O2, Ar; flow: 300 cm3/min; mcat = 2 g; T = 273–673 K | (Ex-coal pitch) NO conversion: AO > ONA > AM > ACF > HT NO conversion sequence dependent on ACF precursor Strong inhibitory effect of water | [84] |
| Deashed AC and chemically modified commercial | Heated under vacuum at 1023 K/3 h {AC-h}; oxidized with HNO3 {AC-ox}; NH3 1073 K {AC-h-N or AC-ox-N}, promoted by Fe3+ | Gas velocity: 100 mL/min, mass of catalyst: 400 mg, gas pressure: 1 bar, gas composition: 800 ppm NO and NH3, 3% O2 and the rest helium | NO conversion: T: 413–493 K: AC-ox-N > AC-ox > AC-N > AC-h T: 520–573: AC-ox-N > AC-N > AC-h > AC-ox | [83] |
| AC | HNO3 in 363 K{C90}, NH3 at 1073 K {C-ox-N or C-N}; followed by urea, curing in air or N2 {C/U or C-ox-U} | SCR: 800 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, flow rate 100 cm3/min, mass catalysts: 400 mg, GHSV 8000 h−1 | NO conversion: C-ox-U > C-ox-N > >C, C-U, C-N | [34] |
| AC | HNO3 in 343 K followed by urea, curing in air {P2} or N2 {P3}; pure AC {N} with urea, curing in air {P1} | SCR: 800 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, flow rate 100 cm3/min, mass catalysts: 400 mg, GHSV 8000 h−1 | NO conversion: P2 > P3 > P1 > >N | [155] |
| AC | Oxidation by HNO3 (or not oxidized) Heating at 1073 K, NH3 at 1073 K, promotion MnOx | SCR: 800 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, flow rate 100 cm3/min, mcat: 200 mg | – H2O decreases NO conversion (inhibition is reversible) – H2O decreases N2O formation | [134] |
Ahmed et al. [85] for commercial active carbons (AC) modified by concentrated H2SO4 at room temperature, followed by treatment in N2 flow at 373, 473 or 573 K. There was a correlation between NO conversion and oxygen bulk content. H2SO4 treatment led to the formation of acidic surface functionalities, which increased NH3 sorption capacity and played a positive role in SCR.
Mochida et al. [86] for polyacrilonitrile-based activated carbons oxidized by H2SO4 and subsequently heat-treated at 623–873 K. There was an optimum temperature of heat treatment which left surface oxidized but removed sulfur species.
Ku et al. [87] for activated carbon modified by the solution of sulfuric acid, or first ammonium sulfate and then sulfuric acid, followed by heat treatment in nitrogen at 673 K. This sort of treatment, as previously found in many studies, eliminates carboxylic species, and this was then rationalized in the mechanism, as carboxylic species not being the main reaction sites.
Teng et al. [80], who studied activated carbon fibres (ACF) oxidized by HNO3 or H2SO4. Depending on the presence or absence of oxygen in reaction mixture, activity formed a sequence:
- • with O2: ACF oxidized by HNO3 > ACF oxidized by H2SO4 > ACF;
- • without O2: ACF oxidized by HNO3 > ACF > ACF oxidized by H2SO4.
Marbán et al. [22] oxidized activated carbon fibres with HNO3 solutions of different concentrations at 323 or 363 K for 1 or 2 h and with air at 573 K. Activity sequence was the same as that of the acidity of the obtained catalysts.
Szymański et al. [83] who also found the positive affect of HNO3 treatment.
Sousa et al. [70] confirmed the positive effect of HNO3 treatment on catalytic properties. Nitric acid modified samples exhibited a ca. 10% higher NO oxidation ability versus untreated samples.
Huang et al. [20] handled the case of viscose-based activated carbon fibres treated with HNO3 or oxygen plasma and untreated, as discussed in Section 2.1. NO conversion in SCR increased by ca. 10–15% after HNO3 treatment and by ca. 5% after oxygen plasma treatment. This was ascribed to the formation of oxygen functionalities, which was higher in the former than in the latter case.
Higher influence of pre-oxidation by HNO3 in comparison to that carried out by oxygen plasma was also registered by Zhu et al. [88], who compared the performance of DeNOx for CeO2 supported on viscose-based activated carbon fibres so pretreated. It was observed that NO conversion was higher for the former than for the latter catalysts, which was explained by the positive role of oxygen functionalities in active phase dispersion and the formation of adsorption sites for NH3.
Boyano et al. [24], who studied the effect of oxidation of carbon layer formed on ceramic honeycomb. They noticed that the acidic character of the surface is of key importance for achieving a high NO reduction efficiency.
The mechanism of SCR with NH3 was discussed based both on DeNOx results, studies of NO interaction with carbonaceous materials (i.e. the reaction of direct reduction of NO by carbon) and NO sorption studies [89].
Sorption of NO and NH3 on the surface was connected to acidic–basic character of the surface. NO sorption was ascribed to the presence of basic sites. Ahmed et al. [85] observed that oxidation of AC with H2SO4 led to the decrease in adsorption of NO from a mixture containing 15% O2. This may be attributed to the increase in the amount of acidic groups with the simultaneous depletion of basic groups.
Xue et al. [89] studied (by TPD) NO sorption on activated carbon untreated or oxidized by HNO3, H2O2, and H2SO4. The sequence of amount adsorbed was the same as the sequence of the number of basic sites.
Mochida et al. [86] studied reduction of NO by carbon oxidatively pretreated. The increase in NO reduction was explained by the increased adsorption of NO on surface vicinal O-species. The reaction between such two groups was found to be a rate-determining step. N2 desorbed from the surface and the initial sites were recovered when CO2 desorbed from the surface.
There is a general agreement that acidic functionalities are sites for ammonia adsorption, although, as discussed below in more detail, the appropriate surface species were not yet identified without doubt.
Zawadzki and Wiśniewski [90] investigated by IR sorption of NH3 on carbon films prepared by carbonization of cellophane. NH3 was chemisorbed as NH4+ on carboxylic species. At higher temperatures (473–673 K), additionally amide and imide structures were formed.
Marbán et al. [91] analyzed the mechanism of low-temperature selective catalytic reduction of NO with NH3 on Mn3O4/carbon–ceramic monolith by step-response and temperature-programmed desorption experiments. Although most conclusions concern the role of the active component, NH3 interaction with AC was also considered. AC surface devoid of carboxylic groups due to pretreatment in N2 at 673 K showed the following NH3 surface species:
- • NH3 adsorbed through hydrogen band to anhydrides;
- • NH3 adsorbed as NH2 (with H abstraction);
- • NH4+ ions linked to phenolic surface groups.
Teng et al. [80] proposed the overall mechanism for DeNOx reaction, basing on the comparison of activity order for carbonaceous materials pretreated with HNO3 and H2SO4, as described above. The number of species evolving CO2 in TPD formed the same sequence as NO conversion for a mixture NO + NH3. The number of species decomposing to CO formed a sequence that was the same as for NO conversion from a NO + NH3 + O2 mixture. Thus, two types of sites for NH3 sorption, different for a mixture with or without oxygen were postulated: phenolic groups for the latter and OH part of carboxylic group for the former. According to the authors, adsorption of NO occurs either (i) in the nearest vicinity of NH4+ species i.e. on the CO part of carboxylic group (for a mixture without oxygen) or on basic species of pyrone-like type groups (for a mixture with oxygen). The two types of mechanisms are illustrated by the reactions in Fig. 3a–c.
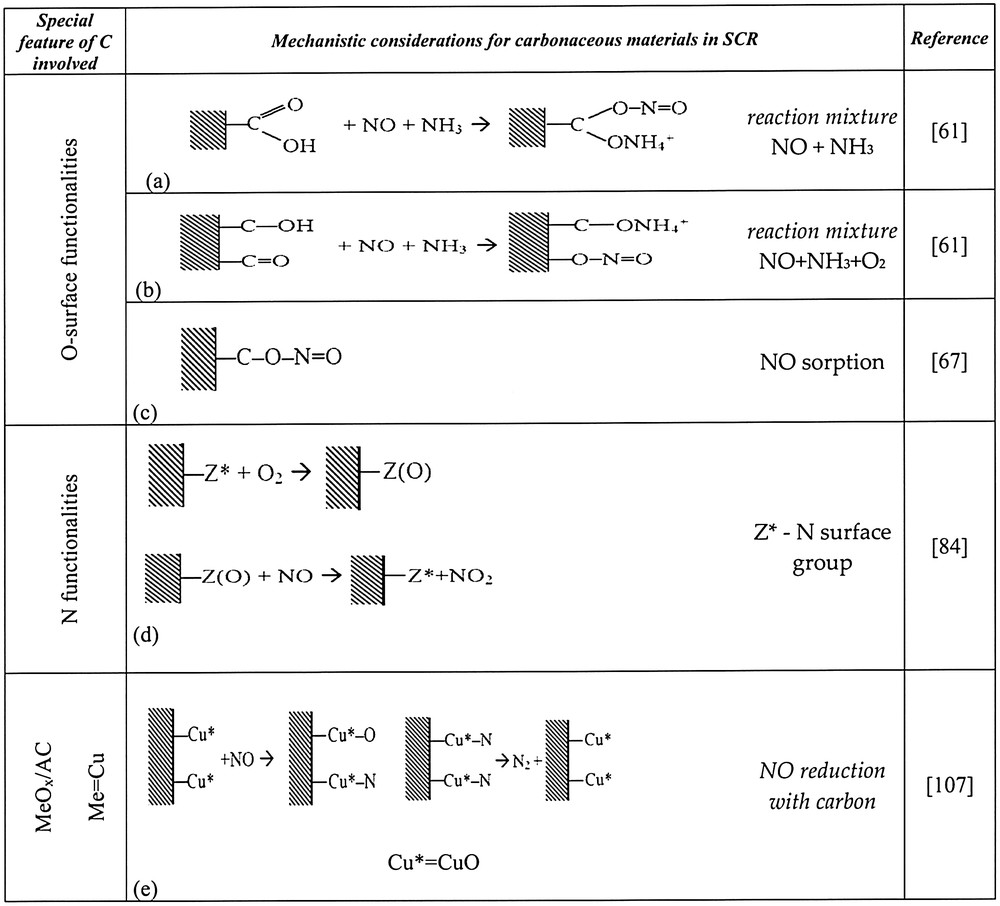
Elements of mechanism of SCR-NH3 on carbonaceous materials or MeOx on carbon supports.
