

1 Introduction
Nitrous oxide (N2O) is a harmful greenhouse gas, which stays in the atmosphere for 150 years and contributes to ozone layer destruction [1]. Nitric acid production has been recognized as the biggest industrial source of N2O emissions. The catalytic decomposition of N2O into N2 and O2 represents a possible way of N2O emissions reduction. Different materials were examined as catalysts for the N2O decomposition reaction [2], among which the most promising groups are Fe-zeolites [3,4], Co-spinels [5,6] or calcined layered double hydroxides [7,8].
Sufficient activity at the temperatures usable for practical applications (low temperature application up to 500 °C), stability in the presence of other components (water vapor, NOx, etc.) and the price of the prepared catalysts represent the main target of today's research effort. Depending on the structure and method of production, oxide catalysts can be divided into two main groups: bulk catalysts and supported catalysts. Bulk catalysts are mainly produced when the active components are cheap. Since the preferred method of their production is precipitation, they are also known as precipitated catalysts.
In the last years, also supported catalysts have been extensively investigated. One of the best-known methods for producing supported catalysts is the impregnation of porous support materials with solutions of active components. After impregnation, the catalyst particles are dried, and the metal salts are decomposed to the corresponding oxides by heating. Especially catalysts with expensive active components, such as noble metals, are employed as supported catalysts. However, impregnated catalysts have many other advantages apart from price compared to precipitated catalysts. Their pore structure and specific surface area are largely determined by the support, so the priority of the supported catalysts could be their high surface area and good mechanical strength together with the reduction of the risk of sintering [9]. The catalyst support generally has two main functions: (i) it provides a thermally stable and mechanically robust platform for active phase dispersion and (ii) it provides a suitable environment for mass transfer (large pores, hydrophilic/hydrophobic surface). The widely applied support materials are Al2O3, SiO2, TiO2, ZnO, ZrO2, CaO, and CeO2 [10–27].
The most common support Al2O3 has also been often tested as a support of catalysts for N2O decomposition [10–12,14]. Mostly noble and transition metals were used as active components [11,14,10], however, other metal oxides also possess promising activities [28,29]. Dimitrova and Mehandjiev [29] found out that in addition to the calcination temperature and impregnation solution concentration, the amount of Co3O4 formed and its surface area are strongly affected by the texture of the support used. The specific surface area of the support and its pore size distribution play an important role with respect to the amount of cobalt bonded to Al2O3.
TiO2 is another available support tested for N2O catalytic decomposition. The TiO2–sepiolite matrix appeared to play a key role in the process: it facilitates the migration of oxygen species through support vacancies and their final recombination [15–17], the step that is demanding during N2O decomposition.
Mesoporous materials represent a special type of nanomaterials with ordered arrays of uniform nanochannels. These materials have important applications in a wide variety of fields, such as separation, catalysis, and adsorption or as advanced nanomaterials [21]. Mesoporous silica, SBA-15, which contains larger pores, thicker walls and higher thermal stability in comparison with other mesoporous silicas as a support was also tested for N2O decomposition by several authors [25,30]. Du et al. [25] postulated that the trend in the activities change corresponds well with the dispersion status of the Rh species on SBA-15: the better the dispersion was, the higher was the activity.
Apart from commonly used supports, scientists are also searching for other materials, especially for inexpensive and easy accessible ones. Such a type is represented, e.g., by different slags from iron and steel production. The blast furnace slag processes a good adsorption capacity of transition metals ions, which enables the modification of the surface [31,32].
Since supported catalysts seem to be promising materials for N2O catalytic decomposition, we decided to prepare and test a series of catalysts loaded on different supports with the same active component. The guiding principle for support selection was variable in kinds and easy accessibility of the support. For these reasons, commercial supports and also waste materials from steel production were chosen to examine the possibility of utilization of local low-cost and abundant waste materials as catalyst supports. A CoMnAl mixed oxide was chosen as the active component on the basis of the research provided previously [33,7,34].
Co3O4 as active phase deposited on different kind of support was studied by Shen et al. [35]. The cobalt loadings in Co/ZnO, Co/MnxOy, Co/Al2O3 and Co/CeO2 were studied. Co/MgO with cobalt loading of 15% showed the best activity for N2O decomposition. The active phase of cobalt species in Co/MgO catalysts was Co3O4 highly dispersed in the matrices of MgO. Supported Co3O4 on MgO showed higher catalytic activity than unsupported Co3O4. Recently, the stainless steel wire mesh was also employed as support for potassium-doped Co3O4 nanowires and tested for N2O decomposition [36]. The activity of this novel structured catalyst was compared with reported results obtained on grains of cobalt-containing catalysts. Due to a different reactor concept and operation conditions, the kinetic constants were used for comparison with literature data. Based on this comparison, stainless steel wire mesh-supported potassium-doped cobalt oxide was found to be the most active among the catalysts reported in the literature.
To the best of our knowledge, only two research groups reported on manufacturing of shaped cobalt spinel based catalysts in pilot plant scale conditions [37–39]. In both cases, the conventional packed beds with pelletized cobalt spinel based catalysts were used for N2O decomposition. During scale up, troubles accompanying formation of Co–Mn–Al mixed oxide into larger pellets and relatively low mechanical strength significantly decreased its practical application in industry. Other important problem is that the catalytic reaction takes place in a narrow surface region of the pellets because of a high influence of internal diffusion effects during the catalytic reaction, which causes low utilization of the pellet volume.
Supporting of active phase of Co–Mn–Al mixed oxide on the suitable support would eliminate these troubles. As a support Al2O3, SBA-15, SiO2, Na-montmorillonite, and two slags were chosen together with TiO2, which was already investigated in our previous paper [40]. The aim of the present work is to study the effect of Co–Mn–Al mixed oxide deposition on selected supports on catalyst properties (reducibility, texture, phase composition) and N2O decomposition activity and to elucidate the observed phenomena. The supported catalyst with the best N2O decomposition performance could be used for scaling up.
2 Experimental
2.1 Catalyst preparation and characterization
Co–Mn–Al mixed oxide on different supports in powdered form was prepared by pore filling impregnation method. Supports were crushed, sieved to the fraction with particle size of 0.160–0.315 mm and water absorbability was found out. Then, the wet impregnation was carried out in aqueous saturated solutions of Co(NO3)2, Mn(NO3)2 and Al(NO3)3. At first, saturated solution of Mn(NO3)2 was prepared and then saturated Co(NO3)2 and Al(NO3)3 were added in order to have molar ratio of Co:Mn:Al = 4:1:1. After drying of impregnated supports at 105 °C, the prepared samples were calcined for 4 h at 570 °C in air. The whole procedure of impregnation, drying and calcination was repeated twice. The prepared catalysts were again sieved to obtain a fraction with particle size of 0.160–0.315 mm. Catalysts were labeled as active phase/support. For comparison, an unsupported Co–Mn–Al mixed oxides was prepared by calcination of corresponding nitrates keeping the molar ratio of Co:Mn:Al = 4:1:1. Marking of catalyst supports and prepared catalysts are summarized in Table 1.
