

1 Introduction
EU directive 2010/75/EU requires the drastic reduction of NOx emission in flue gases from the 1st of January 2016. Until now the norms could have been fulfilled in many cases with appropriate primary measures, such as, e.g., flue gas recirculation, air staging, low excess air technique, low NOx burners, etc. The BAT methods for coal-fired power stations, allowing the fulfilment of the new norms, require now (with the exception of fluidized bed combustion) that either selective non-catalytic reduction SNCR or selective catalytic reduction SCR be introduced in large stationary sources.
Selective catalytic reduction (SCR) of NOx with ammonia is the most efficient method of NOx emissions reduction and it is widely used in EU, USA and Japan. The most often applied catalyst consists of vanadia on titania support, with the addition of WO3 to increase the temperature window, SiO2 to stabilize the structure and MoO3 to make the catalyst more resistant towards poisoning by SO2 [1]. The catalysts are usually integrated into the flue gas cleaning chain in the so-called “high-dust” position, upstream of an electrostatic precipitator. This leads to catalyst deactivation, which is a major drawback.
There are several mechanisms of deactivation, resulting from the presence of pollutants other than NOx in the cleaned flue gases. Of those, especially ash and SO2, which may lead to fouling and poisoning, are important. The problem of deactivation by SO2 has been well-recognized and led to the improvement of the catalysts, by the addition to the industrial catalysts of small amounts of WO3 or MoO3 which decrease the rate of SO2 oxidation to SO3 and thus the extent of poisoning.
Current climate policy requires that the emissions of pollutants, other than typically considered NOx, SO2 and particulate matter (PM) are also drastically reduced, with special attention paid to CO2. In coal-based economies this may be, among others, realized by co-combustion of coal and biomass. Tillman [2,3] reported that every ton of co-fired biomass reduced CO2 emissions by over 1 ton, and in case this biomass had to be landfilled without methane collection and flaring, the balance would be 3 tons of fossil CO2 equivalent. The USA experience showed the reduction of fossil CO2 emissions typically of the order of 2.7 to 3.15 fossil CO2 avoided per ton of biomass burned [4]. From the point of view of the SCR process, however, the change in the conditions for the DeNOx catalysts may be expected as a result of co-combustion because of different compositions of coal and biomass. Tillman [3] quotes typical numbers for some US coals and compares them with biomass. For N and S contents, the numbers are:
- • coals: 0.35 to 2.79% S and 0.68 to 1.40% N;
- • different types of biomass, S and N, respectively: 0.01 and 0.26 (sawdust); 0.07 and 1.00 (urban wood waste); 0.16 and 0.65 (switchgrass); 0.09 and 1.83 (alfalfa stalks).
Thus, the contents of elemental N may be of similar order and those of S, even much smaller for certain types of biomass. However, several other polluting species are present in biomass. Sahu et al. [4] published a review on coal–biomass combustion and its influence on the emissions of CO2, SO2 and NOx but the subject of SCR catalysts was not discussed. It must be taken into account, however, that for a typical SCR installation in the high-dust configuration, not only the differences in S and N contents, but also the different compositions of fly ash, are of great importance. Especially relevant is the alkali content (CaO, MgO, Na2O, K2O) leading to different base/acid ratios, e.g.: 2.08 (sawdust), 0.59 (black coal) or 0.44 (Illinois No. 6 coal [3]). K and Na are the main constituents of aerosols [5] and they were recognized as the potential source of deactivation for V2O5–WO3–TiO2 SCR catalysts at biomass-fired heat and power plant [6,7].
Despite the general agreement on the importance of potassium for SCR catalyst deactivation, the major drawback is that the published data were, in most cases, obtained under the conditions far from those in industrial installations co-firing coal and biomass. This is very important, as large industrial installations usually co-fire different types and amounts of biomass over longer periods, and Obernberger et al. [5] showed that different kinds of biomass affect not only their thermal utilization but also flue gas cleaning. Of the very few articles that would be, at least partly, relevant for industrial conditions in a large power plant co-firing coal and biomass, the articles of Wieck-Hansen et al. [8], Larsson [7], Strege et al. [9] and Kling et al. [10] should be mentioned.
Wieck-Hansen et al. [8] published the results of 2-year demonstration program for a 150 MWe power plant co-firing coal and straw. The plant was not equipped with SCR facility but the tests were carried out in a test drawer with undisclosed catalysts coming from three different manufactures. The catalysts were exposed to high-dust and low-dust conditions. For the former, after 2860 h, the activation levels decreased by ca. 35% i.e. roughly 8%/1000 h, which was much higher than for coal-fired installations. At low-dust position deactivation was ca. 15% after 2350 h. Electron microscopy revealed the deposition of elements coming from fly ash, mainly Si, S, Al, but also K and P.
Kling et al. [10] studied three types of V-W-Ti catalysts, one typical for coal-fired power station, the second with increased content of V for application with low SO2 content in flue gases and the third tested for flue gases with high alkali content, containing very high amounts of vanadium. The catalysts were exposed to flue gas in three power stations burning wood and/or peat. The time of exposure was between ca. 100 to ca. 2900 hours. It was found that the increased content of V decreased the deactivation rate and ultra-fine fly ash particles (< 10 nm) were mainly responsible for alkali accumulation. It was not possible to decrease the deactivation rate with the use of an S-containing additive.
Larsson [7] compared the performance of the V2O5–WO3/TiO2 catalyst modified by the deposition of KCl and K2SO4 particles with catalysts samples used in a commercial biomass-fired power plant for a period of 6500 h. The former catalyst was tested under the following conditions: T = 350 °C, 3% H2O, 150 ppm NOx and SO2 concentration < 1 ppm. The observed deactivation was similar in both cases.
Strege et al. [9] mixed samples of ash collected from a full-scale utility boiler co-firing wood and waste with ground SCR catalyst and exposed the samples to simulated flue gas containing 14% CO2, 8% H2O, 4% O2, 400 ppm SO2 and 100 ppm NH3 at TGA equipment at 316, 371 and 427 °C. As the major mass gain, the alkali sulfate formation and the blocking of pores of the SCR catalyst was recognized.
Apart from the size of the test installation (laboratory versus industrial scale) and the differences in flue gas compositions (with or without SO2 and ash), another possible difference between the purely laboratory studies of deactivation of SCR catalysts and those on industrial installation may arise from different mechanisms of poisoning, as studied under the former and the latter conditions. As an example, the article of Kamata et al. [11] can be cited. The authors discussed the potassium poisoning mechanism for a honeycomb industrial catalyst impregnated with an aqueous solution of KNO3, thus being the poisoning element artificially introduced. Other results reported by Larsson [7] showed different deactivation degrees for catalysts poisoned by impregnation with K and those exposed to K-containing real flue gas. They ascribed such differences to dissimilar ways of poison penetration into the catalyst's pores.