The dependence of SCR activity on the presence of acidic phenolic species rather than carboxylic moieties may be also derived from the results of Ku et al. [87]. Two important findings should be mentioned in this case: (i) the number of CO2-evolving groups decreased and those evolving CO increased after treatments with H2SO4 or (NH4)2SO4·H2SO4 while SCR activity increased, and (ii) NO conversion depended linearly on the amount of NaOH used for neutralization i.e. on acidity. Thus, acidic phenolic groups which evolve CO on decomposition were identified as the most likely sites in the reaction.
On the other hand, the study of Szymański et al. [83] indicates that mechanism (b) may not be the only possible route for a reaction mixture containing oxygen. The studies were carried out on active carbon oxidized with HNO3, which leads predominantly to the formation of acidic (carboxylic, anhydrides, lactones and phenolic species). The reaction procedure was: the measurement of NO conversion in an increasing temperature sequence (413, 453, 493, 533 and 573 K) and afterwards at a decreasing temperature sequence (533 and 493 K). A clear decrease in activity for the same temperature was observed for the “downward” curve in the comparison to the “upward” one. According to the literature [25,92,93], carboxylic groups decompose at 373–673 K and anhydrites and lactones around 883 K (both to CO2), while phenol, carbonyl, quinone and pyrone groups decompose to CO at respectively 923, 973, 1073 and 1123 K. Taking into account that the highest temperature in the experiment of Szymański et al. [93] was 573 K, the decrease in activity observed on the “downward” curve should be ascribed rather to the removal of carboxylic species than phenol groups, which are much more stable. Possibly both routes (a) and (b) are valid in the presence of oxygen in an SCR mixture, the former being the secondary one and the latter the primary one.
4 The influence of nitrogen functionalities or catalytic performance in DeNOx
4.1 Activity and selectivity
Apart from oxygen functionalities, surface groups containing nitrogen have been considered as beneficial for DeNOx reaction [32,34,70,80,87,94–96]. Generally, it was found that the modification with N-compounds resulted in a much higher increase in activity than that observed for oxidatively pretreated activated carbons, as illustrated by Szymański et al. [83] who compared NO conversion for activated carbons treated under vacuum at high temperature (DHT), activated carbon oxidized with HNO3 (D-ox) and DHT and D-ox treated with NH3 at 1173 K. For low temperature (413 K) and high temperature (573 K), NO conversion formed a sequence: D-ox-N > D-HT-N > D-ox > DHT. At medium temperatures, D-ox was slightly better than DHT-N but still less active than D-ox-N.
On the whole, however, the direct comparison of the effects of modification procedures and compounds is rather difficult, because of varying DeNOx testing conditions. The procedures used for modification and their influences on catalytic performance in SCR-NH3 are summarized in Table 2.
One of the first articles published by Singoredjo et al. [94] described DeNOx properties of carbonaceous materials obtained by co-carbonization of sucrose with glucosamine, uracil, picolylamine or 2-hydroxy-methyl-pirydine or impregnation of activated carbons with glucosamine. The latter led to the highest SCR activity.
Ku et al. [87] compared NO conversion for activated carbons modified either by O2–NH3, or H2SO4, or (NH4)2SO4/H2SO4. The last of the mentioned proved the most efficient.
When activated, carbon composites were treated under vacuum at high temperature (1073 K, sample HT), with NH3 at 1073 K (sample AM), with HNO3 and subsequently with NH3 at 523 K (sample ONA) or ammoxidized at 723 K (sample AO), NO conversion at 373–673 K formed a sequence: AO > ONA > AM > untreated sample > HT [84].
Grzybek et al. [34,97] and Szymański et al. [83] compared, under the same conditions, the behavior of N-promoted activated carbons obtained by the treatment with (i) NH3 at 1073 K [34,83], (ii) aqueous solution of urea followed by curing in air [34] and ammoxidation at 623 K [98]. The latter samples were prepared in the following way: activated carbon N either untreated or pre-oxidized with concentrated HNO3 at 363 K for 1 h were ammoxidized with a mixture of NH3 + O2 (1:3) at 623 K for 4 h, then cooled under argon (respectively further addressed as C/350/NH3-O2 or C90/350/NH3-O2). These samples were characterized by low temperature argon sorption (SBET), XPS (N/C ratio and chemical state of N-functionalities and TPD of surface oxides). Fig. 2 compares DeNOx performance of selected so-modified active carbons [34,97–101]. Oxidative pretreatment prior to the modification with urea or NH3 at 1073 K [83] or ammoxidation [98] resulted in more active catalysts than the same procedures applied directly to untreated AC. The beneficial effect was explained by the formation of acidic surface sites, which preferentially interacted with N-precursors. The formed N functionalities were believed to be sorption sites for NO molecules [96], thus leading to the improvement in NO conversion. However, urea treatment and ammoxidation resulted in considerable increase in NO conversion between 413 and 573 K, while high-temperature NH3 treatment was less efficient in this temperature range. No direct correlation with the amount of introduced nitrogen was observed at low DeNOx temperatures. The urea-treated sample (ca. 3.7 wt% N) showed activity only slightly higher than the ammoxidized sample C90/350 NH3-O2 (ca. 7 wt% N) while AC oxidized with NH3 at 1073 K with ca. 1.4 wt% N was much less active at lower temperatures. A possible explanation may be derived from quantum mechanics calculations of Strelko et al. [102] who suggested that N-doping changes the catalytic properties of carbon by modifying electron mobility and semiconductor properties, to an extent depending on the amount of N, with a certain optimum content. Additionally, apart from N-species, oxygen functionalities present on carbonaceous materials whose DeNOx performance is given in Fig. 2 may play a positive role. Such oxygen groups are depleted on the surface during the high-temperature NH3 treatment in contrast to ammoxidation or urea/air treatment.