Summary of supports and catalysts marking.
| Support marking | Support type | Support preparation/producer | Catalyst marking |
| S1 | Steel slag | ArcelorMittal | Co–Mn–Al/3APZ |
| Al2O3 | Commercial support | Eurosupport | Co–Mn–Al/Al2O3 |
| S2 | Granular blast furnace slag | ArcelorMittal | Co–Mn–Al/B3 |
| MMT | Na-montmorillonite | Crook Country Wyoming | Co–Mn–Al/MMT |
| SBA-15 | Mesoporous silica | laboratory prepared [41] | Co–Mn–Al/SBA-15 |
| SiO2 | Commercial support | Eurosupport | Co–Mn–Al/SiO2 |
| TiO2 | Commercial support | Eurosupport | Co–Mn–Al/TiO2 |
| – | – | Laboratory prepared (this work) | Co–Mn–Al |
The supports used and prepared catalysts were characterized by several methods. Chemical analysis of the calcined catalysts was performed by atomic adsorption spectrometry (AAS) after milling of the sample and its dissolution in aqueous solution of HCl (1:1).
Measurements of N2 adsorption/desorption isotherm using Sorptomatic series 1990 (TermoFinnigan) were used for determination of surface area by BET method and for pore analysis using Horvath Kawazoe and BJH methods. Prior to the measurement, the samples were dried at 120 °C for at least 12 h and then evacuated until the pressure 10−6 Pa was achieved.
For volume and density determination of the support, the pycnometry method was applied (Pycnomatic ATC, Surface Measurement Systems Ltd).
Powder X-ray diffraction patterns were recorded using Bruker D8 Advance equipment with Co Kα radiation. For phase identification, the PDF 2 Release 2004 database was used.
The distribution of the Co, Mn and Al was studied by scanning electron microscopy (Philips XL 30, EDAX) by the elements mapping.
Reducibility of prepared samples was investigated by temperature programmed reduction (TPR-H2) measurement: 0.025 g of catalyst was reduced with 10% H2/N2 mixture. Flow rate 50 mL/min and linear temperature increase of 20 °C/min from 25 °C up to 1000 °C were applied. Water evolved during reduction process was eliminated by freezing out at –78 °C and a change in H2 concentration was detected with catharometer or a mass spectrometer Omnistar 300 (Pfeiffer Vacuum). Reduction of the grained CuO (0.16–0.315 mm) was performed to calculate absolute values of the hydrogen consumed during reduction.
2.2 Catalytic measurements
Catalytic measurements of N2O decomposition were performed in an integral fixed bed stainless steel reactor of 5 mm internal diameter in the temperature range of 300–450 °C and under atmospheric pressure. Total flow rate was 50 mL/min (NTP). The catalyst bed contained 0.3 g of sample with particle size 0.160–0.315 mm (GHSV = 10 l g/h). Inlet gas consisted of 0.1 mol% N2O balanced by helium or of mixture of 0.1 mol% N2O with other gases: 5 mol% oxygen, 3 mol% water vapor and/or 0.02 mol% NO to simulate real waste gas from HNO3 production. Before each run, the catalyst was pre-treated in the He flow at 450 °C for 1 h. Then, the catalyst was cooled to the reaction temperature and the steady state of N2O concentration level was measured.
A mass spectrometer RGA 200 (Stanford Research Systems, Prevac) was used for N2O analysis (mass/charge ratio = 44). Argon (0.3 mol%) was applied as an internal standard for instability elimination of mass spectrometer. Data was acquired with UMS-TDS software. The content of the water vapor in inlet gas was determined from temperature and relative humidity measurements.
For comparison of the catalysts, specific activity expressed as the number of N2O moles reacted per second at given reaction temperature related to the weight of Co + Mn (Eq. (1)) supported on the carrier was determined:
| (1) |
3 Results and discussion
3.1 Catalyst characterization
The composition of the support was determined by the type of the used support (TiO2, Al2O3, SBA-15, SiO2, MMT); the slags were composed from lot of different oxides. Steel slag, S1, contained more than 10 wt% of SiO2, CaO and Al2O3 and 5–10 wt% of Fe2O3, MgO and MnO. Granular blast furnace slag, S2, contains primarily SiO2, CaO, MgO and Al2O3. Other oxides, like TiO2, P2O5, K2O and Na2O exist in these slags in amounts around or lower than 1 wt%. Some physicochemical properties of the prepared catalysts are summarized in Table 2.
Physicochemical properties of the prepared supported catalysts.
| Catalyst | Co (wt%a) | Mn (wt%a) | Molar ratio Co:Mn | SBET (m2/g) | Smeso (m2/g) | Vmicro (mm3liq/g) | Water absorbability |
| Co–Mn–Al/SiO2 | 25.7 | 6.6 | 3.63 | 135.0 253.7b | 98.1 166,1b | 20.3 46.6b | 0.68 |
| Co–Mn–Al/SBA-15 | 32.2 | 9.3 | 3.23 | 241.0 527.4b | 229.9 381.0b | 10.3 81.0b | 1.25 |
| Co–Mn–Al/Al2O3 | 19.0 | 4.8 | 3.69 | 132.7 216.7b | 105.6 137.3b | 16.2 40.8b | 1.36 |
| Co–Mn–Al/S2 | 15.6 | 4.2 | 3.46 | 11.6 60.2b | 5.5 29.0b | 3.5 15.9b | 0.69 |
| Co–Mn–Al/MMT | 30.3 | 7.8 | 3.62 | 33.6 42.6b | 14.8 22.5b | 9.6 10.0b | 2.72 |
| Co–Mn–Al/S1 | 15.1 | 8.0 | 1.76 | 9.8 8,0b | 9.8 5.4b | 0 1.4b | 4.90 |
| Co–Mn–Al/TiO2 | 16.4 | 4.5 | 3.40 | 74.1 132b | 48 85.3b | 15 27b | 5.67 |
| Co–Mn–Al | 47.3 | 11.0 | 3.73 | 34.8 | 23.5 | 6.5 | – |
a Determined by AAS.
b Free support (without impregnated active species).
Helium pycnometry was used for support density determination. The highest density 3.1 g/cm3 was found out for the S1 support and the lowest value of density 1.79 g/cm3 for SBA-15 support. Water absorbability differs significantly from sample to sample.