Moreover, the information on the combined effect of potassium and sulfur dioxide (both present in real flue gases) is scarce. Li et al. [12] have proven lately on the laboratory scale that the presence of SO2 in flue gases may influence the alkali-induced deactivation, but again the studied catalysts were pre-poisoned with K+ by impregnation and subsequently exposed to the reaction mixture containing NO, NH3, O2, H2O, SO2, and N2.
The aim of this work was to determine the influence of deactivation on the structural properties of a commercial SCR catalyst kept under real conditions in power plant co-fired with coal and biomass and thus exposed to simultaneous poisoning with alkali cations and SO2. Such information may be useful when predicting:
- • possible modifications of industrial catalysts, and/or;
- • regeneration strategies, such as, e.g., the ones proposed and tested on a laboratory scale by Khodayari and Odenbrand [13].
2 Experimental
2.1 Catalysts
The catalyst was a commercial honeycomb (proprietary one, undisclosed). It was exposed to flue gas in the industrial pulverized coal boiler at the position schematically shown in Fig. 1, operating at the parameters summarized in Table 1. Over the studied period, the boiler co-fired coal and biomass (share of biomass varied between 4 and 13% in energy and 122 days within the 12 months period were without biomass). The types of biomass used were wood pellets and sunflower pellets, with typical composition shown in Table 2. The exposure time was 2270, 4527 or 9405 h, i.e. ca. 3, 6 and 12 months, and in the text, these samples are designated as 3M, 6M and 12M, respectively. The starting (fresh) catalyst is designated 0M.
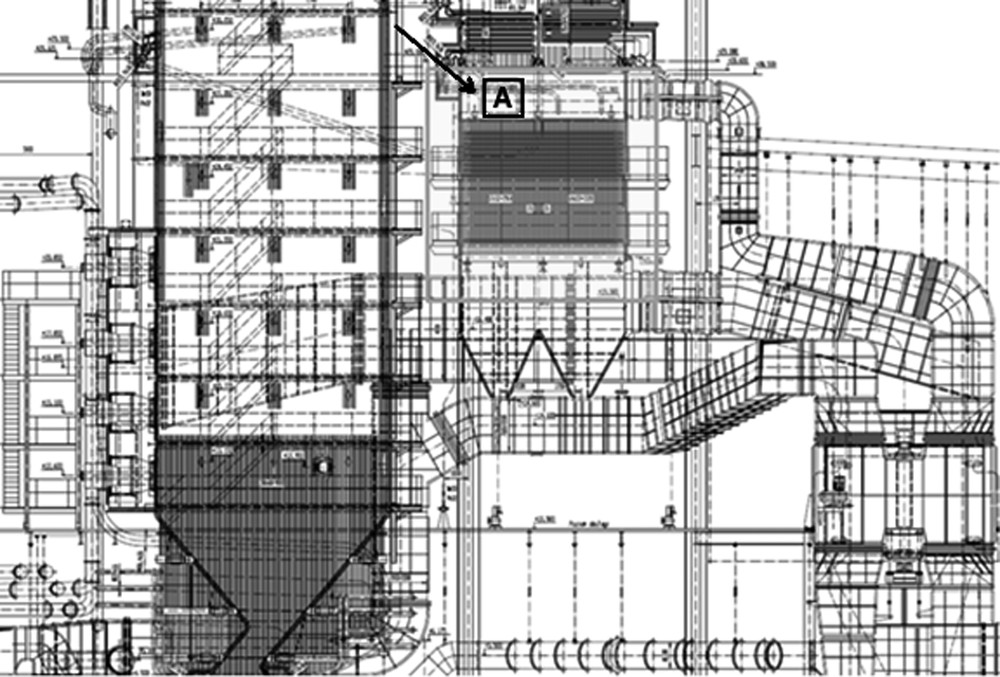
Scheme of flue gases route with marked place (A) of catalyst samples.
The operation parameters of the boiler.
| Parameter | Value |
| Boiler type | Steam, pulverized coal, drum |
| Circulation | Natural |
| Maximum capacity | 182 kg/s |
| Life steam temperature (outlet) | 540 °C |
| Life steam pressure (outlet) | 13.5 MPa |
| Reheated steam temperature (inlet/outlet) | 320/540 °C |
| Reheated steam pressure (outlet) | 2.3 MPa |
| Feed water temperature | 242 °C |
| Boiler efficiency | 91.50% |
| Type of fuel | Hard coal |
| Fuel calorific value | 23.1 MJ/kg |
Typical composition of biomass used for co-combustion [mol %].
| Biomass | C % | H % | N % | S % | O % | Fe2O3% | SiO2% | Al2O3% | K2O % | Na2O % | CaO % | MgO % |
| Wood | 51.03 | 6.06 | 0.08 | 0.02 | 42.31 | 1.71 | 13.20 | 2.89 | 11.00 | 0.51 | 27.70 | 6.41 |
| Sunflower | 49.87 | 5.82 | 1.06 | 0.19 | 38.16 | 0.77 | 9.52 | 1.07 | 27.20 | 0.25 | 16.80 | 10.40 |
The mean concentrations of NOx, SO2 and O2 in the flue gases, measured on the exit of the boiler during its normal operation, for the considered periods of catalysts exposure, was equal to 515 [mg/Nm3], 1795 [mg/Nm3] and 6.65 [%] respectively for NOx, SO2 and O2. Flue gas contained also dust, in the concentration of 20–25 g/Nm3. The average reaction temperature was 420 °C.
The fresh catalysts and those exposed to flue gas are designated as 0M, 3M, 6M and 12M, respectively, throughout this work. The spent catalysts showed considerable accumulation of fly ash with some channels totally blocked, as illustrated in Fig. 2. The ash samples scraped from the catalysts are designated throughout this work by giving the name of the appropriate catalyst and adding the word “ash”, e.g., 3M-ash.

The monolith catalyst after 9405 h of exposure (12M) under industrial conditions.
2.2 Characterization
Before characterization, the honeycomb catalysts, after ash removal, were crashed and reduced to powder using a porcelain mortar. Each ground sample consisted of inner and outer parts of the monolith in the appropriate proportions. Ash samples scraped from the catalysts were additionally tested, in order to compare the content of certain relevant elements.