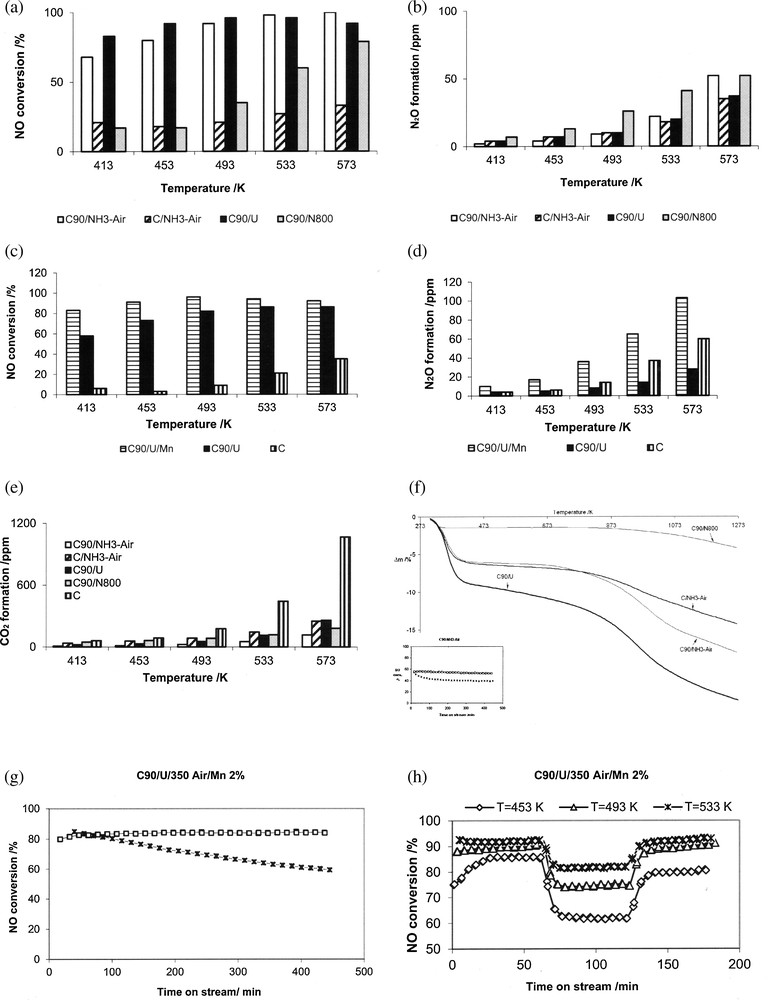
The comparison of catalytic performance and stability of activated carbon modified by the introduction of nitrogen surface functionalities and transition metal oxides/hydroxides. a–b: catalytic performance (NO conversion and N2O formation) for active carbons differently pretreated to change the number of oxygen and/or nitrogen functionalities (based on data from [34] and [98]); c–d: catalytic performance (NO conversion and N2O formation) for FeOx or MnOx on carbon support pretreated by oxidation and urea (based on data from [95] and [98]): e: TGA results for activated carbons modified by oxidative and/or nitrogen-pretreatments. The conditions of TGA: the experiments were carried out at NETSCH; mass of the sample 20 mg; 20 mg of Al2O3 as a reference. At the beginning of each experiment, the sample was evacuated to 1 mbar in order to remove air and physically adsorbed gases. Then, it was linearly heated at the rate of 10 K/min up to 1273 K in a stream of helium. The small figure included in (f) shows NO conversion for ammoxidized sample in the absence of H2O during 8 h on stream at temperatures 413 K (dashed line) and 453 K (solid line); g–h: NO conversion for MnOx supported on active carbon pretreated by oxidation and urea; (g) versus time on stream (without H2O added to reaction mixture), (h) with H2O sequentially added and removed from the reaction mixture [97]. Designation of the catalysts: C – untreated activated carbon; C90 – activated carbon oxidized with HNO3; U – modified with urea solution, followed by curing in air at 623 K; NH3-Air – ammoxidized at 623 K; N800 – treated in NH3 at 1073 K, Fe or Mn – promoted with FeOx or MnOx (2 wt% Me) by wet impregnation. CR conditions: calc. 523 K for 2 h in helium, 100 mL/min; mass of catalyst 200 mg; reaction mixture: 800 ppm NO, NH3, 3% O2, He, 100 mL/min, H2O, if added, 0.3 g/h.
The second type of procedures leading to N-containing activated carbons was based on carbonization and activation of N-containing precursors: polymers [103] or resins [96].
Muñiz et al. [103] studied N-promoted activated carbons obtained by carbonization and activation of polyaryloamide fibers (Nomex and Kevlar). The applied carbonization and activation conditions leading up to 25% burn-off influenced NO conversion only to a small extent.
Huang and Teng [63] used phenol–formaldehyde resin as the starting material to prepare N-containing activated carbons. Then, 10, 20 or 30 wt% of m-phenylenediamine solution in alcohol was added to the resin, which was subsequently carbonized in nitrogen by heating from room temperature to 1073 K and then gasified in oxygen at 673 K, resulting in carbonaceous materials containing 3.7 to 8.8 wt% N (elemental analysis) and SBET of ca. 309 to 372 m2/g. Pyridinic and pyrrolic or pyridonic groups accounted for the majority of nitrogen functionalities. SCR-NH3 experiments carried out between 383 and 503 K revealed a considerable increase in NO conversion in comparison to activated carbon obtained from the resin without the modification with the above-mentioned N-compound. Again no direct correlation between the N content (as determined by elemental analysis) and activity was observed.
4.2 Stability of carbon materials in DeNOx
Apart from activity and selectivity to N2, stability is also of the highest importance. DeNOx catalysts operate at elevated temperatures and in oxidative atmosphere, as flue gas from stationary sources contain typically 5–6% O2. Fig. 2f presents thermal stability of N-modified activated carbons whose activity was addressed in Section 4.1. The TGA curves show three distinct regions: a considerable mass loss up to ca. 473 K (water desorption), a small mass loss up to 623 to 673 K and again a considerable mass loss for temperatures over 623 to 673 K.
The second region is especially interesting from the point of view of the DeNOx reaction, as it is relevant to stability of the studied carbonaceous materials when used as SCR catalysts. The mass loss between 453 and 673 K is small and forms a sequence: oxidized AC treated by aqueous solution of urea > untreated AC, ammoxidized > oxidized AC ammoxidized > oxidized AC treated by NH3 at 1073 K.
Thus, it may be concluded that the stability of AC treated in NH3 at 1073 K is excellent. Similar conclusions were drawn by Biniak et al. [26]. Ammoxidised and urea-treated catalysts have somewhat worse, but still satisfactory, thermal stability in the DeNOx low temperature window (453 to 573 K). As additionally no N-species were found in TPD, the overall conclusion is that the N-functionalities are fairly stable at the temperature window relevant for DeNOx reaction. Fig. 2f [98] further shows NO conversion versus time on stream (TOS) for the ammoxidized sample with a very good stability at 453 K. A small decrease in NO conversion with TOS for 413 K is most probably connected with competitive adsorption of water (reaction product) and NH3 (substrate). Similar results were obtained for doubly promoted AC–N–Mn [97] and AC–N–Fe (Fig. 2g) [98], although the loss of activity with TOS at 413 K was more pronounced for the former than for the latter catalyst.
Apart from the stability of N-surface groups, the presence of oxygen may lead, especially at higher temperatures, to considerable CO2 formation and, in consequence, to the loss of a catalyst. Mang et al. [43] showed that the nitrogen incorporation in carbons resulted in a decrease of the rate of their oxidation (with 5% O2 in helium) at temperatures below 873 K as compared to N-free carbons. Gasification of carbonaceous materials containing N additives was much slower but no direct correlation between nitrogen content and gasification rate was observed. It was concluded that pyridine-like nitrogen resulted in the stronger inhibition than other N-functionalities. This was also confirmed by Matzner and Boehm [104]. A similar effect was observed in SCR-NH3 [98]. Fig. 2e [98] compares CO2 formation for the N-modified active carbons with that for the untreated sample and proves that N-modification is beneficial for stability. However, no explanation of the increased stability of N-containing carbons observation was given as yet.
4.3 The role of N-functionalities in DeNOx mechanism
The generally agreed-upon mechanism of SCR on nitrogenated carbons does not exist. The role of N-surface functionalities was explained by the basic character of such groups. The changes in reductive character of graphitic carbons should also be taken into account.
There is a general agreement that N-doping of carbonaceous materials introduces changes of their electron density. Pollak et al. [105] studied oxygen chemisorption on cellulose- or Kapton-based carbons, as well as cellulose-based carbon with the addition of ammonium chloride, obtained by temperature-programmed pyrolysis in nitrogen or additionally activated by carbon dioxide. Both cellulose-based materials treated with N-compound and Kapton-based activated carbons contained N-groups (pyridinic and quaternary N), which facilitated oxygen chemisorption. A direct correlation between the amounts of nitrogen in the structure and the number of chemisorption sites for oxygen was observed. Similarly, Strelko et al. [102], Stőhr et al. [56], Matter et al. [106] and Maldonaldo [107,108] showed that increased electron donating by nitrogenated carbon materials influenced the reductive adsorption of oxygen.
Huang and Teng [96] concluded that the electron from N atoms substituted in the aromatic rings were delocalized and could be easily transferred to form reactive surface species and thus N-groups were adsorption sites for NO below 413 K and catalytic sites over this temperature. Kinetic studies revealed the negative value of the reaction order with respect to NO which was interpreted as the large amounts of NO adsorbed and stabilized on the surface. This according to the authors could retard ammonia adsorption on acidic sites.
Muñiz et al. [103] analyzed the adsorption behavior of polyarylamide-based activated carbon fibres in the presence and absence of oxygen. In the absence of oxygen, no NO adsorption was registered, in contrast to the case of the NO + O2 mixture. This was explained by the fact that the adsorbed species was NO2 instead of NO. The mechanism proposed by Muñiz et al. [103] was:
| Z(O) + NO → Z* + NO2 |
The conclusion of the findings of Muñiz et al. [84] was that basic surface groups are sites for NO2 formation on the Z* sites and NH3 adsorbs on acidic sites. Taking into account the above-mentioned arguments, an additional route for N-containing carbon materials will be presented (Fig. 3d).