According to the results from AAS analysis, the highest amount of active metals (Co, Mn) was observed in the case of Co–Mn–Al/SBA-15 and Co–Mn–Al/MMT catalysts. The lowest content of these metals was found out in Co–Mn–Al/S2 and Co–Mn–Al/TiO2. Molar ratios of Co:Mn calculated from AAS are little bit less than target 4:1, only in the case of Co–Mn–Al/S1, the required ratio is much more lower, because used support already contained MnO.
The specific surface areas of supported Co–Mn–Al mixed oxide ranged from 9.8 to 241 m2/g; the highest value of surface area was observed for Co–Mn–Al/SBA-15. For Co–Mn–Al/SiO2 and Co–Mn–Al/Al2O3 catalysts, the value of specific surface area was approximately a half of the maximum value, 135 m2/g. The lowest surface areas were observed for Co–Mn–Al mixed oxides on slags Co–Mn–Al/S2 and Co–Mn–Al/S1. These were three times lower than the surface area of unsupported sample Co–Mn–Al prepared by calcination of nitrates.
Impregnation of the active species on the support caused significant decrease of surface area of all samples with the exception of S1 support, which was very poorly porous, compared to free supports. Pore blocking of the support can be seen from the decrease of surface area of mesopores and from the changes in micropore volumes.
The phase composition of all samples was determined by XRD analysis. Diffractograms of Co–Mn–Al mixed oxides deposited on different supports are shown in Fig. 1. Individual phases found in the samples are marked in the graph. It is evident from the diffractograms that all catalysts contained Co–Mn–Al spinel phase (S). The diffractogram of the Co–Mn–Al/TiO2 catalyst showed also TiO2 as crystalline anatase (A) modification. Co–Mn–Al/SBA-15 and Co–Mn–Al/MMT included quartz crystalline phase (Q), γ-Al2O3 (g-Al) was observed in the diffractogram of Co–Mn–Al/Al2O3 sample and calcite modification CaCO3 (C) was identified in Co–Mn–Al/S2 and Co–Mn–Al/S1.
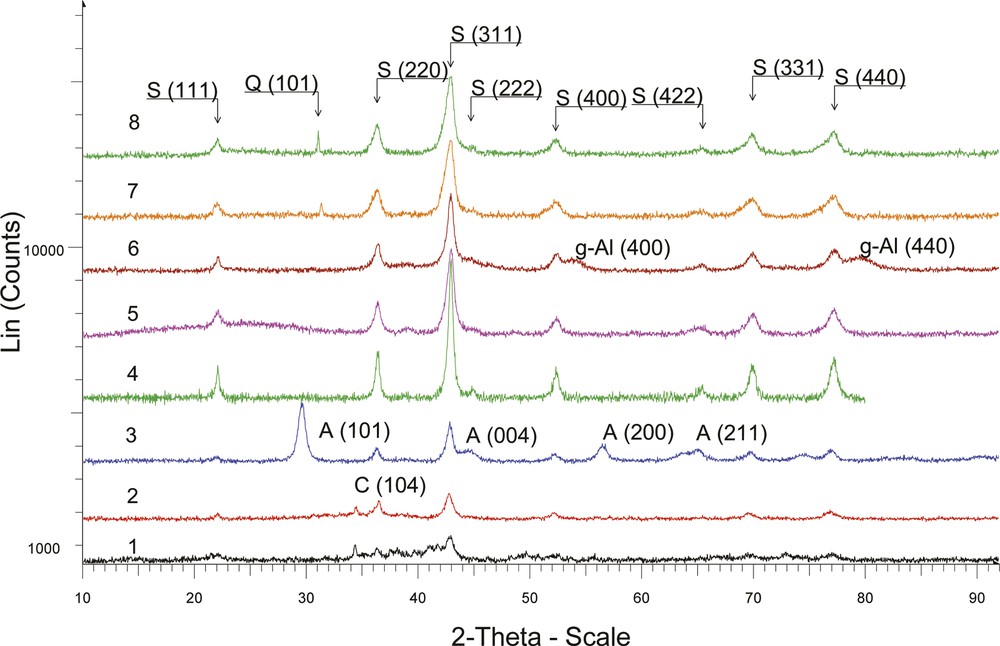
(Color online.) Powder XRD patterns of the Co–Mn–Al mixed oxide on different supports. 1 – Co–Mn–Al/S1, 2 – Co–Mn–Al/S2, 3 – Co–Mn–Al/TiO2, 4 – Co–Mn–Al, 5 – Co–Mn–Al/SiO2, 6 – Co–Mn–Al/Al2O3, 7 – Co–Mn–Al/SBA-15, 8 – Co–Mn–Al/MMT, (A) anatase, (Q) quartz crystalline phase, (g-Al)γ-Al2O3, (C) CaCO3.
From the mapping of the Co, Mn and Al by the SEM–EDAX, it was found out that the distribution of mentioned individual elements is uniform (not shown).
Temperature programmed reduction of prepared catalysts by hydrogen was performed in the temperature range from 25 to 1000 °C. Amount of consumed hydrogen and temperature peak maxima are shown in Table 3. The catalysts were reduced in two temperature regions, namely 150–550 °C and 550–1000 °C (Fig. 2). Both reduction peaks consisted of several overlapping peaks, which can indicate simultaneous reduction of several different components. The assignment of the oxidation states was done on the basis of literature data and on the basis of the measurement of the TPR of Mn oxides standards in our previous works [42]. Generally, presence of kinetics effects during TPR measurements makes assignment of the TPR peaks to individual chemical compounds extremely difficult in the case of Mn oxides as was discussed recently [43]. The main low temperature peak (418–550 °C) could be ascribed to the reduction of Co3+ to Co2+ and Co2+ to Co0 together with reduction of Mn4+ to Mn3+. A deep reduction of Mn3+ to Mn2+ together with reduction of Co–Mn–Al spinel like phase takes place at higher temperatures [33,44]. The total amount of H2 consumed in temperature range 25–1000 °C is directly proportional to the content of loaded amount of cobalt and manganese (Fig. 3, rhombus), which is directly connected with water absorbability of support (Fig. 3, empty circles). The exception is Co–Mn–Al/S1 catalyst, where the amount of H2 consumed is higher than expected (not shown in Fig. 3, rhombus), which led us to the conclusion that also species from support were reduced in this case.