The characterization of the fresh and poisoned catalysts, as well as ash samples (where stated) was carried out by:
- • X-ray diffraction, to determine phase analysis. The measurement was performed for all studied catalysts (fresh and exposed) and ash samples with PANanalytical Empyrean diffractometer using Cu Kα radiation (λ = 154.05 pm) at room temperature;
- • atomic absorption spectroscopy AAS. The concentrations of major elements such as SiO2, Al2O3, CaO, Fe2O3, MgO, K2O and Na2O were determined by atomic absorption spectrometry with use of flame atomization by Hitachi Spectrophotometer Z-2000 with Zeeman effect background correction and HCL (hollow cathode lamp) as the source of radiation. For the measurements, the samples were digested in concentrated HNO3 and HF acids in Berghof microwave digestion system–Speedwave Four;
- • SEM/EDS. The morphology of the fresh and exposed catalysts was assessed by SEM using a Joel JSM7001F FEG-SEM with an Oxford energy dispersive X-ray high-vacuum detector (E = 20 kV) for elemental microanalysis. One of the collected ash sample (6M-ash) was also examined. The powder samples were fixed on double face carbon adhesive (analytical grade) and covered with a thin film of graphite;
- • UV–vis. The diffuse reflectance UV–vis spectra of fresh, 3M, 6M and 12M catalysts, were collected using a Cary 5000 Varian equipment with a DRA 2500 diffuse reflectance accessory (integration sphere) to identify changes in the active phase after exposure to industrial flue gas.
The surface acidity of the fresh and post-reaction catalysts was characterized using the standard reaction of skeletal isomerization of 1-butene to cis and trans 2-butene [14]. The catalytic tests were carried out in a conventional flow apparatus for catalytic tests using 500 mg of powdered catalysts. The reacting mixtures were prepared by diluting 1-butene (43% V/V) in N2 (total gas flow 2.64 NL/h). The reactor effluent was quantified by chromatography (FID detector). The isomerization activities were evaluated in the temperature range 180–250 °C. The reaction conditions were selected in order to obtain measurable activities. The reaction temperatures were in the range of those used for V2O5-TiO2 catalyst by other researchers [15].
The activity of fresh and post-reaction catalysts was determined additionally by EDF Polska S.A. outside the power station under the following conditions: catalysts in the form of the original monolith; gas composition: NOx [ppm wet at 6% O2]: 167–215; NH3/NO = 1.01–1.05; H2O [vol %]: 9.39–10.60; O2 [vol %]: 5.9–8.37; CO2 [vol %, wet] 7.99 to 9.83; CO [ppm, wet]: 1221-463; flow [m3/h]: 63 (fresh catalyst) and 66 (catalysts after exposure to flue gas at industrial unit); temperature [upstream/down steam in °C]: 374/343 (fresh cat.); 361/335 (3M); 345/319 (9M); 364/335 (12M); pressure drop [Pa]: 53 (fresh catalysts); 106 (3M); 85 (9M); 93 (12M). The relative loss of activity discussed in this work is defined as:
3 Results and discussion
3.1 Measurement results of the catalyst's activity
The catalysts exposed to flue gas from the industrial boiler partly lost their activity, as illustrated in Fig. 3.
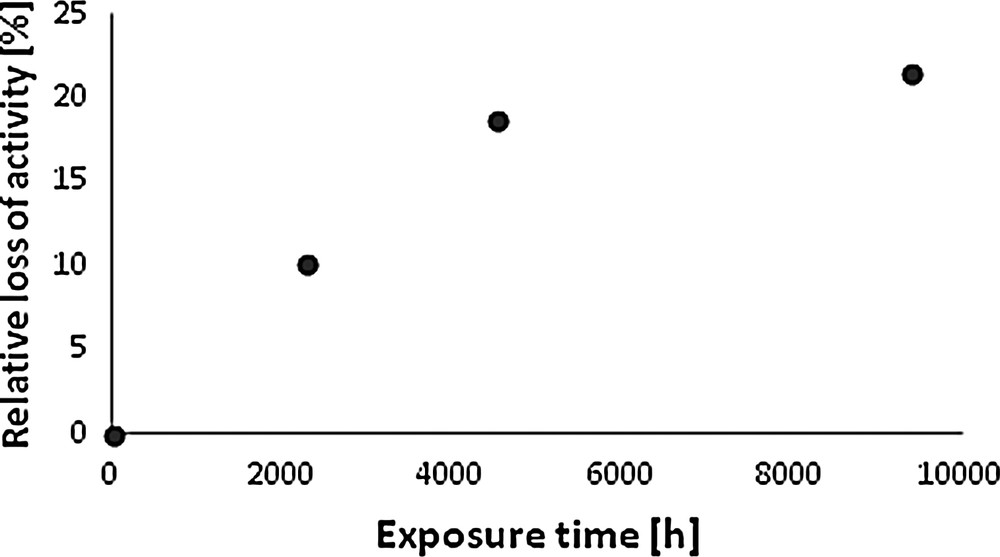
Relative loss of activity for a commercial selective catalytic reduction catalyst exposed to flue gas in power station co-fired with coal and biomass.
The major loss was observed for the first period of exposure of ca. 6 months (ca. 18.7% out of total 21.5% during 12 months). The activity decrease in the next 6 months was smaller. It is possible to draw a straight line through the first three experimental points, with R2 = 0.9969, including the fresh catalyst with relative decrease of activity equal to zero, while there is no correlation for all four experimental points (R2 = 0.7378). A similar tendency was observed by Wieck-Hansen et al. [8] for the SCR catalyst poisoned in a coal- and straw-co-firing boiler, where the total loss of activity (expressed as k/k0) of ca. 35% was observed after 2860 h of exposure, with 15% loss during the first 500 h.
Taking into account the tendency registered for the first 6 months, the average relative loss of activity for the catalysts studied in this work was ca. 4.2%/1000 h. For comparison, Wieck-Hansen et al. [8] registered the decrease in activity, defined as the ratio of k/k0 of ca. 8%/1000 h for the installation co-firing coal and straw. Even taking into account that during the last 2 months of the operation of the installation only coal was fired (without the addition of biomass), the 12M catalyst showed the decreasing effect of poisoning in comparison to 3M and 6M.
3.2 Ash characterization
As the decrease in activity of the SCR catalysts, used in the power stations firing biomass or co-firing coal with biomass, is mainly ascribed in the literature to the influence of fly ash, the samples of ash collected from the catalysts after exposure to industrial flue gas for 3, 6 or 12 months were characterized by atomic absorption spectrometry AAS and XRD and in the case of one sample (6M-ash), also by SEM/EDS.
3.2.1 Atomic absorption spectroscopy AAS
The most relevant elements according to the literature, i.e. K, Na, Mg, Ca, as well as Fe, Si and Al, were determined by AAS for all ash samples and are presented in Table 3.
The content of K, Na, Mg, Ca, Fe, Si and Al (presented as the % of appropriate oxides in the sample) in the studied ash samples.
| Composition (w/w) by AAS | Na2O | K2O | CaO | MgO | Fe2O3 | SiO2 | Al2O | ∑alkalia | |
| Ash | 3M | 0.52 | 2.42 | 7.47 | 2.72 | 6.37 | 41.73 | 20.13 | 13.13 |
| 6M | 0.65 | 2.43 | 4.37 | 2.63 | 6.87 | 41.11 | 16.99 | 10.08 | |
| 12M | 0.67 | 2.71 | 3.16 | 2.20 | 6.09 | 49.32 | 20.44 | 8.74 |
a Oxides of K + Na + Ca + Mg.