5 O- or N-modified carbonaceous materials as supports for transition metal oxides/hydroxides in DeNOx
Transition metal oxides supported on typical carriers, such as Al2O3, SiO2 or TiO2 have maximum activity at medium temperature range around 523 to 723 K. Silica-aluminas, such as zeolites or layered clays, promoted with transition metal oxides/hydroxides, may be applied at higher temperatures, over 573 to ca. 873 K, depending on the material [109–112]. In contrast, promoted activated carbons proved to have high activity in the low temperature region, below 573 K. This is illustrated by the article of Yoshikawa et al. [3], who compared MnOx supported on activated carbon and alumina. At temperatures between 320 and 523 K, NO conversion was several times higher for the former than the latter catalyst. Similarly, Chuang et al. [2] proved that Cu supported on activated carbon was more active than on Al2O3. The effect was ascribed to a better distribution of Cu. Gálvez et al. [4] compared commercial V2O5/WO3/TiO2 catalysts with active carbon briquettes differently pretreated, with petroleum coke ash (main component V) as the active component. Under 573 K, NO conversion was 3–5 times lower for commercial catalyst. The extent of the improvement for carbon-based catalysts depended on the type of their pretreatment.
Several types of active components supported on activated carbons or other carbonaceous materials, such as e.g., nanotubes or semi-cokes were proposed as DeNOx catalysts. The most often studied were oxides/hydroxides of: vanadium [16,23,27,113–119], manganese [3,18,22,97,113,120–123], iron [3,81–83,121,124] or copper [122,125–127]. Rare earth (Ce, La, Pr, Nd) oxides supported on activated carbon fibres or semi-coke were also tested [88,128]. Several facts for MeOx supported on carbonaceous materials were well established. One of them is that the chemistry of surface carbon is of primary importance for catalytic performance of the MeOx-AC system. Table 3 summarizes the most important experimental information on MeOx/carbon-supported DeNOx catalysts. In this work, only catalysts with carbonaceous supports with the chemistry modified by the change in oxygen or nitrogen surface functionalities are considered. This choice was made based on the fact that the treatments leading to such changes resulted in improvements of SCR-NH3 catalytic performance.
MeOx supported on modified carbonaceous materials as catalysts for NO reduction with NH3 (if not stated otherwise).
| Carbonaceous material | Modification | Active component | Reaction conditions | Information on catalytic activity | Reference |
| AC | HNO3 | V 3% | SCR: 450 ppm NO, 500 ppm NH3, 5% O2 3% H2O, rest Ar | H2O inhibiting NH3 sorption on Lewis sites of V2O5/AC | [130] |
| AC | HNO3 {AC-Nconc, AC-N}, H2O2 {AC-H} and air {AC-air} | 3 wt%V + 3% petroleum coke ash (23% V, 3.5% Fe, 3% Ni) | SCR: 1000 ppmv NO, 1500 ppmv NH3, 3,5% O2, rest Ar, T = 150–325 °C | – NO conversion dependent on oxidative pretreatment of AC tor the same T: AC-Nconc > AC-N > AC-H > AC-air | [129] |
| Carbon–ceramic monoliths | Impregnation, calcination, carbonation in N2 at 700 °C. Oxidation by 40 wt% HNO3 at 90 °C | V, Mn | SCR: 700 ppmv NO, 800 ppmv NH3, 3 vol%, rest He | – Deactivation by SO2; higher for Mn then V – Reversible deactivation by H2O – Considerable catalysts aging the power plant over 200 days | [113] |
| AC | HNO3 | V 1–5% | SCR: 500 ppm NO, 560 ppm NH3, 3.3 vol% O2 and 400 ppm SO2 in Ar 300 mL/min; GHSV = 36,000 or 90,000 h−1 | NO conversion increased by SO2 (at 250 °C) for low V contents | [115] |
| Carbon nanotubes (CNT) | HNO3 | V | 800 ppm NO, 800 ppm NH3, 5% O2, rest He, 500 cm3/min, GHSV = 35,000 h−1 | NO conversion of V/CNT increased with the increase in CNT mean diameter | [17] |
| AC | HNO3 | V | 1000 ppm NO, 1500 ppm HN3, 3.5% O2 in Ar 22.2 cm3/min, mcat = 0.4 g | NO conversion higher for oxidized samples | [152] |
| AC | HNO3, H2SO4 | V | 1000 ppm NO, 1500 ppm HN3, 3.5% O2 in Ar 22.2 cm3/min, mcat = 0.4 g | – A correlation between carboxylic groups concentration and the amount of chemisorbed NH3 – A rough correlation between apparent kinetic rate constant and the amount of chemisorbed NO – Preparation steps (wet oxidation, calcinations) influenced catalytic performance | [4] |
| AC | HNO3 | 1, 3, 5, 6 or 8% V | 1000 ppm NO, 1500 ppm HN3, 3.5% O2 in Ar 100 cm3/min, mcat = 0.4 g | – NO increased for oxidized samples – The influence of V2O5 loading on NO conversion | [134] |
| AC | HNO3, 333 K | 5% V additives: W, Mo, Zr, Sn {V–M/AC (where M = W, Mo, Zr and Sn)} | 0.5% NO in Ar, 0.56% NH3 in Ar, 15% O2 in Ar rest Ar, mcat = 500 mg, 300 cm3/min | Effect of SO2 on NO conversion: – without SO2: V > V–Zr > V–W > V–Sn > V–Mo – with SO2: all additives, expect Mo increased activity | [148] |
| ACFs prepared from Nomex or powdered phenolic resin | HNO3, H2SO4, H2O2 or air | Mn | 700 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, 11,000–17,000 h−1 mcat = 0.3 g | NO conversion of Mn/ACF dependent on ACF pretreatment – linear dependence on acidity (exception treatment with H2SO4) – highest after HNO3 treatment Selectivity to N2 ca. 100% at 423 K, 92% at 472 K Gasification negligible at 423 K, more significant for 473 K (correlation with Mn content) | [22] |
| carbon–ceramic monolith | HNO3 | Mn | 700 ppm NO, 800 ppm NH3, 3% O2, 3.3% H2O, rest He, mcat = 1 g, flow 150 cm3/min | [91] | |
| AC | HNO3 {C or C90}, followed by: (i) NH3, 1073K–{C/N800/N or C90/N800/N/}; (ii) urea sorption and then curing in air 623 K {C/U/350 Air or C90/U/350 Air} | 5 % wt Mn | SCR: 800 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, flow rate 100 cm3/min, mcat = 400 mg | – NO conversion dependent on support pretreatment; C90/N800/N/Mn >> C90 C/N800/N/Mn C90/U/350 air >> C/U/350 Air/Mn – N2O formation much lower for MeOx on pre-oxidized AC | [95] |
| AC | As above | Mn | SCR: 800 ppm NO, 800 ppm NH3, 3 vol%O2, rest He, flow rate 100 cm3/min, mcat = 200 mg | – NO conversion dependent on MnOx distribution and difference in chemical state of active component – H2O decreasing NO conversion but increasing N2 selectivity | [97] |
| Carbon–cellular monolith | Air 387 K | Mn | 700 ppm NO, 800 ppm NH3, 3 vol% O2, rest He, 300 cm3/min, mcat = 300 mg, 3500–4500 h−1 | Pre-oxidation of support enhances Mn loading and thus improves activity | [151] |
| Carbon-based honeycomb monoliths | HNO3 | Cu2+, Mn2+ | 3000 ppm NO, 6000 ppm NH3, 2 vol% O2, rest He and N2 GHSV: 6390 h−1, 0.90 ± 0.05 cm edge square section and 4-cm long monolith used for each test | – Drying method (compared: conventional and microwave) has critical effect on final redox state and distribution of Cu – NO conversion not influenced by acid pretreatment of the support | [122] |
| Coconut shell-derived AC | HNO3 | Cu 3 wt% | SCR: 400 ppm NO, 400–800 ppm NH3, 6% O2 rest N2 500 cm3/min, different weights (0.2, 0.4, 1.0, and 1.5 g) | NO conversion dependent on: – oxidation of the support – method of active component introduction (microwave heated polyol process, polyol process impregnation) | [131] |
| AC | HNO3 | Fe3+ | 900 ppm NO 1080 ppm NH3 1% O2 rest He, mcat = 500 mg | NO conversion depends on the conditions of oxidation of AC | [156] |
| Deashed commercial AC | HNO3 | Fe3+ | 100 cm3/min, mcat = 400 mg, gas composition: 800 ppm NO and NH3, 3% O2, rest helium | NO conversion for Fe/AC-ox much higher than for NH3 or HNO3 treated AC | [83] |
| AC | HNO3 {AC-N}; H2O2 {AC-H}; H2SO4 {AC-S} | CuO | mcat = 500 mg, GHSV = 20,000 h−1, 2000 ppm NO (reduction C) | NO conversion: T < 225 °C: CuO/AC-H > CuO/AC-N > CuO > AC-S≈CuO/AC-S T > 225 °C CuO/AC-H > CuO/AC-N > CuO/AC-S > CuO/AC | [89] |
| AC from cotton stalk | Activation H3PO4 + H2O {CACS} Co-activation in N2 | Mn, Ce | mcat = 500 mg, NO 0.06 vol%, NH3 0.06 vol%, O2 3 vol%, rest N2 | NO conversion (max. 68%) depended on Mn/Ce ratio: CACS0.75-Mn/Ce > CACS1.5-Mn/Ce > CACS1.5-Mn/Ce> | [156] |
| ACF | 40% HNO3, 3 h, refluxing at 90 °C, 2 h; oxygen plasma | CeO2 | 1000 ppm NO and NH3, 5% O2, Ar; GHSV 11,000 h−1; mcat = 0.2 g | NO conversion: CeO2/ACF-HNO3 > CeO2/ACF-plasma | [88] |
| CN | 68% HNO3 | MnOx loaded on outer or outer and inner surface of CN | 800 ppm NO and NH3, 5% O2, rest Ar; GHSV = 35,000 h−1; mcat = 0.2 g | NO conversion higher for MnOx confined in channels | [18] |
| CN | HNO3 + H2SO4, 60 °C, 2 h | MnOx (8–14 wt%) | 900 ppm NO and NH3, 5% O2, He; H2O (when present) 2.5%; SO2 (when present) 100 ppm; GHSV = 30,000 h−1; mcat = 0.3 g | NO conversion depended on MnOx content (max. for 12%); above 200 °C, sel. To N2 decreased to an extent depending on the MnOx content; activity depended also on calcination T and time | [123] |
5.1 Activity
5.1.1 The influence of the oxidation of carbonaceous supports
As one of the first, Grzybek [124] and Grzybek and Papp [82] observed that oxidation with HNO3 prior to the impregnation with Fe(NO3)3 led to more active catalysts than those prepared with untreated support. This was explained by the presence of oxygen surface species influencing the distribution of active component. Based on XPS spectra, it was shown that the impregnation of non-oxidized activated carbons with solutions of metal precursor led to the formation of bigger aggregates of MeOx mainly on the outer surface of support particles. On the other hand, oxidation with HNO3 resulted in a more even distribution of smaller entities, present also in the pores. Similar conclusions were drawn for MnOx/active carbon catalysts [120]. A strong dependence on oxidation conditions was observed. The application of concentrated HNO3 at boiling temperature, 353 or 343 K followed by impregnation with Fe(NO3)3 resulted in XPS O/C ratio respectively 0.69, 0.37 and 0.29 and the increase in NO conversion by ca. 15% in each case at temperatures between 453 and 533 K.