TPR-H2 results of Co–Mn–Al mixed oxides on different supports.
| Catalyst | R1: 25–500 °C (mmol H2/g) | R1 + R2: 25–1000 °C (mmol H2/g) | Tmax (°C)a |
| Co–Mn–Al/MMT | 2.96 | 8.16 | 450, 660 |
| Co–Mn–Al/S1 | 1.20 | 10.11 | 288, 550, 753, 960 |
| Co–Mn–Al/SBA-15 | 3.43 | 9.72 | 292, 432, 641, 765 |
| Co–Mn–Al/S2 | 1.12 | 5.55 | 527, 599, 774 |
| Co–Mn–Al/Al2O3 | 1.46 | 5.01 | 223, 491, 670, 731 |
| Co–Mn–Al/SiO2 | 2.53 | 7.50 | 298, 434, 644, 806 |
| Co–Mn–Al/TiO2 | 2.04 | 5.83 | 156, 318, 465, 603 |
| Co–Mn–Al | 6.30 | 13.71 | 418, 595, 697 |
a The maximum of the main peak in the low temperature region is highlighted.
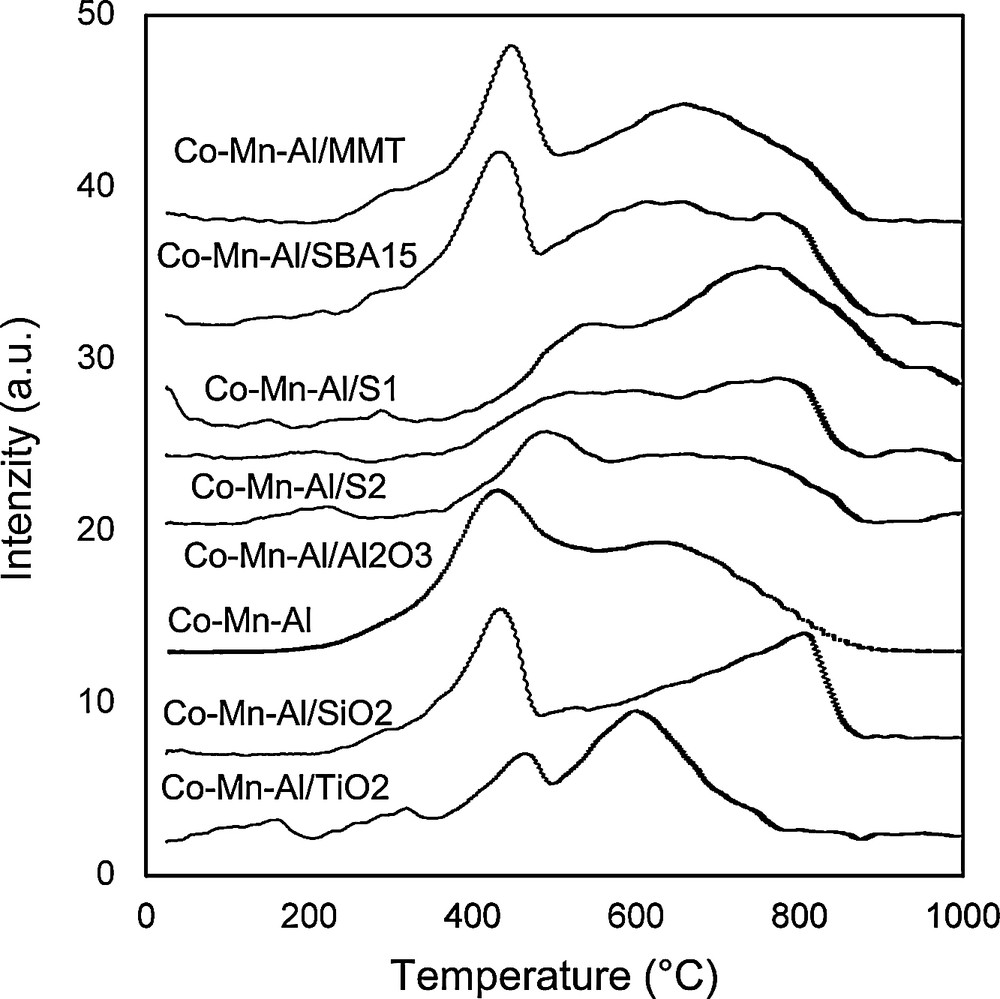
TPR-H2 patterns of Co–Mn–Al mixed oxides on different supports.
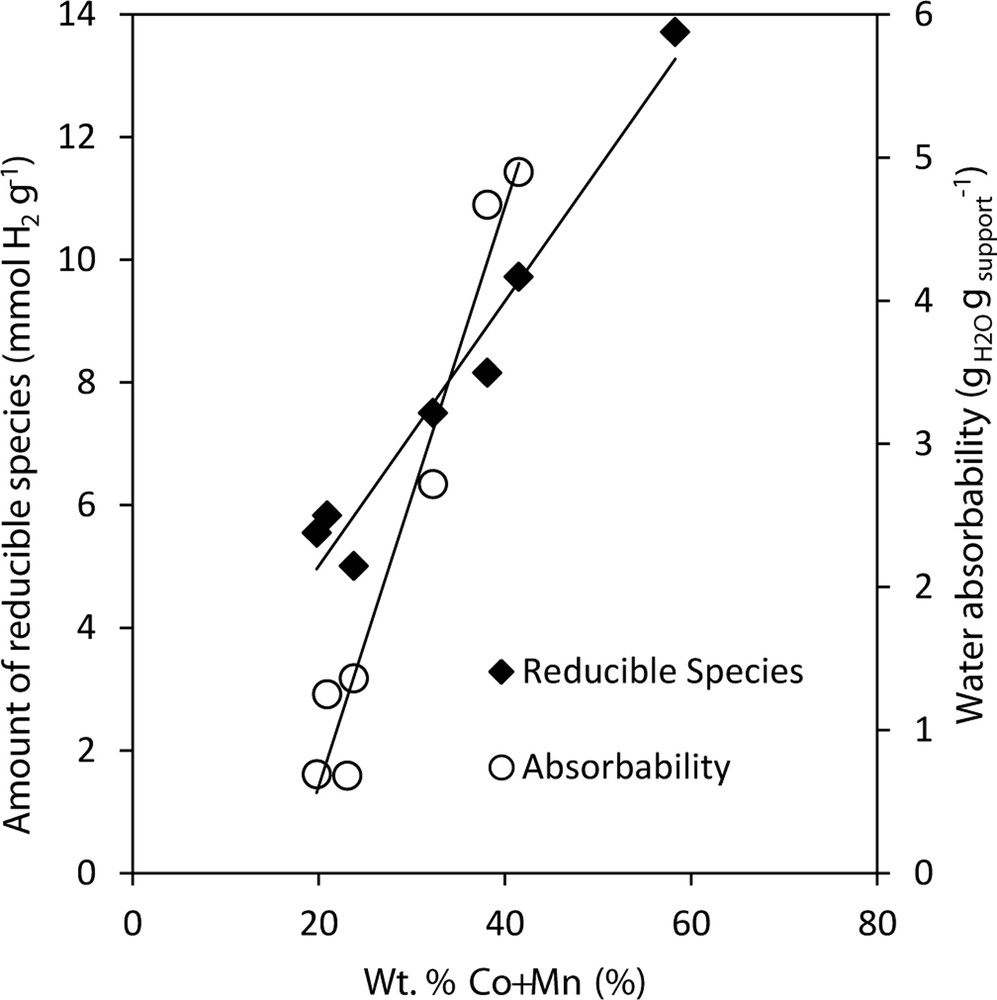
Dependence of amount of reducible species in the catalysts expressed as H2 consumption during TPR in the temperature region between 25 and 1000 °C and the water absorbability of the support on the amount of (Co + Mn).