From Table 3, it may be concluded that ash contained alkali elements (K, Na, Mg, Ca), as well as Fe, and high amounts of Si and Al. The composition of ash collected from the catalysts after 3, 6 and 12 months of exposure differed somewhat where alkali and Fe content was concerned. The sum of alkali was higher for 3M-ash and 6M-ash and smaller for 12M-ash sample. This may be explained by the fact that during the whole studied period different amounts of biomass were co-fired, and during the last 2 months of exposure, only coal was fired, without the addition of biomass. On the other hand, during the first 3 months of exposure the average consumption of the fired biomass was of the highest level during the whole testing period (ca. 18% of mass share).
3.2.2 SEM/EDS
The micrographs of ashes, collected from the catalyst monoliths, showed spherical particles, with a wide size distribution (< 200 μm), studded with small catalysts particles. The large spherical particles seem to be formed by a thin wall being hollow. These glassy particles, mainly alkaline and iron-rich aluminosilicates (EDS analysis – table included in Fig. 4) are reported in the literature as cenospheres [16,17].
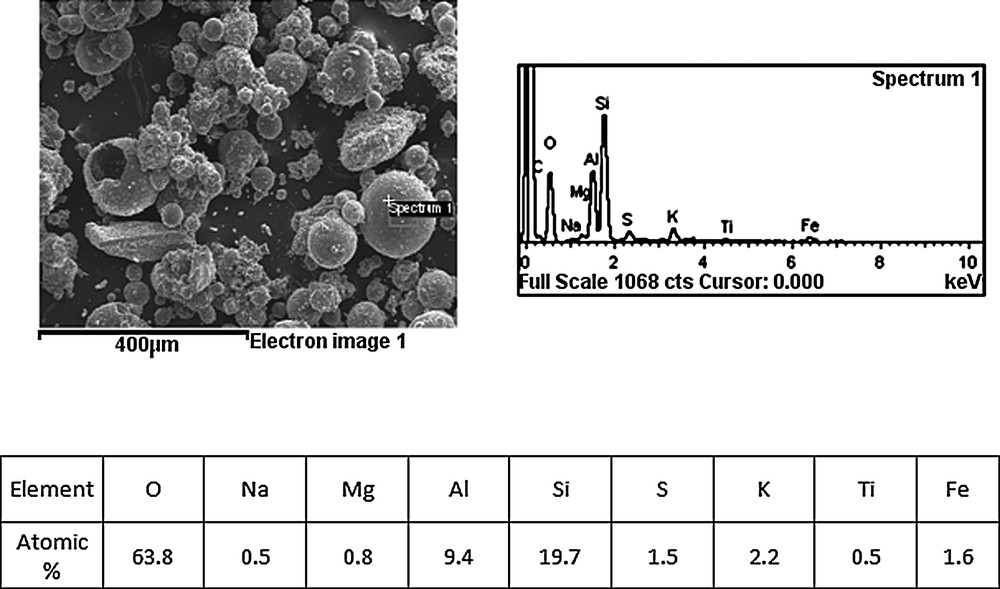
SEM micrograph of 6 months ash and EDS spectrum of the elemental analysis of a cenosphere (spectrum 1).
The image shows the presence of spherical objects in the collected ash of the size of 15 to ca. 150 μm. One of them is broken, revealing that it has very thin walls and is empty inside, thus identifying it as microsphere. This allows us to assume that other spherical objects are also microspheres. Apart from these objects, several irregular crystallites are observed. EDS analysis was taken from one of the supposed microspheres, as well as from one of the irregular crystallites. The irregular crystallite contained [at. %]: 7.47 Si, 4.50 Al, 1.55 S, 1.20 K, 0.38 Fe, 0.27 Ti, 0.23 Mg, 0.21 Ca and 40.95 O. As the sample was attached to a carbon film, the C content is not quoted here.
Gosal and Self [18] reported that submicrometer components in ash resulted from the condensation of mineral vapors. The elements observed in ash collected from 6M catalyst are typical of fly ash from coal combustion as observed, e.g., by Matsunaga et al. [19], but also for fly ash from solid biofuels [4]. The composition points to the presence of different types of deposits of small size.
The composition of the supposed microspheres, although most probably not directly related to the chemical poisoning of SCR catalysts, was also taken and showed the presence [in at. %] of: 13.31 Si, 6.33 Al, 1.49 K, 1.07 Fe, 0.98 S, 0.56 Mg, 0.34 Na, 0.34 Ti and 43.01 O. The observed objects contain typical elements, similar to those found for microspheres collected from ash from coal-fired power stations [20].
3.2.3 X-ray diffraction
The XRD patterns of the ashes collected from the catalyst monoliths are displayed in Fig. 5. All the ash diffractograms showed lines ascribable to titania (main line at 24.6°), the major component of the catalyst.
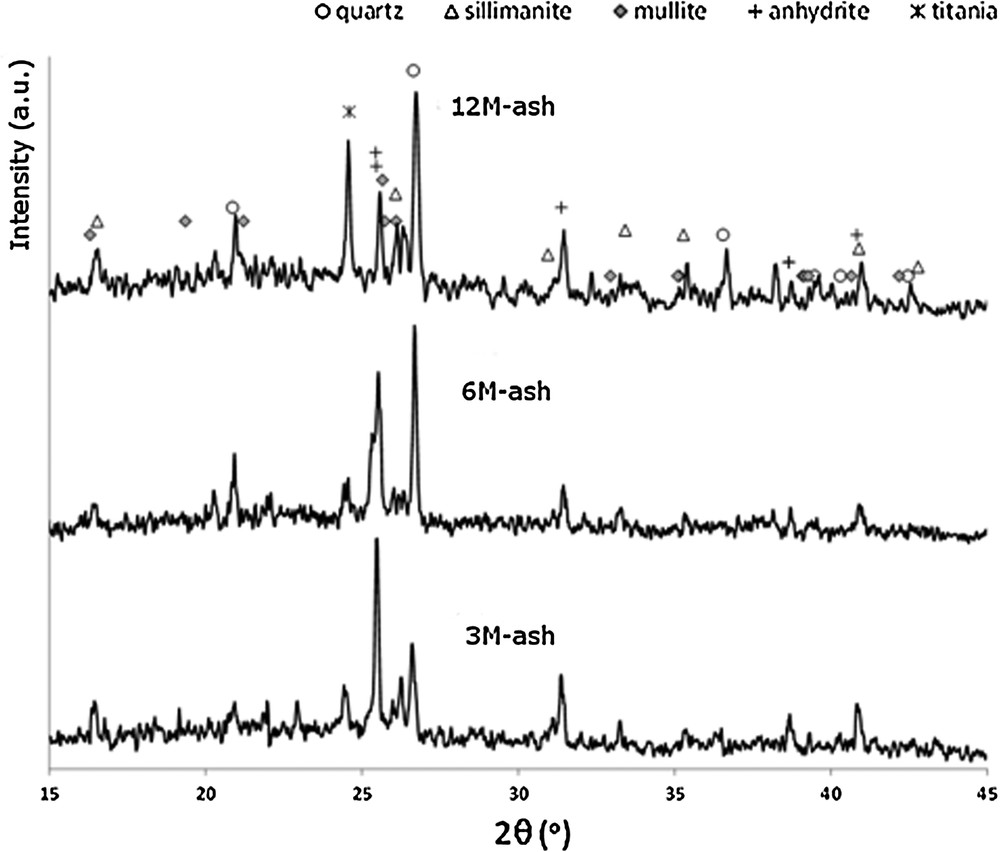
XRD patterns of ashes collected from catalysts monoliths.