The extent of the improvement of SCR activity and/or selectivity is dependent on the type and amount of surface functionalities, as proven by Marbán et al. [22] who studied MeOx on active carbon supports oxidized by different oxidizing agents H2SO4, H2O2aq, HNO3aq at varying air temperatures at temperatures between 473 and 573 K. The application of air was much less efficient than the use of HNO3. Activity could be correlated with surface acidity of the supports, with the exception of application of H2SO4. Although the latter oxidant increased acidity more than H2O2, SCR activity was low, due to the poisoning effect of S-species.
Xue et al. [105] compared NO reduction by carbon for CuO/activated carbon untreated or pretreated with HNO3, H2O2 or H2SO4 (designation here, respectively: AC–O, AC-N, AC-H, AC-S). They found the following sequences:
- • under 498 K: CuO/AC-H > CuO/AC-N > CuO > AC-S ≈ CuO/AC-S;
- • over 498 K: CuO/AC-H > CuO/AC-N > CuO/AC-S > CuO/AC.
Again there was a correlation between NO conversion and the number of oxygen surface groups. The effect was ascribed to the changes in CuO dispersion.
Gálvez et al. [129] found that oxidization of carbon supports with HNO3 and H2O2 led to V2O5/AC catalysts more active than those with non-oxidized supports. Oxidation with air at 473 K, however, did not increase NO conversion. The enhancement resulting from HNO3 treatment was attributed to the creation of a higher number of acidic surface groups and thus of more sites for NH3 sorption after HNO3 or H2O2-oxidative procedures.
Huang et al. [24] studied viscose-based ACF oxidized by HNO3 or oxygen plasma as supports for V2O5 (3-12 wt%). The types of groups formed by these treatments influenced the performance of resulting V2O5/ACF catalysts. There was a correlation between the amount of introduced O-species and NO conversion.
The possible gasification problems led to the idea of the application of more stable carbonaceous materials (carbon nanotubes) as supports. Pourkhalil et al. [123] studied nanotubes functionalized by HNO3 or H2SO4 and loaded with 8–14% MnOx. High activity and selectivity to N2 were registered at temperatures of 100 to 200 °C for catalysts containing 8–12% of MnOx. However, stability as a function of time or the amount of CO2 in the products was not tested.
Surprisingly enough, Ouizzine et al. [122] who studied Cu or Mn supported on carbon-based honeycomb concluded that HNO3-pretreatment did not affect catalytic performance of CuO catalysts. However, the authors compared monoliths under different conditions (different W/F), so direct correlations are impossible.
The beneficial effect of oxidative pretreatment was so well established that all successful preparation procedures applying activated carbons or other type of carbonaceous material (e.g., nanotubes) are now started with oxidative pretreatment of the support [e.g., 4,18,115,117,125,133-135].
5.1.2 The role of N-functionalities
Carbonaceous materials with the increased number of N-surface functionalities were also considered as supports for MeOx (cf. Table 3) [22,23,95,97].
The preparation procedures consisting of a combination of two or three treatments (oxidative, the modification by N-compounds and the introduction of transition metal oxides/hydroxides) were also reported. As an example, Fig. 2d and e compare the catalytic DeNOx performances of selected MnOx (based on data in [95]). The introduction of both oxides resulted in an additional increase in NO conversion, especially at the lower studied temperature region. Similar results were obtained for MnOx supported on activated carbon treated with NH3 [34].
5.1.3 N2O formation
The introduction of active component onto carbonaceous supports leads on one hand to the increase in activity but, on the other hand, to a certain decrease in selectivity to N2 (cp. e.g., [83] for FeOx/AC; [95,97] for MnOx/AC). The amount of formed N2O depends on the choice of active component, on its distribution, and on the temperature of the reaction. A direct correlation is difficult to establish, because the experiments were carried out under different conditions. Some general conclusions are, however, possible:
- • the amount of formed N2O increased with temperature, but at low temperatures (below 473 K), it was usually negligible. Such results were found for FeOx/AC [83] or MnOx/AC [95,120];
- • there was a strong dependence on the MeOx distribution. The changes in distribution were obtained either by support pretreatment and/or the choice of Me precursor. In contrast to larger aggregates of active component, smaller entities resulted in improved selectivity to N2, with less N2O in the outlet stream [95,97,120];
- • the choice of the precursor for non-oxidized support resulted in different oxidation states of MnOx oxides, which influenced N2O formation for MnOx/AC [120];
- • the presence of H2O influenced N2O formation, as discussed in more details in Section 6.
5.1.4 The influence of preparation parameters of MeOx/carbon materials on DeNOx performance
Apart from oxidative modification of carbonaceous support, other preparative parameters considered for the specific catalysts were: type of active component precursor, the method of the introduction of active component, the influence of calcination of the catalysts before reaction, etc.
Marbán and Fuertes [133] studied the influence of impregnation procedure for MnOx/ACF prepared from Nomex. Two procedures were used: equilibrium adsorption and pore volume impregnation, the former leading to higher NO conversion. Calcination, which followed the introduction of the active phase, revealed that both conversion and selectivity depended on the applied atmosphere, temperature and time. Calcination at 673 K led to the formation of Mn3O4 and MnO. Calcination in 3% O2 in helium was successful at temperatures lower than 473 K, as for higher temperatures, extensive gasification was observed. Oxidation time between 1 and 18 h influenced gasification rate only slightly. NO conversion increased up to 10% with a similar loss in activity for higher calcination times. XRD of oxidized samples revealed only Mn3O4 while TPR experiments pointed to ca. 60% Mn2O3 + MnO2 and ca. 40% of MnO + Mn3O4. The authors concluded that such oxidation procedure had little influence on the oxidation state of active material and thus oxidative treatment was not justified.
Gálvez et al. [4] studied V2O5 or petroleum coke ash supported on active carbon briquettes. Catalytic behavior was influenced by calcination temperature. As the most efficient, calcination in Ar at 473 K or 873 K for 4 h was found. Longer calcination at 473 K (6 h) or higher temperatures, 673 K or 773 K (4 h), were less efficient. The observed effects were explained by the formation of vanadium oxides, which are highly active in SCR, and changes in the type/number of O surface species on the support.
The effect of calcination temperature and time was also studied by Pourkhalil et al. [123] for 12 wt% MnOx supported on carbon nanotubes pre-oxidized by HNO3 and H2SO4. Calcination was carried out in helium at 300 °C for 2, 3, 6 or 12 h or at 400, 500 or 600 °C for 3 h. Calcination at 300 °C led to higher activity and selectivity to N2 than the treatments at other temperatures, which was ascribed to the changes in dispersion and structure of MnOx. Higher calcination temperatures favoured the transition from more active MnO2/Mn2O3 to less active MnO. The prolongation of calcination time at 300 °C decreased activity but did not influence selectivity.