In our evaluation of TPR-H2 results, we focused on the temperature range up to 500 °C (R1), which was important for the practical application of N2O decomposition in this study because this is the temperature range of catalyst testing. Two basic pieces of information can be obtained from TPR – amount of reducible species determined as H2 consumption and easiness of catalysts reduction corresponding to the temperature of TPR peak maximum (Table 3).
In the case of supported Co–Mn–Al mixed oxide, the highest amount of H2 consumed in the R1 region was found out for Co–Mn–Al/SBA-15, Co–Mn–Al/MMT and Co–Mn–Al/SiO2.
When we compare the positions of the main peak in the low temperature region, we can see that loading of Co–Mn–Al mixed oxide on all tested supports led to the shift of TPR peak maxima to higher temperature. The shifts correspond to the worse reducibility of supported Co and Mn oxides compared to bulk Co–Mn–Al mixed oxide. Moreover, differences of reducibility between individual supported Co–Mn–Al mixed oxides were also observed. Different reducibility of supported samples could be caused by interaction of active species with the support phase based on the different nature, concentration and physicochemical properties of the surface sites of each support [45,46]. Decrease of reducibility of supported cobalt species were reported by several authors and was explained by forming of worse reducible silicates, titanates or aluminates [47,48] or by the reduction of the fraction of cobalt that is contained in the inner cavities of the support [47].
3.2 Catalytic activity
The temperature dependences of N2O conversion over Co–Mn–Al mixed oxide impregnated on different supports are shown in Fig. 4. The highest conversions for N2O decomposition were obtained over Co–Mn–Al mixed oxide on SBA-15, TiO2 and MMT. The lowest activity for N2O decomposition was achieved on the catalysts supported on slags and alumina; however, the unsupported sample was the most active one. The higher content of Co + Mn loaded on the support was, the higher conversion was achieved. Beneficial effect of increasing content of catalytic component deposited on the support on the catalytic activity for N2O decomposition was reported by Wang et al. [49], however, the increase in activity was not linearly dependent on the number of the support impregnation cycles, which could be explained by the gradual decrease of the surface area of the catalyst by pore blocking and thus by worse accessibility of the active species or by changing the intensity of the interaction of deposited phase with the support based on different loadings of active species as was reported by Jacobs et al. [50].
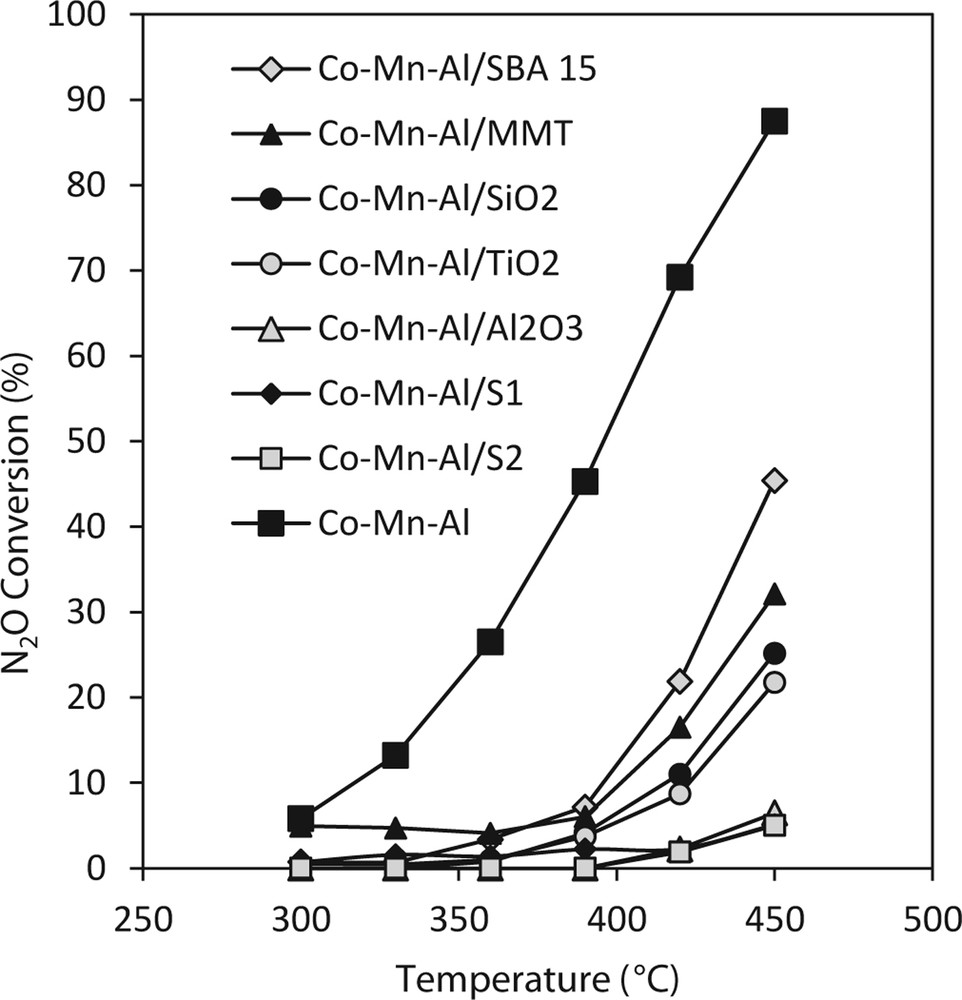
Temperature dependence of N2O conversion over Co–Mn–Al mixed oxides on the different supports. Conditions: 0.1 mol% N2O balanced by He, 0.3 g of catalyst, 50 mL/min.
N2O conversion values are linearly dependent on the amount of species reducible in temperature region 25–500 °C (Fig. 5) where active species for N2O decomposition Co3+/Co2+ and Mn4+/Mn3+ are reduced, which is connected with the amount of deposited cobalt and manganese and thus absorbability. As the N2O decomposition reaction is an oxidation–reduction reaction, the involvement of the species that could be oxidized and reduced in the studied temperature region is supposed.