All the characterized samples showed low crystallinity patterns containing features ascribable to anhydrite (CaSO4), quartz (SiO2), mullite (3Al2O3·2SiO2 or 2Al2O3·SiO2), silimanite (Al2SiO5). Vassilev and Vassileva [21] reported that coal fly ash consisted of amorphous and crystalline mineral matter with the addition of unburned organic material. There is, however, no general agreement concerning the type of crystalline compounds present in fly ashes. The differences were explained by Sahu et al. [4] who claimed that different fuel characteristics, the gas atmosphere and the used combustion technology influenced the composition of fly ash. Anhydrite and minor amounts of quartz were found, e.g., by Strege et al. [22] in ash scraped from the surface of catalyst exposed to flue gas from utility boiler co-firing wood waste and coal and by Koukouzas et al. [23] in fly ash from a pilot-scale fluidized boiler firing coal, wood chips and their blends. Mishra and Das [24] reported, for coal-derived fly ash, mullite, quartz and hematite as the main crystalline phases. Mullite was also registered as the main crystallographic phase in fly ash collected from the wet ash concentration unit of power plant firing Chinese coal. Its presence is explained by high content of kaolinite in raw coal [25]. Anorthite and gelehnite were, among others, found by Ilic et al. [26] in fly ash from power station firing lignite.
3.3 Characterization of catalysts
3.3.1 Atomic absorption spectroscopy
Table 4 summarizes the concentrations of Na, K, Ca, Mg and Fe as measured by AAS for 0M, 3M, 6M and 12M catalysts.
The concentration of Na, K, Ca, Mg and Fe for the studied catalysts, assessed by AAS.
| Poisoning elements (%W/W by AAS) | Na2O | K2O | CaO | MgO | Fe2O3 | ∑alkalia | |
| Catalyst | Fresh | 0.01 | 0.02 | 0.84 | 0.09 | 0.04 | 0.12 |
| 3M | 0.06 | 0.10 | 1.03 | 0.09 | 0.04 | 0.25 | |
| 6M | 0.11 | 0.18 | 1.12 | 0.11 | 0.05 | 0.40 | |
| 12M | 0.44 | 0.63 | 0.80 | 0.44 | 0.53 | 1.51 |
a Sum of oxides of Na, K, Ca and Mg.
Table 4 indicates that the total alkali concentration in the studied catalysts increased with the time of its exposure, with the exception of CaO, which on average did not change significantly in comparison to the fresh catalyst. As discussed above, XRD showed the presence of crystalline calcium sulfate (VI) in ash. The melting point of CaSO4 is 1460 °C and Tammann temperature (1/2 of melting point in [K]) is 594 °C, at which a solid-solid reaction is possible, was not reached under the conditions of catalyst operation at the power plant (ca. 420 °C). Therefore, it may be safely assumed that the reaction between the active sites on the catalyst and Ca from this ash component was limited. This is in good agreement with Krocher and Elsener [27] who reported from laboratory studies that calcium sulfate had very low deactivation potential for SCR vanadia-based catalysts. The small increase in 3M and 6M catalysts may have originated from Ca compounds in ash different from calcium sulfate, which were amorphous and thus not detected by XRD.
On the other hand, KCl and K2SO4, which in most publications were found to be the main reason for the deactivation of SCR catalysts [27], have lower Tammann temperatures (respectively, 247 and 397 °C), as well as much lower Hüttig temperature (0.3 of melting point) at which surface species or atoms at defects exhibit high mobility. The latter is 40 and 129 °C, for KCl and K2SO4, respectively, increasing the possibility of interaction with the catalyst. Zheng et al. [6] postulated based on the Tammann temperature that the deactivation was caused by surface diffusion of potassium deposited from the finest aerosol particles.
The comparison of the relative decrease of NO conversion versus the content of K2O or the total increase in alkali content (Na2O + K2O + MgO) does not reveal any linear relationship. Larsson [7] compared NO reduction for a SCR catalyst exposed to flue gases in a commercial biomass boiler with the catalysts impregnated with potassium compounds prior to flue gas exposure. In this case, for the contents of KCl/K2SO4 of up to ca. 0.1%, a rapid decrease in NO reduction was registered, while for higher contents (up to ca. 0.9%), the rate of deactivation was much slower. However, the deactivation on a commercial installation was lower than in case of the laboratory studies. Bench-scale deactivation studies of Strege et al. [9] carried out for a commercial catalyst and co-firing of 80% biomass and 20% coal revealed that the loss of activity was approximately inverse with time. Possibly, the size of ash particles is responsible for that, as suggested by Kling et al. [10] who reported that mainly alkali in ultra-fine particles, smaller than 100 nm, led to increased alkali accumulation.
3.3.2 SEM/EDS
The morphology of the catalysts observed by SEM is depicted in Fig. 6.

The morphology of the studied catalysts fresh (upper left), 3M (upper right), 6M (lower left) and 12M (lower right).
The details of SEM images under higher magnification are given in Fig. 7 and the results of EDS are summarized in Table 5.

SEM images of the studied catalysts.