MeOx loading was usually found to influence NO conversion. The extent of the improvement depended, however, on the exact nature of the support.
Lázaro et al. [134] compared V2O5/AC (monolithic samples) and catalysts based on powdered active carbon with petroleum coke ash (PCA) containing V as the main constituent. For the former catalysts, NO conversion increased from 12% to ca. 68% for V content, respectively from 0 to 6 or 8%. For the latter, there was a very small difference in NO conversion between 1 and 5% PCA, respectively 45 and 50%.
Huang et al. [23] studied V2O5 on activated carbon fibres (ACF). The support was either non-oxidized or oxidized by HNO3 or plasma. There was a strong dependence of NO conversion on V content (3–12 wt%) for a catalyst with non-oxidized ACF or oxidized by plasma. For the catalyst on the support oxidized by HNO3 the V loading had, depending on temperature, little or no effect on activity. These differences were explained by different dispersion of V2O5.
5.2 Stability of MeOx/modified active carbon systems
The introduction of active component on modified carbonaceous materials may influence stability, and especially the formation of CO2. For some active components and some temperatures, increased gasification of the support was observed. Marbán et al. [22] observed no gasification for MnOx on oxidatively pretreated activated carbon fibers at 423 K. At 473 K, however gasification was registered and the rate depended on Mn content, which was explained by manganese oxides catalyzing reaction between oxygen and carbon.
Gálvez et al. [4] observed that stability of V2O5/AC catalysts supported on oxidized AC was lower than on un-pretreated AC.
Lázaro et al. [134] observed higher stability for supports prepared from polymer blends than from coal. The addition of V2O5 slightly decreased stability.
5.3 Mechanistic considerations for MeOx/carbon systems
Some mechanistic aspects were considered for carbon-based catalysts with the added active component. The most detailed work was published by Marbán et al. [91]. NH3 role in SCR mechanism at 398 K was studied for Mn3O4/AC composite or carbon–ceramic monolith, as is shown in Fig. 3e. The role of active component was discussed in detail and some remarks concerning carbon support were also included (as discussed in Section 3). Two main mechanisms proposed were:
- • without oxygen, consisting of the following steps:
- ∘ NH3 adsorption on O-atoms,
- ∘ formation of aminooxy groups,
- ∘ reaction of aminooxy groups with gaseous NO or NO2 (the latter resulting from oxidation of nitrosyl groups with gaseous O2),
- ∘ dehydroxylation of Mn3O4,
- ∘ re-oxidation of O-vacancies by NO with the formation of N2;
- • with oxygen, consisting of the following steps:
- ∘ NH3 adsorption on –OH groups of Mn3O4 leading to NH4+,
- ∘ NH4+ reaction with gaseous NO2 resulting from nitrosyl oxidation with gaseous oxygen.
It must be mentioned, however, that the mechanism is changed at higher temperatures, which was very shortly addressed by the authors. It was stated that at 448 K (i) NO2 was no longer formed, (ii) the surface of the active component was reoxidized by gaseous oxygen instead of NO/O2 and (iii) nitrate formation did not take place because of the absence of NO2.
Lately an attempt to elucidate the full mechanism for semi-coke-supported V2O5 has been published by Wang et al. [119]. Based on the results of DRIFT studies, the authors postulated that SCR-NH3 over V2O5/semi-coke took place over two pathways, that of SCR and fast SCR. In the first pathways, the reaction proceeds according to the Eley–Rideal mechanism:
| NH3(g) (over Lewis sites) → NH3(a) |
| NH3(a) + O(a) → NH2(a) + OH(a) |
| NO(g) + NH2(a) → NH2NO(a) |
| NH2(a)NO → N2(g) + H2O |
The support provides the possibility for fast SCR, according to Langmuir–Hinshelwood mechanism, through oxidation of NO over active carbon sites:
| 2 NO + O2(g (over AC sites) → 2 NO2(a) |
| NH3(g) (over Lewis acid sites) → NH3(a) |
| NO2(a) + 2 NH3(a) → NO2(NH3)2 |
| NO2(NH3)2 + NO → … → 2 N2 + 2 H2O |
It should be mentioned, however, that NO oxidation is faster at lower temperatures while at higher temperatures, the third mechanism – direct reduction of NO by carbon material – cannot be excluded. As an example, the article of Bhaduri and Verma [135] may be quoted. The authors studied nitrogen-doped carbon micro/nanofibers with supported CeO2 and copper nanoparticles as catalysts for NO reduction without the addition of a reducing agent at 400 °C. NO conversion for unpromoted carbonaceous material was low (ca. 10%) and ascribed to the presence of oxygen- and nitrogen-containing surface species, according to the reactions:
| C(O) + NO → C(N) + COx (removal by oxygen groups) |
| Cf + NO → C(N) + COx (removal by free active sites) |
| C(N) + NO → N2 + COx (reduction step). |
The presence of copper increased NO conversion, according to the reactions:
| 2 Cu + NO → Cu2O + ½ N2 |
| Cu2O + NO → 2 CuO + ½ N2 |
| 2 CuO → 2 Cu + O2 |
The addition of CeO2, playing the role of O2 pool, increased the number of oxygen surface functionalities.
For other systems, only partial mechanism information is available. It is obvious from the literature data that the introduction of active component increases the number of sites for NH3 sorption in comparison to the support, as well as may positively influence reducibility of the system. The former was supported by experimental data for MnOx/AC [91,95], V2O5/carbon nanotubes [17], V2O5/carbon-coated ceramic monoliths [16].
Gálvez et al. [4] found a correlation between the concentration of carboxylic groups and of the apparent kinetic rate constant with the amount of chemisorbed NH3 for V2O5/AC. IR and transient kinetic studies showed NH3 adsorbed on Brønsted acid sites. The authors concluded that carboxylic groups accumulated NH3, which could migrate to vanadia, where reaction was completed.
García-Bordejé et al. [16] reported that the addition of a second metal oxide led to further changes in the number of NH3 sorption sites. When 1% Me was added to 3%V, the following sequence was found: V–Fe ≈ V–Mn > V–Cu > V > V–Cr. However, the role of a second metal was not limited to the formation of NH3 sorption sites, as NO conversion leads to different sequence: V–Fe > V–Mn > V–Cr > C–Cu > C. The authors concluded that, the second metal increased reducibility.
Much less information is available on the role of NO in DeNOx.
As an example, Huang et al. [17] found that NO sorption for V2O5/carbon nanotubes was very low and concluded that gaseous NO interacted with NH3 adsorbed on Lewis sites.
On the other hand, Wang et al. [127] confirmed the role of rare earth (La, Pr, Nd) oxides and CeO2 on semi-coke in the oxidation of NO to NO2, which led to the increased NO conversion via increased oxygen vacancies. The authors concluded that NO and NH3 adsorbed on different sites, the former on active oxygen atoms stored in oxygen vacancies of Ce3+/Ce4+ system and the latter on Lewis sites, thus leading to the overall process following Langmuir–Hinshelwood mechanism in the temperature range of 150–300 °C.
Xue et al. [89] found from TPD NO that NO adsorbed on CuO sites in two forms, desorbing as NO or N2. The amount adsorbed was dependent on the type of pretreatment of the support: AC-ox H2O2 > AC ox. HNO3 > AC ox. H2SO4 > AC. The authors postulated the following mechanism for NO reduction with carbon (Fig. 3f):
| 2 Cu* + NO → Cu*–O + Cu*–N |
| 2 Cu*–N → 2Cu* + N2, |
6 H2O and SO2 influence on catalytic performance
The presence of water in SCR mixtures may lead to a larger or smaller decrease in activity, depending on the catalyst type. Only a few studies address the problem of water for carbon-based catalysts [16,97,113,123,136–138].
At low temperatures, H2O had an inhibitory effect on DeNOx performance even when water was not added to the reaction mixture, as found for SCR reaction carried out for ca. 8 h at 413 K for N-promoted activated carbons prepared by urea treatment [97] or ammoxidation [98] (cp. Fig. 2f), as well as for doubly promoted (N and MeOx) carbons [97]. This is illustrated by Fig. 2 g for N- and MnOx-promoted active carbon (cp. Fig. 2g [97]). At 453 K, all the above-mentioned activated carbons proved stable without water online and this, in connection with TPD showing the extensive peak of water desorption up to ca. 423–453 K, suggests that probably NH3 (substrate) and H2O (product of the reaction) compete for adsorption on hydrophilic sites at lower temperatures of SCR process.
On addition of 10% water to SCR mixture, Muñiz et al. [103] registered that activated carbons obtained by carbonization and activation of Nomex fibers underwent deactivation at temperatures below 523 K. At 373 K, the decrease in NO conversion was ca. 80%. Over 573 K, there was no inhibitory effect [103]. Similar temperature dependence was observed for active carbon fibres oxidized and then treated with NH3 at 523 K, although in this case some small deactivation was registered, even at higher temperatures [131]. Deactivation upon addition of H2O was also found at 453–533 K for active carbons doubly promoted with urea and MnOx [96] or FeOx [139], but the extent of deactivation was smaller than, e.g., for Mn/Al2O3 [138]. Moreover, it was totally reversible after removing H2O from the feed, similarly as found for vanadium on AC [137], or V, Fe or Cr oxides supported on mesoporous carbon covered cordierite monolith [121]. For V2O5/AC, that inhibition effect was reported to increase with increasing H2O content [16].