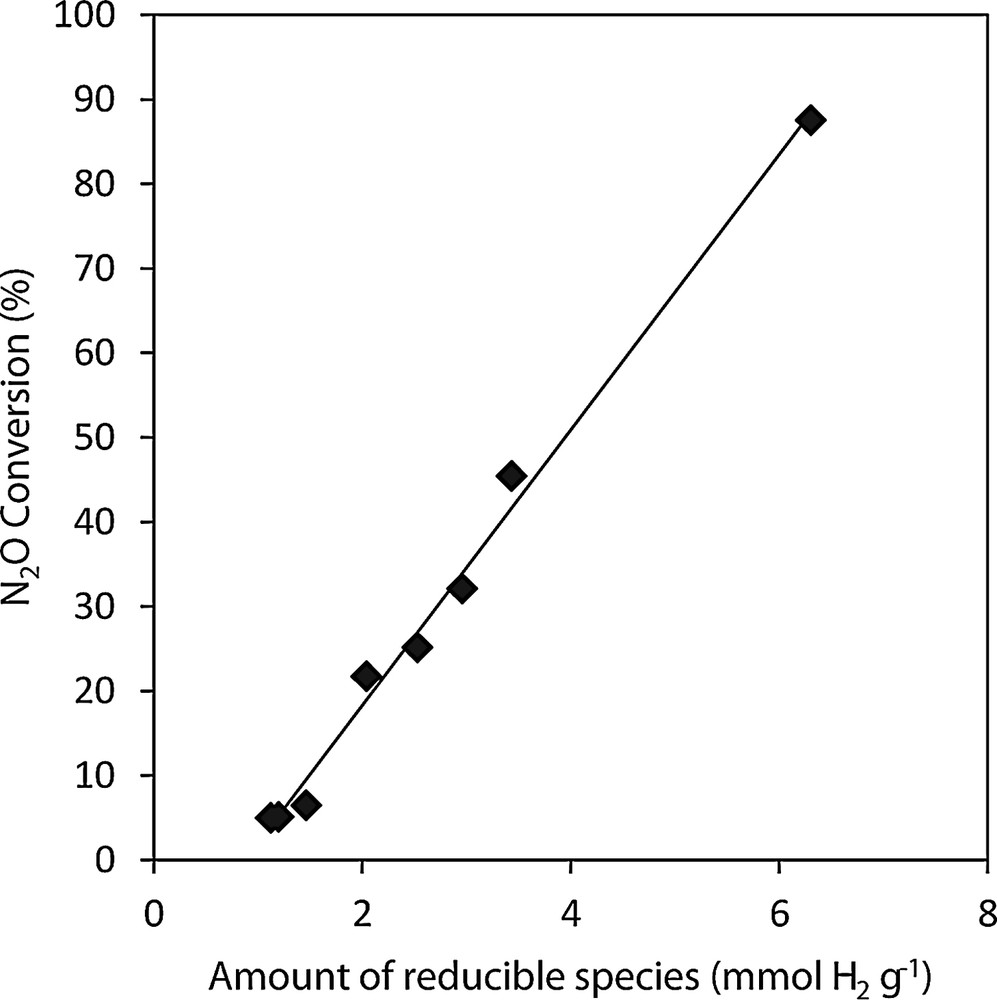
Dependence of N2O conversion (450 °C) on the amount of species reducible in low temperature region (25-500 °C) over Co–Mn–Al mixed oxides supported on the different supports.
In order to find whether deposition of active spinel phase on supports influenced the catalytic efficiency of unit amount of deposited cobalt and manganese, the comparison on the basis of specific activities (equation (1)), providing information about the number of N2O moles reacted per unit amount of Co and Mn in the samples, was done. In Fig. 6, the specific activities obtained at 450 °C in an inert gas and in simulated waste gas from nitric acid plant containing with N2O also O2, H2O and NO are shown. If the specific activities were the same, the effect of the support would be just the active species dilution compared to bulk Co–Mn–Al mixed oxide. Different specific activities could mean that deposition of active species on the support caused changes in catalytic properties of the active site due to some kind of chemical interaction of the species with the support or ensured different distribution or accessibility of the active species.
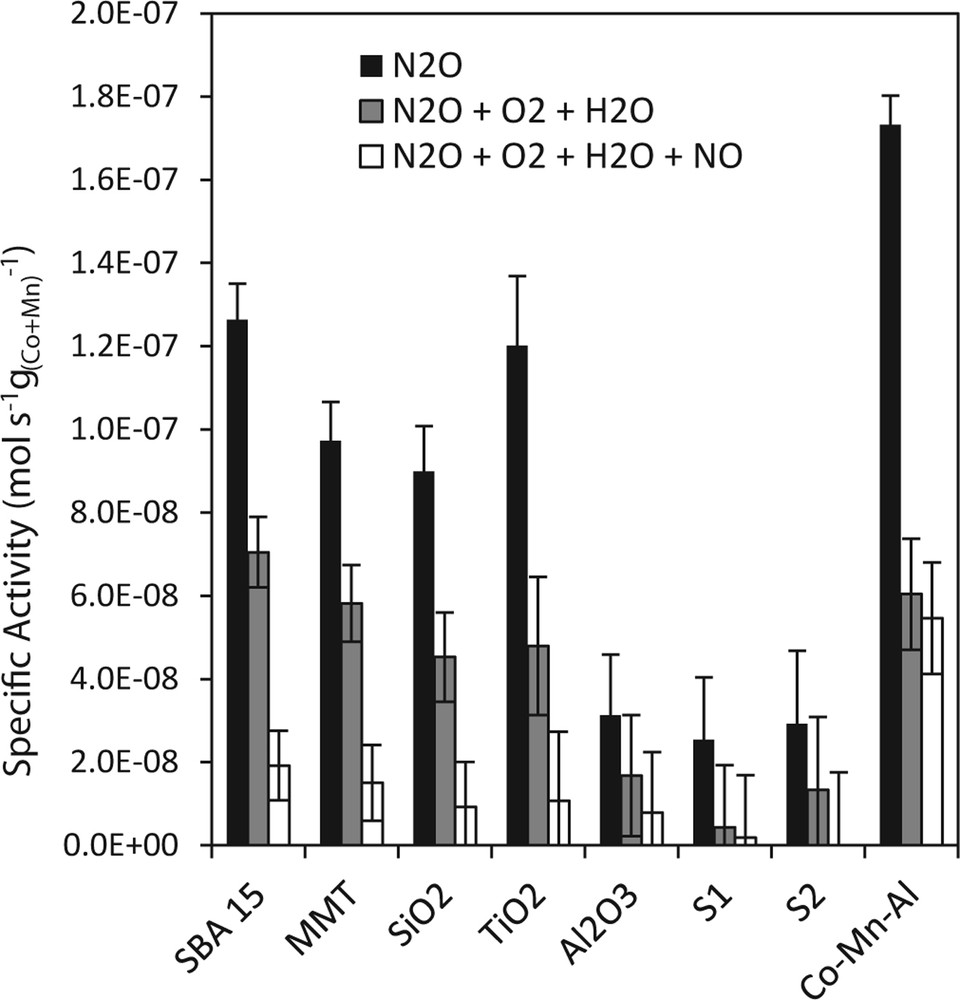
Specific activities for N2O decomposition at 450 °C over Co–Mn–Al mixed oxides supported on the different supports.