The registered elements and their content at the selected points of SEM images for the studied catalysts.
| Sample | Spectrum | Element [at. %] | |||||||||
| Ti | V | W | Si | Al | Mg | Ca | K | S | O | ||
| Fresh | 1 | 32.01 | 0.69 | 0.59 | 0.36 | 63.1 | |||||
| 2 | 24.55 | 0.19 | 0.12 | 0.45 | 0.43 | 69.38 | |||||
| 3M | 1 | 23.37 | 0.38 | 0.39 | 0.72 | 0.45 | 1.14 | 47.13 | |||
| 2 | 31.25 | 0.2 | 0.39 | 0.39 | 0.83 | 61.29 | |||||
| 6M | 1 | 36.13 | 0.36 | 0.75 | 0.29 | 60.09 | |||||
| 2 | 1.74 | 19.75 | 19.75 | 5.42 | 1.89 | 7.73 | 63.46 | ||||
| 3 | 25.33 | 0.76 | 0.76 | 0.34 | 0.44 | 73.36 | |||||
| 12M | 1 | 26.6 | 0.51 | 0.15 | 0.35 | 1.16 | 45.68 | ||||
| 2 | 26.53 | 0.54 | 0.28 | 0.98 | 64.05 |
From Table 5 and Figs. 6 and 7 it may be derived that:
- • catalyst particles of irregular shape are present for the fresh catalyst (0M) ranging from under 10 to ca. 150 μm size. The elongated particles are most probably binder fibers, as indicated by the presence of Si and Al. Similar morphology was registered by Lisi et al. [28] for the ground commercial monolith catalysts supplied by ENEL Produzione. The morphology of the catalysts 3M, 6M and 12M did not change significantly after exposure to flue gas (cf. Fig. 6);
- • titanium, vanadium and tungsten were observed in all catalysts, both fresh and exposed to flue gas. The additional elements registered for the exposed catalysts indicate that fly ash either stuck to their surface or reacted chemically with them;
- • small amounts of sulfur (ca. 0.4%) were registered for the fresh catalyst. The presence of S, as well as additionally small amounts of Ca and Mg, were reported by Lisi et al. [28] for the commercial catalysts. For the exposed catalysts in our work, the content of S increased from 0.43% (0M) to ca. 0.8–1.1% (3M) and ca. 1.2% (12M), at the selected analyzed spots. The highest content of S was registered in the analyzed spots with the increased content of K (cf. Table 5 – spectra No. 1 for 3M and 12M), indicating a possible connection between these two elements. If the average amount for our fresh catalyst is subtracted (ca. 0.40%), the atomic ratio of S/K was ca. 1.6 and ca. 2.2, for 3M and 12M, respectively. This is much higher than the stoichiometric ratio of 0.5 for K2SO4 and points to the presence of other S-compounds on the surface of the exposed catalysts. An additional proof could be spectrum 2 of 12M where no alkalis were found and still there was the increased amount of S in comparison to the fresh catalyst.
The atomic ratio of Ca/S in spectrum 3 of 6M is not very far from 1 and, taking additionally into consideration the atypical morphology of the crystallite whose composition was analyzed, it may be connected with CaSO4, which as discussed above, was observed in XRD patterns of fly ash. Different amount of alkalis (K, Ca, Mg), which were additionally found on the exposed samples, arise from fly ash. Si and Al may have originated from fly ash, or belong to the catalyst binder.
3.3.3 X-ray diffraction
Fig. 8 shows X-ray patterns for the fresh and exposed catalysts. They reveal only the presence of anatase (TiO2). No changes were registered after exposure to flue gas in the power station, proving the stability of the support under the discussed conditions. This is in agreement with Lisi et al. [28] who studied a commercial catalyst poisoned by impregnation with KNO3 and NaNO3 solutions. A similar conclusion was reached by Li et al. [12] who studied XPS for a laboratory-prepared catalyst poisoned by K and observed no change in the binding energy of Ti 2p peaks after exposure to a simulated flue gas containing NO, NH3, SO2, O2 and H2O.
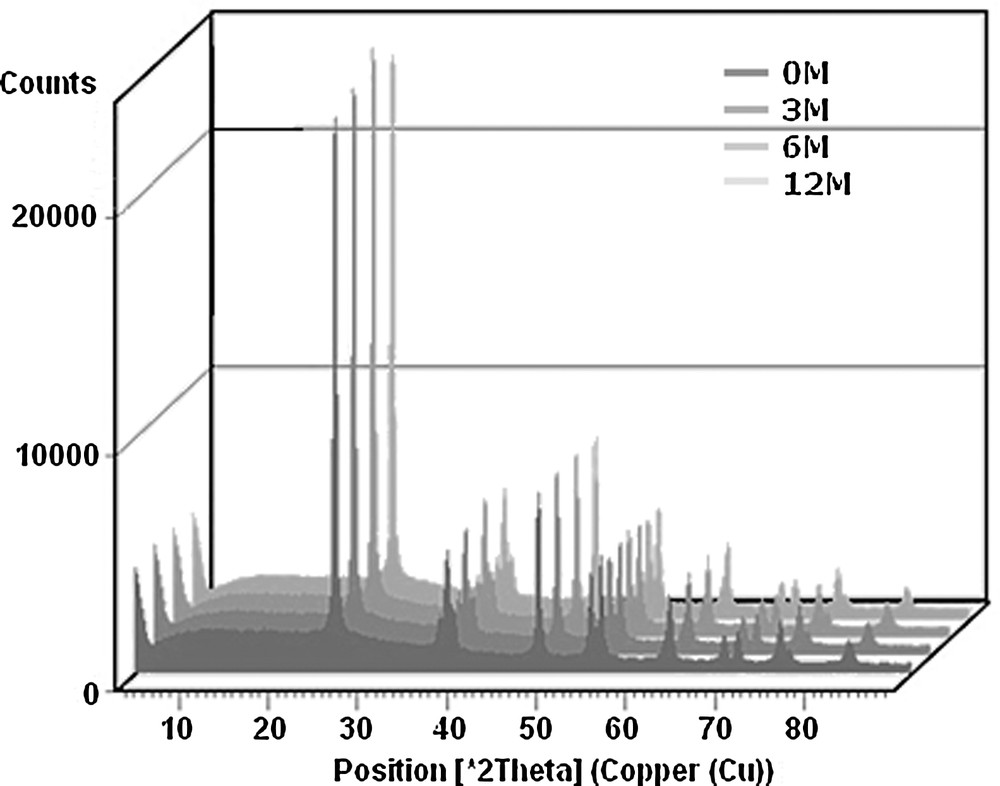
X-ray diffraction patterns for the studied catalysts.
3.3.4 UV–vis spectroscopy
Fig. 9 depicts UV–vis spectra for the studied catalysts.