It should be stressed, however, that the presence of water in the feed positively influenced selectivity in SCR reaction. For Mn/N/AC catalysts [97], the amount of N2O in the products diminished, especially at higher temperatures (493, 533 K) to a much higher degree, than would be expected from the decrease in NO conversion. Similar results were registered by some authors for other (non-carbon-based) catalysts: MnOx supported on γ-Al2O3 at 598 K [138], iron-vanadyl phosphate catalyst [141] or on a commercial V2O5/WO3/TiO2 catalyst [142].
A consistent mechanism including the role of H2O has not been offered as yet. As discussed in detail in [97], it is not even concluded without doubt if NH3 or NO sorption sites are affected.
Huang et al. [137] showed for V2O5/AC that H2O did not influence NO sorption, while sorption of NH3 was even higher in the presence of water due to the increased Brønsted acidity. Bagnasco [141] also reported increased NH3 sorption at 423 K in the presence of H2O for iron-vanadyl phosphate SCR catalysts and attributed it to the formation of new Brønsted acid sites.
Valdés-Solís et al. [113] attributed the inhibitory effect of H2O to its competition for adsorption sites with NH3 (Mn/carbon–ceramic catalysts). Similar explanation was offered by Amiridis et al. [143] and Klijstra et al. [140] for non-carbon-based catalysts: MnOx supported on V2O5/TiO2 [143] and MnOx/γ-Al2O3 [140]. Amiridis et al. [143] confirmed this based on kinetic measurements and Raman spectra. Klijstra et al. [140] offered for MnOx/γ-Al2O3 a more complicated mechanism explaining H2O inhibition. The model takes into account molecular adsorption of H2O and the formation of stable surface hydroxyls. TPD studies showed that H2O competed for sites with both NH3 and NO in the presence of O2. However, kinetic measurements revealed that the reaction order in NH3 was not affected, in contrast to reaction order in NO. This was interpreted as inhibition of NO sorption being relevant for SCR. NH3 was sorbed on the surface in excess and thus its sorption inhibition was believed as not important for the overall NO reduction.
The explanation of the decrease of N2O formation is also not totally understood. Taking into account that: (i) the increase in selectivity to N2 was more pronounced for catalysts with high amounts of labile oxygen [97] and (ii) at least part of N2O originates from oxidation of ammonia, it may be speculated that the decrease in N2O formation is due to the limitation of available oxygen by H2O sorption. This may be supported, e.g., by Curtin et al. [144] who claimed that water decreased the number of labile oxygen species on the surface and thus restricted the access of NH3 molecules to oxidation sites.
The presence of SO2 in flue gases may also lead to deactivation. On the whole, limited effort has been made to address the problem of sulfur tolerance. Liu et al. [145] published an extensive review on the subject of deactivation of different types of catalysts by SO2, which shows that there are still a lot of open questions in this matter and in some cases the experimental information is contradictory, even for similar catalysts.
SO2 interaction with active carbon-based catalysts was considered only in a few publications. The influence of SO2 on activated carbon modified by N-species was discussed by Samojeden et al. [100,146]. Their research showed that the decrease of NO conversion on catalysts prepoisoned by SO2 containing mixture [100]. There was a possibility of partial or total recovery of activity after regeneration by heating at helium [146].
Valdés-Solís et al. [113] reported the influence of SO2 on MnOx or VOx supported on carbon–ceramic monolith. First, SCR was carried out on a fresh catalyst, which was then subjected to a stream of SO2 (350 ppm) and O2 (3%) in nitrogen, followed again by SCR measurement at 423 K. The poisoning procedure was repeated for different periods of time until no change in the following SCR reaction was observed. Higher deactivation was found for Mn than for V-containing catalysts, due to the formation of manganese sulfates on the surface. For V/AC, the loss of catalytic activity was ca. 20% of the initial value. As a possible reason for the relatively small loss in activity, the formation of ammonium sulfate NH4HSO4 or (NH4)2S2O7, which may react with NOx, was claimed.
On the other hand, Zhu et al. [115,116] reported that V/AC catalysts showed an increase in activity in the presence of SO2 at temperatures over 453 K. This was explained by (i) the decomposition of NH4HSO4 being promoted by AC and only slightly by V2O5 [115], or (ii) the increase of surface acidity [116]. The positive effect was observed for low V contents only. The authors proposed thus two different mechanisms for SO2 promotional effect of SO2 for vanadium supported on mesoporous carbon-coated monoliths. Similar behavior was also observed by García-Bordejé et al. [16]. The so-increased activity was stable for at least 20 h at 423 and 453 K and was ascribed to the increased acidity. Additionally, mesoporosity of carbon material played a positive role; for typical carbon supports there is an accumulation of NH4HSO4 in the pores while no plugging was found for mesoporous carbons.
These observations were only partly confirmed by Hou et al. [114] who studied the influence of different loadings of V2O5 on the extent of deactivation by SO2. No deactivation by SO2 was observed for active carbon support and activity in SCR reaction increased for ca. 50 minutes to reach a steady state of ca. 90% for 0.5 or 2 wt% V2O5 AC. For higher vanadium loading (5 wt%), a small negative effect was observed. The introduced SO2 showed a promoting effect even after the removal of sulfur dioxide from the feed, suggesting a positive role of S containing surface species (most probably sulfate anions) on the surface, which form additional sites of acidic character and thus may promote ammonia adsorption.
Certain additives were found to improve catalytic performance in the presence of SO2. As an example an industrial V2O5/TiO2 catalyst with WO3 additive is most often quoted. Zhu et al. [147] studied the influence of oxides of W, Mo, Zr and Sn on the catalytic activity of V2O5/AC. In the absence of SO2, NO conversion formed a sequence: V2O5/AC > V–Zr/Ac > V–W/AC > V–Sn/AC > V–Mo/AC. When SO2 was present in the feed, all additives, except MoO3, increased SCR activity. For Sn addition, the increase was considerable and stable for several hours at temperature as low as 473 K, although SnO2/AC showed no activity in SCR. NO conversion for V–Sn/AC was stable for several hours, depending on the details of the preparation procedure, which was attributed to different interactions between V and Sn species. Deactivation which followed after some time on stream (20 h to ca. 50 h, depending on the preparation technique used) was explained by pore plugging by deposited sulfates.
Tang et al. [148] compared the deactivation of Mn, Cu, Fe and V oxides supported on ceramic support covered with activated carbon, either promoted or unpromoted. All catalysts showed irreversible deactivation after exposure at 473 K to SO2 containing feed. The loss in activity formed a sequence: MnOx > CuO > FeOx> V2O5. Doping of MnOx with cerium oxide had a negative effect in contrast to Mn/ZSM-5 catalyst. The latter upon contact with SCR feed containing H2O and SO2 was reported to show only minimal decrease in DeNOx activity [127]. On the other hand, doping of MnOx-based monolithic catalysts with Fe or V decreased the extent of deactivation to reach ca. 51 and 56% of the initial value, respectively [148]. SEM showed that deactivation was due to the formation of oxides of active material and the deposition of ammonium sulfate on the surface, the former effect being irreversible and the latter possible to remove by washing.
The simultaneous presence of H2O and SO2 was studied for V2O5/AC by Huang et al. [138] who reported a strong deactivation of V2O5/AC catalysts, in contrast to the effects observed when only H2O or only SO2 was present in feed. H2O deactivated the catalysts at 523 K only slightly while the presence of SO2 led to the increase in activity, as discussed above. Two reasons for deactivation by H2O + SO2 were given: (i) competitive adsorption of H2O and NH3 and/or NO, and (ii) pore blocking by NH4HSO4 and (NH4)2S2O7 whose deposition rate is increased in the presence of water. In the absence of H2O, those salts could react with NO, which resulted in stable catalytic activity. Deactivation strongly increased with V2O5 loading, which was due to the increased SO2 oxidation.
The simultaneous addition of SO2 and H2O to the reaction mixture was also studied by Pourkhalil et al. [123] for 12% MnOx loaded carbon nanotubes. The decrease in NOx conversion was observed which was partly reversible after the removal of these poisons. The deactivation was ascribed to the formation of ammonium sulfate salts while no MnSO4 was registered by XRD. The full initial activity could be regained after heating of the catalyst in helium at 350 °C for 2 h.
7 The application of monolithic supports
Industrial application of DeNOx catalysts requires an appropriate configuration due to high flows of flue gases. The preferred solution, realized on industrial scale in case of V2O5/WO3/TiO2 catalysts, is the application of a monolithic or a plate catalyst. For carbonaceous catalysts, the following possibilities are taken into account: (i) carbon honeycombs [24,121,122], or carbon–ceramic monoliths [16,24,117,150], or (ii) carbonaceous materials fixed on plates [149], or (iii) activated carbon briquettes (for smaller stationary sources) [4,132,152,153].