In inert gas, the specific activity of the unsupported Co–Mn–Al mixed oxide was the highest; among the supported samples, the highest activities were obtained over Co–Mn–Al mixed oxide supported on SBA-15 and TiO2, the lowest on slags and alumina. It means that specific activity of Co–Mn–Al mixed oxide was negatively influenced by deposition on all supports.
The possible decrease of the inhibition effect caused by adsorption of O2, H2O and NOx on active sites or their surroundings was another expected effect of active spinel phase deposition on suitable carrier. From Fig. 6, it is evident that N2O specific activity over all tested catalysts decreased after gradual addition of inhibiting compounds. However, in the presence of oxygen and water vapor, the specific activities of the active group of supported catalysts (Co–Mn–Al mixed oxide deposited on SBA-15, MMT, SiO2 and TiO2) decreased less than activity of unsupported sample, therefore, the activities of the active supported samples were in this case comparable with the unsupported one. This clearly indicated a beneficial effect of the mentioned supports on the degree of O2 and H2O inhibition, which is important for potential practical application of the supported catalysts for N2O decomposition in wet waste gas.
The addition of NO had detrimental effect, the specific activity for N2O decomposition was only around one tenth of original value, or nearly zero on less active samples. The unsupported sample did not suffer so significantly from NO inhibition, which makes supported samples especially applicable in N2O decomposition when waste gas does not contain NOx (e.g., downstream the DeNOx reactor). It follows that the deposition of the active phase on the carrier was beneficial to reduce the O2 and H2O inhibition. In contrast, the presence of the carrier did not allow reducing the NO inhibition.
The inhibiting effect of water, oxygen and nitric oxide on N2O decomposition is well known, however the detail explanation of the effects of these compounds over different supports requires a more detailed study. On the basis of the theory of adsorption of the inhibiting components on the catalyst surface, it is supposed that different supports change the surface characteristics of the catalyst and thus the energetic affinity of the compounds presented in the feed gas towards investigated surface which means that when both molecules (N2O and other compound) are simultaneously present in the feed, a higher inhibition effect will be seen in the case where the inhibiting compound has higher energetic affinity towards the catalyst surface [5].
The specific activities were also correlated with the amount of reducible species in low temperature region R1 and with the ease of catalyst reduction. However, amounts of reducible species are dependent on the amount of active species in each sample. Therefore, the obtained amounts of species reducible in the temperature range R1 were recalculated into the amount of H2 consumed during TPR run per unit amount of Co + Mn in the sample (Fig. 7). Specific activity increased with increasing amount of such expressed reducible species. It means that in the case of high specific activities more moles of N2O decompose on the same amount of Co + Mn and this amount possesses more reducible species, which are needed for N2O decomposition, namely for the slowest step of N2O decomposition mechanisms, i.e. O2 desorption [51]. In other words, active species in more active samples are able to ensure more N2O decomposition cycles due to the higher content of reducible species that are formed either during synthesis or are better available from the same amount of Co and Mn deposited on the support.
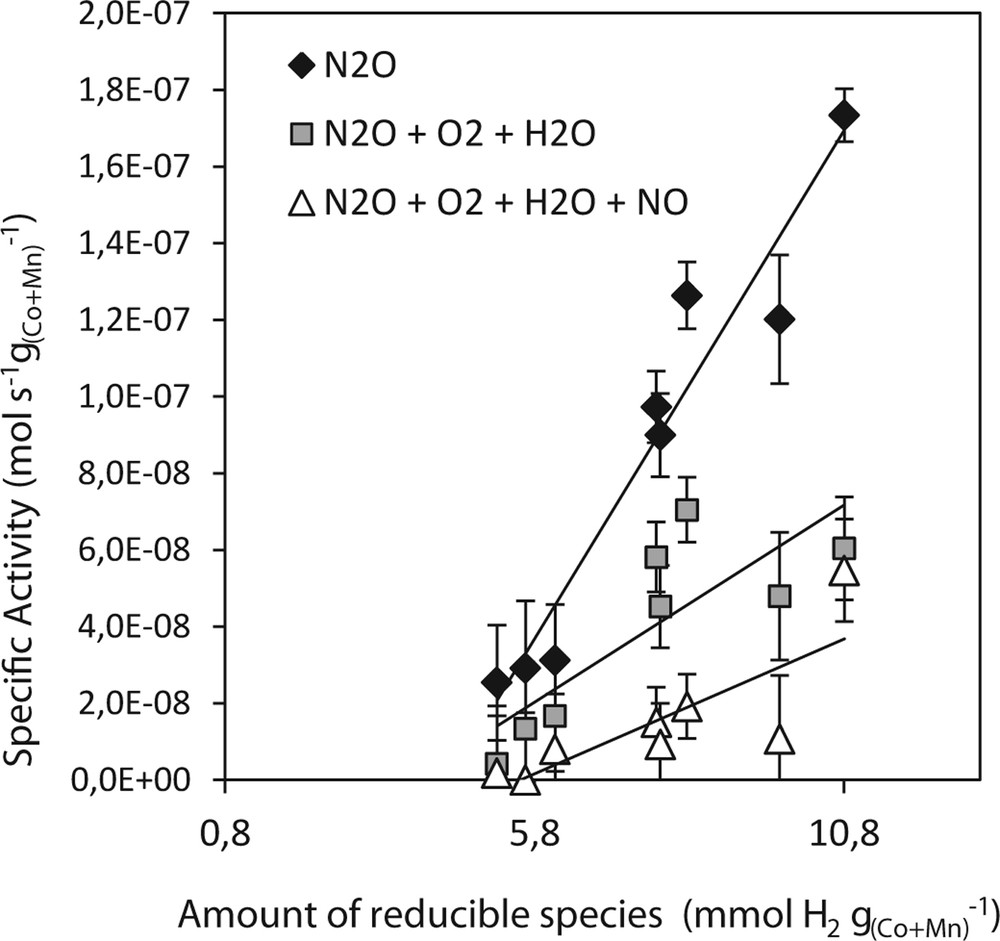
Dependence of specific activities for N2O decomposition at 450 °C over Co–Mn–Al mixed oxides supported on the different supports on the amount of reducible species (low temperature region) relative to the amount of Co + Mn.