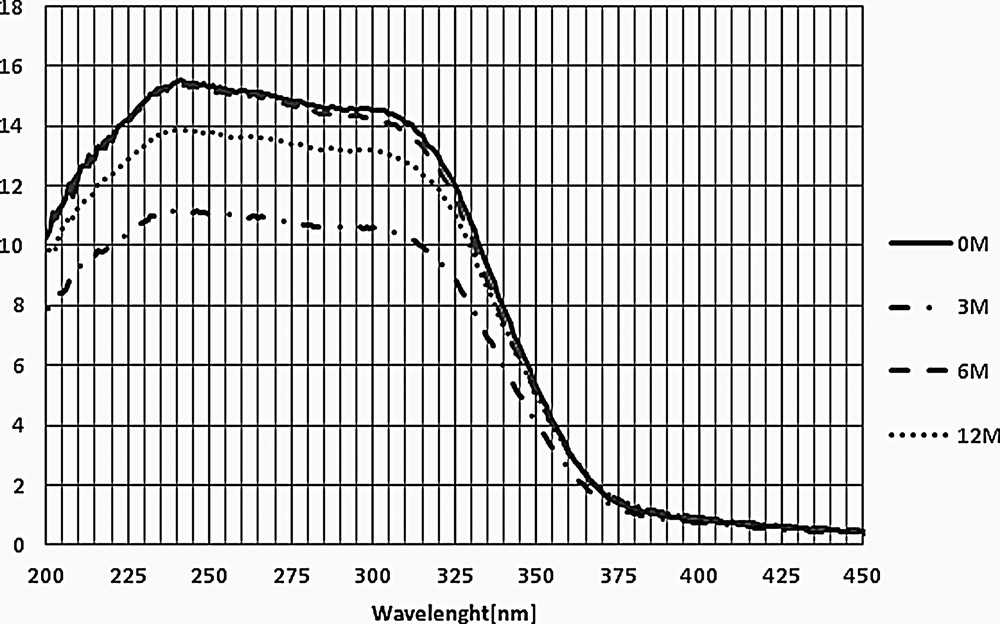
UV–vis spectra for the catalysts after exposure to flue gas in the power station (3M, 6M, 12M) in comparison to the fresh catalyst (0M).
Two bands can be distinguished for all studied samples. The band at around 240 nm may be attributed to Ti4+→O2− charge-transfer transition of catalyst support [29], whereas the band at around 320 nm is related to vanadium species occurring most probably as isolated species in tetrahedral position [30,31]. The absence of bands in the region of 375–425 nm suggests that polymeric vanadium species (cluster or oxide) are not present in either fresh or exposed catalysts [32]. Furthermore, the lack of signals of d–d transition in the range over 425 nm indicates no formation of V3+ or V4+ species under reaction conditions [33]. This is only partly in agreement with Li et al. [12]. These authors confirmed by XPS the stability of TiO2 and the absence of poisoning of this support with K+ for a laboratory-prepared catalyst. However, their studies of the state of V are not consistent with the results of the authors of this paper, but it should be mentioned that they registered the presence of different oxidation states (V5+, V4+ and V3+) both for the starting and exposed catalysts. On the other hand, only minor changes in relative amounts of V4+ and V5+ after poisoning were reported by Li et al. [12], which agrees with the herein obtained results where the influence of exposure on the oxidation state of vanadium is concerned. The possible reason for the differences in oxidation state may be different preparation procedures in both cases.
3.3.5 Acidity measurements
The surface acidity of V2O5-WOx/TiO2 catalyst plays an important role in the SCR mechanism, since acid sites adsorb and activate ammonia [34], thus it is crucial to evaluate such characteristics for fresh and post reaction (deactivated) catalysts. The main data on the 1-butene skeletal isomerization of catalysts is displayed in Fig. 10 and was taken as an indirect measure of acidity (Brønsted).
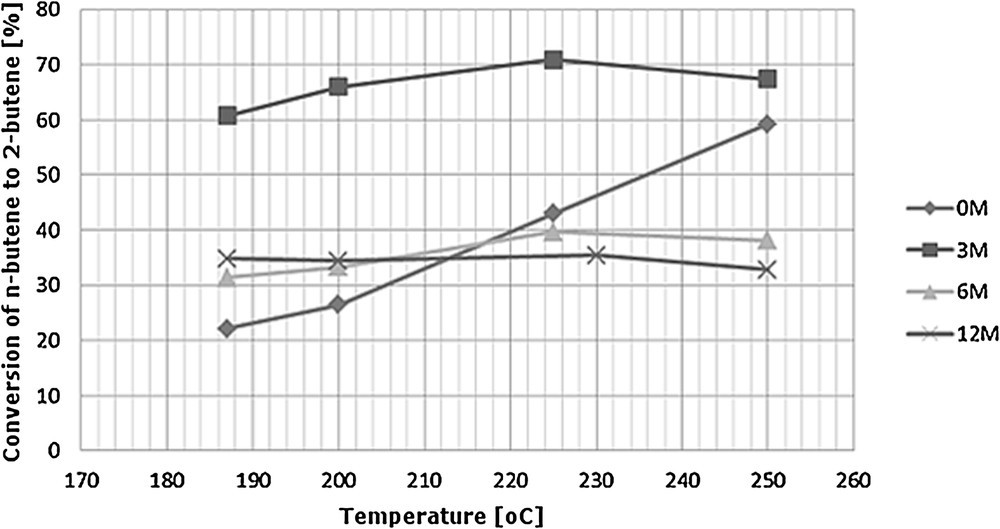
Conversion of 1-butene to 2-butene as a function of temperature.
In Fig. 10, it may be seen that for the fresh catalyst (0M), the reaction rate increased with rising temperature almost linearly from 20% to 60%. All three catalysts exposed to flue gas in the power station show a different dependence of 1-butene conversion on temperature, with a relatively small increase in conversion up to 230 °C for 3M and 6M, followed by a slight decrease at higher temperatures and almost no dependence on temperature for 12M. This indicates that acidic sites present for the exposed catalysts are at least partially different from those for the fresh catalyst. Additionally, sample 3M shows much higher conversion of 1-butene to 2-butene than all other studied samples, both exposed and fresh. This points to the formation of new acidic sites under the conditions of high-dust configuration, where flue gas contains not only high amounts of fly ash but also elevated SO2 concentrations, which may result in the formation of sulfate anions on the surface. Thus, two phenomena must be taken into account, the increase in acidity arising from the newly created sulfate species and the alkali poisoning of Brønsted acidic sites. The acidity results presented in Fig. 10 indicate in combination with the decrease in activity observed for 3M (cf. Fig. 3) that the these new acidic sites are different from those present on a typical SCR catalyst and possibly not taking part in NO reduction by NH3.
3.4 Discussion of the combined influence of alkali-containing ash and SO2 on SCR-NH3
The analysis of the influence of flue gas composition on the performance of SCR catalysts, operating under industrial conditions of co-combustion of coal and varying types and amounts of biomass must include the following facts established in the course of this paper, as well as discussed in the literature:
- • the physical deposition of ash with pore blockage;
- • the change in ash composition when biomass is added, especially the increase of alkali content;
- • the deposition on the surface, among others, of compounds with relatively low Tammann temperature and their increased mobility and possibility of reaction of with SCR active sites;
- • the presence of SO2, resulting in the formation of new acidic sites of properties different from those for the fresh catalyst.