The preparation of carbon monoliths was described e.g. by Marbán et al. [154]. The monoliths were prepared from Nomex rejects fibres carbonized in N2 at 1123 K, milled and dry-mixed with powdered phenolic resin, formed in a mold, cured and carbonized again, followed by activation in steam at 1023 K, resulting in composites with specific surface area up to ca. 1300 m2/g. Other support-forming carbon fibres (Rayon, phenolic resin-, pitch and PAN-derived) were also studied [121]. As active materials oxides of manganese, iron, vanadium, chromium or nickel were chosen. The DeNOx activity order was found to be: Fe > Mn > V > Cr > Ni [121].
Ouzzine et al. [122] reported the preparation of carbon monoliths formed from volatile bituminous coal with the following additives: silicate clay, glycerin, methylcellulose, and aluminum phosphate dissolved in o-phosphoric acid. The extrudates were dried, carbonized and activated. As active components, Mn or Cu was used. High NO conversion over a fairly broad temperature window was registered.
Carbon–ceramic materials are classified into two types: integral and coated. The former are obtained by mixing the carbon precursor with binders and other substances but they often have low mechanical strength, while the latter are prepared by dipping of a ceramic honeycomb in a polymeric solution, followed by the procedures of active carbon layer formation: carbonization and activation [132]. Valdés-Solís et al. [132] used cordierite monoliths and phenolic resin, furanic resin and polysaccharides in appropriate solvent as activated carbon precursors. Carbonization was carried out in nitrogen stream up to 973 K (heating rate 5 °C/min) and then physical (steam/nitrogen mixture at 973 K) or chemical activation followed (zinc chloride). Up to 14 wt% C could be introduced, deposited carbon was found to be uniformly distributed and carbon-coated monoliths were claimed to have higher mechanical strength than the uncoated materials [132]. Carbonized monoliths activated by gasification in steam/nitrogen stream, with MnOx as active components were tested in SCR-NH3 [150]. The best catalytic results were obtained for catalyst support with 40% burn-off, oxidized with nitric acid and Na-exchanged prior to impregnation with Mn precursor. NOx reduction was 73% at 423 K at space velocity of 4000 h−1. At higher temperatures loss of selectivity was observed [150].
Cordierite monoliths covered with a blend of two polymers (Furan resin and polyethylene glycol) were proposed by García-Bordejé et al. [16,117]. As active component vanadium oxide was used. Dispersion and structure of vanadium species determined activity and selectivity, respectively. Loading higher than a monolayer resulted in a considerable decrease of both [117]. The addition of a second metal (Fe, Mn, Cu or Cr) led to a small increase in specific activity which was attributed to better reducibility of bimetallic catalyst in comparison to a monometallic one [16]. Boyano et al. [24] studied the influence of oxidative pretreatment of polymeric carbon covered monoliths with VOx as active component. NO reduction activity followed the trend: V/AC/ox HNO3 > V/AC/H2SO4 > V/AC/H2O2 and was related to the type of oxygen surface species created in the oxidation process. Li et al. [151] tested carbonaceous catalysts modified with Cu, which were pasted on aluminum plates with organoclay, and inserted into a fixed-bed flow reactor.
A different approach to the problem was presented by Boyano et al. [152], Lázaro et al. [132,153], and Gálvez et al. [4]. Carbon-supported catalysts were prepared in the form of briquettes from Spanish low-rank coal, which was first pyrolized, then blended with tar pitch and pressed into briquettes. Pressed briquettes were pyrolized and activated by steam or carbon dioxide at different temperatures and residence times [3,153]. Vanadium was chosen as the active component. As one of the possible sources of V, petroleum coke ash was considered [4]. The so-prepared catalysts were active in SCR-NH3 at low temperatures (423 K) [4,134]. Catalytic briquettes showed also good thermal stability while mechanical strength depended strongly on activation and oxidation processes [134].
8 Conclusions
Carbonaceous materials (activated carbons, carbon fibres, carbon cloth or nanotubes) are promising catalysts for low-temperature SCR with ammonia. Several facts have been well established.
The surface chemistry of carbonaceous material plays a decisive role in the preparation of an efficient catalyst. The most important preparative factors are:
- • the presence of oxygen functionalities; they increase activity only to a small extent, but more importantly, influence the distribution of active component for MeOx/C systems, thus improving both NO conversion, and in some cases also selectivity. Two effects play a role:
- ∘ the increase in polarity of carbonaceous materials which allows a better contact with aqueous solution of the precursor,
- ∘ the direct interaction (exchange) of Men+ cations with acidic surface groups.
On carbonaceous materials whose surface is characterized by higher acidity, smaller aggregates of active component are formed in contrast to bigger aggregates deposited mainly on the outer surface of unoxidized support particles. Smaller aggregates result in lower amounts of N2O in the product mixture.
The introduction of nitrogen functionalities considerably increases activity, as well as the stability of carbon materials towards oxidation. The extent of the improvement depends on both the type of N-compound used and the pretreatment (oxidative treatment prior to the introduction of N-species is beneficial). The appropriate materials may be also obtained starting from N-containing polymers or other carbon precursors modified by N-species, followed by carbonization and activation.
Several transition metal oxides/hydroxides supported on activated carbons result in the increased activity at low temperature region of SCR-NH3.
Vanadium, manganese, iron and copper were identified as the most efficient oxides/hydroxides.
The performance depends strongly on the chemistry of the support, as well as on other preparation details, such as the method of introduction of the active material, MeOx loading and calcination temperature. It is especially important to control the type and amount of oxygen surface functionalities of the support, as explained above. In that respect, wet oxidation, especially HNO3, proved to be the best choice. However, the overall effect depends also on the concentration of the oxidant, the duration of the process and its temperature.
The introduction of MeOx usually results in higher conversion, but also in decreased selectivity and stability. The latter two effects depend on temperature and are not very important at low temperatures (T < 473 K).
Methods were established to prepare carbonaceous systems in the form of monoliths – either integral or carbon–ceramic, or carbon briquettes, thus making it possible to use such materials in SCR-NH3 on an industrial scale.
There are full or partial mechanistic models for modified activated carbons. It was recognized that NH3 is adsorbed on acidic sites, either phenolic, or carboxylic and lactonic. There are some arguments that phenol groups form the main site, but other mentioned groups also play some role. The sites for NO are less certain. It is agreed that they are basic in character. For unmodified or oxidized carbon materials, CO surface groups are believed to form the reaction sites. The increase in basicity by N-functionalities is responsible for increased NO conversion. On the other hand, some models assume that SCR on carbon materials follows Eley–Rideal mechanism with NO interacting from gas phase.
There are still several open questions that need to be considered.
- • a detailed analysis of the relation between the type of the formed nitrogen functionalities and DeNOx improvement is necessary, in order to optimize catalytic performance. Especially, the role of such functionalities has to be further studied. The experimental information suggests that they increase NO sorption due to their basic character. Special role of pyrrolic/pyridinic group is suggested. However, there are also indications that N-functionalities, due to their electron donor properties, may be sites for NO oxidation to NO2. In turn acidic NO2 molecule sorbs on basic sites;
- • more experimental data on H2O and SO2 are needed. In most cases, H2O leads to deactivation, the higher, the lower the temperature. Upon removal of H2O, full activity is usually recovered. Additionally, N2O formation is decreased. It was not decided, however, without doubt, which adsorption sites are influenced, those for NO or NH3. SO2 may influence DeNOx performance in several ways: (i) by the formation of new Brønsted sites, leading to the increase in activity (as found for V2O5/AC catalysts), (ii) by poisoning of active component, decreasing conversion, or (iii) by the formation of ammonium sulfates which plug the porous system. The latter may be decreased by changes in porosity, which was proven by higher stability of mesoporous carbons derived from polymer blends in comparison to typical microporous activated carbons;
- • although mechanistic models have been elaborated for carbonaceous materials, only partial solutions exist for more complicated catalysts, e.g., MeOx/AC or V2O5/activated semi-coke. The most detailed model of Marbán et al. [91] for manganese oxides on carbon support may not necessarily be applicable for typical DeNOx case, as it concerns temperatures as low as 398 K. The model of Wang, on the other hand, suggests both Langmuir–Hinshelwood and Eley–Rideal mechanisms in SCR-NH3 on V2O5/activated semi-coke. For higher temperatures, direct reduction of NO by carbon cannot also be totally excluded. Thus, more mechanistic studies are necessary. The established fact for MeOx/C systems is that the active component may increase the acidity and/or the reducibility of the system. Thus (i) more NH3 is sorbed on the surface, and (ii) NO may dissociatively sorb on MeOx sites, which are then replenished by oxygen from gas phase. NO may also form nitrosyls. From the studies of Wang et al. [119,127], it may be also derived that NO oxidation on the support may play an important role.
Acknowledgments
The work was supported by AGH grant No. 11.11.210.213.