Specific activity was also correlated with easiness of catalyst reduction (Fig. 8). The easiness of the catalyst reduction was expressed as the temperature maximum of the main peak in the low temperature region R1 of the TPR reduction profiles. Apart from the amount of the reducible species, it is seen that the most active catalysts contained reducible components, which can be reduced at lower temperatures than reducible species from less active ones. Thus, the specific activity of the samples was determined by type and amount of the reducible species formed from cobalt and manganese. Decreasing amounts of reducible species from R1 region are connected with shifting of the TPR peak to higher temperatures and therefore shifting of maximum of the TPR peak to the high temperature region as well. Dominant peak shift to higher temperatures was found out especially for the samples containing Al3+ ions in the support (Al2O3, S1, S2) and this was also the group of less active samples for N2O decomposition. It is known that during the catalyst preparation the cobalt ions diffuse into the structure of Al2O3 occupying octahedral or tetrahedral positions. As a result, mainly two kinds of cobalt-containing phases are formed: (i) surface phases of Co bonded to the support, which are difficult to reduce and (ii) an easily reducible Co3O4 phase. The phase, where Co ions occupy the tetrahedral position in the Al2O3 lattice, is usually assumed to be CoAl2O4 [52], which is why we suppose that in these samples aluminates reducible with great difficulties are formed instead of more active Co3O4 species. Formation of worse reducible species after deposition of cobalt oxides on TiO2 and SiO2 supports was also reported and this was explained by the presence of titanates and silicates [41,42]. In our case, based on values of specific activities, the following order was found for the N2O catalytic decomposition activity: Co–Mn–Al/SBA-15 ≈ Co–Mn–Al/TiO2 ≥ Co–Mn–Al/MMT ≈ Co–Mn–Al/SiO2 > Co–Mn–Al/Al2O3 ≈ Co–Mn–Al/S1 ≈ Co–Mn–Al/S2.
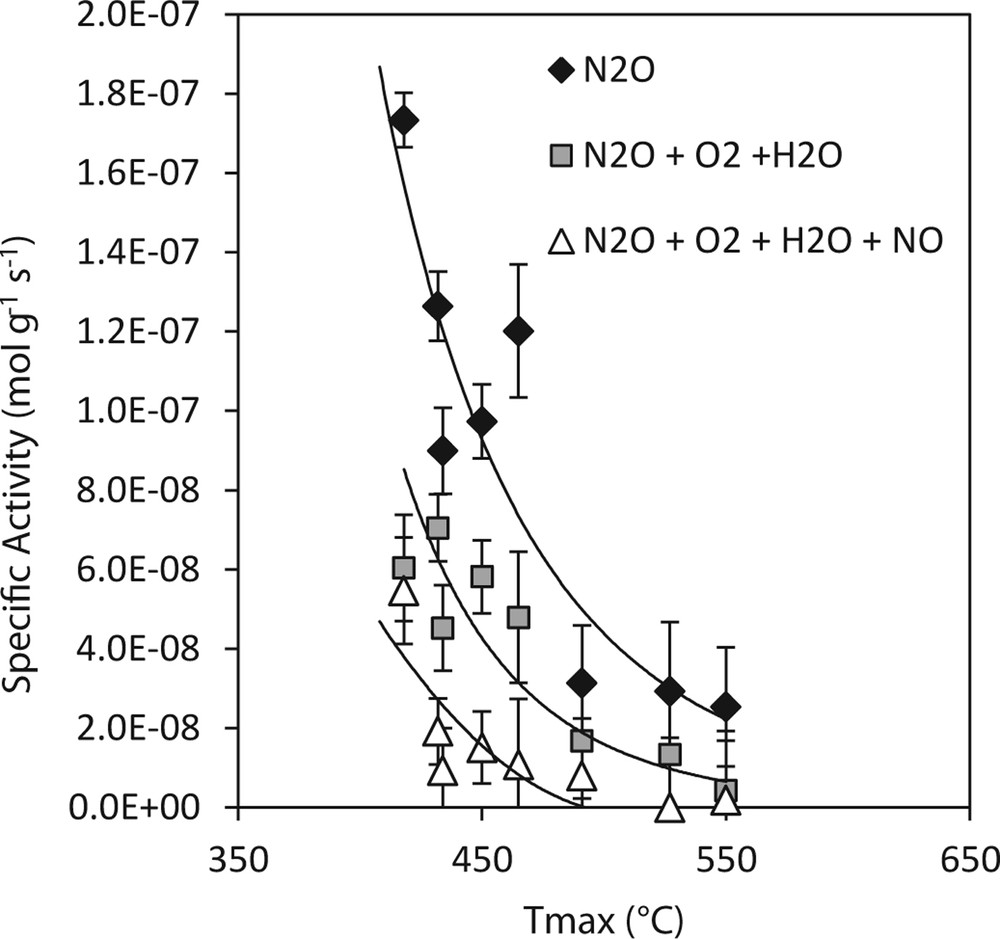
Dependence of specific activities for N2O decomposition at 450 °C over Co–Mn–Al mixed oxides supported on different supports at Tmax from TPR-H2.
4 Conclusions
The Co–Mn–Al mixed oxide catalysts on different supports were prepared, characterized and tested for N2O catalytic decomposition. The highest conversion was observed on Co–Mn–Al mixed oxide deposited on SBA-15, MMT, SiO2 and TiO2 and these catalysts contained the highest amounts of cobalt and manganese, which was dependent on water absorbability of the supports. The highest specific activity in inert gas was achieved over Co–Mn–Al mixed oxide supported on TiO2 and SBA-15, the lowest over Co–Mn–Al mixed oxide on alumina and slags. The same trend was observed also in the simulated waste gas. Deposition of the Co–Mn–Al oxide on supports decreased specific activity compared to that of unsupported one. However, in the presence of O2 and water vapor, the specific activities of the most active supported samples and unsupported one were comparable. The dependence of the specific activity on the catalyst reduction characteristics (amount of reducible species and easiness of reduction) was observed, what means that tested supports affected properties of active components.
Acknowledgement
This work was financially supported by the Czech Science Foundation (project GA14-13750S) and by Ministry of Education, Youth and Sports of the Czech Republic, project NPU I LO1208 “TEWEP”. The authors would also like to thank M. Heliová, V. Matějka and S. Študentová from VŠB–TU for help with the SEM–EDAX, XRD and nitrogen physisorption measurements.