The first, major effect, leading to the deactivation is undoubtedly connected with the blockage of pores by fly ash. Strege et al. [22] showed under industrial conditions that the main ash accumulation was at the entrance of the pores. In the case of the catalysts studied in this paper, some channels of the monolith were totally closed (cf. Fig. 2). On the other hand, it may be speculated that, although the pore blockage by fly ash may restrict diffusion through the channels, it may have still allowed the small molecules of NO and NH3 to approach the sites inside the channels where the direct contact with the poisons present in fly ash was less probable. Larsson [7] proved that deactivation by aerosols of KCl and K2SO4 resulted in NO reduction similar to that observed for a commercially exposed catalyst, but was lower than that registered for the catalysts deactivated prior to the SCR reaction by K+ impregnation. Zheng et al. [6], on the other hand, showed that a part of potassium penetrated the walls and was deposited in the channels due to surface diffusion. The increase in K/V ratio was observed up to the depth of 1200 μm. This may suggest that the sites deeper in the catalyst may still be available, as the total pore volume as measured by N2 sorption by Zheng et al. [6] was affected to a very small extent. It should be mentioned, however, that Zheng et al. studies were carried out for the combustion of straw which did not contain S; thus no SO2 was present in flue gas.
The second major effect is connected with the presence of SO2. Several authors reported that SCR catalysts require acidic sites for ammonia sorption. According to the generally approved SCR mechanism, both Brønsted sites (V–OH) groups and V = O species are necessary for the reaction, the former for NH3 sorption and the latter favoring the fast decomposition of the intermediate product NH2NO. K+ is a well-recognized poison for Brønsted V–OH sites, so its deposition will lead to the decrease in the number of sites necessary for the reaction. Additionally, Nicosia et al. [35] based on IR investigations and DFT calculations on V-W-Ti-O laboratory-prepared catalyst poisoned by impregnation with K or Ca salts showed that oxygen atoms of V5+ = O sites were also affected. Thus, the presence of small amounts of potassium or calcium should result in strong deactivation.
However, the combined effect of potassium salts and SO2 must be taken into account. There is actually very little information on such interaction, as either usually the catalysts were studied under industrial conditions but without SO2 in flue gas (e.g., for biomass combustion) [7] or catalysts pre-poisoned with potassium were studied under simulated, but industrially relevant conditions [12]. The latter conditions do not fully simulate those at the power station, as proven by Larsson [7] who reported that the SCR activity reduction was much higher for the catalyst impregnated with KCl or K2SO4 solutions than for the catalyst exposed to the flue gas in the commercial biomass power plant for a period of 6500 h. As the reason, the differing mechanisms of poisoning penetration were given.
Therefore, the role of both species (K+ and SO2) must be assessed simultaneously, taking into account the role of the deposition of additional sulfate groups on the surface of a SCR catalyst.
Several facts indicate that sulfate species, formed on the catalysts exposed to flue gas containing SO2, may play a positive role and to a certain degree protect an SCR catalyst from the alkali influence.
Baraket et al. [36] reported increased SCR activity for the catalyst containing sulfate species and explained the effect by both the changes in acidity and the improvement of redox properties of vanadates. Kristensen et al. [37] studied V2O5–TiO2 (sulfated or non-sulfated) SCR catalysts prepared by sol-gel method and showed that both total acidity and the strength of acidic sites was increased due to sulfation of the support, and, more importantly, the so-modified catalysts showed increased resistance towards K+ poisoning. This may be due, as suggested by Putluru et al. [38], to the preferential reaction of K+ with stronger acidic sites (sulfates), thus to a certain extent protecting V–OH SCR active species. The latter studies reported that after doping with 100–130 μmol K/g catalyst relative activity at 400 °C was 31.8% for the commercial V2O5–WO3/TiO2 and 86.8% for the nano-V2O5-sulfated-TiO2 catalyst.
However, it should be stressed that the problem is by no means solved – Kling et al. [10] studied alkali deactivation of high-dust SCR catalyst exposed to flue gas from 100 MW-scale biofuel and peat fired boiler and concluded that the addition of S-compound (decomposing in situ to SO3) did not influence the deactivation.
The results of the study on the simultaneous influence of alkalis and SO2 opens thus two possible routes for the improvement of catalyst performance under industrial conditions of co-combustion of coal and biomass:
- • modification of catalysts by pre-sulfation of the support of the catalyst;
- • regeneration by washing, as alkali compounds are well soluble in water, and perhaps potassium sulfate (VI) could be periodically removed, leaving room for the formation of new sulfate groups which could bind some of poisoning K+ species.
4 Conclusions
A commercial honeycomb V2O5/WO3/TiO2 catalyst was exposed to flue gas for a prolonged time (up to ca. 9400 h) in the power station co-firing coal and different types and amounts of biomass. The fresh and the exposed catalysts were tested outside the power station and the relative decrease in activity of ca. 4.2%/1000 h was found during ca. 4500 h. Afterwards the degree of deactivation slowed down.
The catalysts and the ash collected from the catalysts were characterized by AAS, SEM, XRD, UV–vis and acidity measurements. From the obtained results it may be concluded that:
- • alkalis were the main poison arising from co-combustion. The increased amounts of K+, Na+ and Mg2+ were found on the exposed catalysts, as well as in the fly ash collected from the catalysts. The content of Ca was not significantly increased. The main reason for potassium poisoning are relatively low Tammann temperatures of KCl and K2SO4 in the aerosols present in flue gas, leading to the deposition of these compounds on the particles of fly ash;
- • the morphology of the catalysts was not influenced;
- • the support (anatase) structure was stable under the industrial conditions;
- • the vanadium-isolated species were registered in both fresh and exposed catalysts. In neither of them polymeric species were found. The exposure to real flue gas did not change the oxidation degree of vanadium;
- • the acidity of the catalysts was increased during the first ca. 2300 hours, due to the formation of sulfate groups. They may have played a positive role at the initial period of exposure to flue gas, decreasing the extent of deactivation by potassium. Sulfate groups are characterized by stronger acidity than V–OH sites of SCR catalysts and thus may preferentially bind potassium. For the longer periods of exposure, poisoning of V–OH and V = O sites by potassium plays a more and more dominant role, resulting in the deactivation of the catalysts.
The present study offers an alternative to the commercial catalyst's modification by the controlled sulfation of the support prior to the SCR process, as suggested by the laboratory studies on catalysts on TiO2(or ZrO2)–SO42− supports described in the literature [37,39–41].
Acknowledgements
This work was realized under the agreement on cooperation between the EDF Polska S.A R&D Department and AGH-UST. Research for this paper was carried out within the framework of KIC InnoEnergy MSc studies “Clean Coal Technologies”. The support of these studies by EiT is greatly acknowledged. T. Grzybek would like to acknowledge the support of AGH-UST under grant No. 11.11.210.213.