

1 Introduction
Triarylamine-type molecules are very efficient functional units widely used in a number of devices.[1] In particular, they display efficient hole-transport mobility and have thus been inserted in organic solar cells,[2] organic light emitting diodes (OLEDs),[3] organic field effect transistors (OFETs),[4] non-linear materials,[5] and even as photoconductors for the Xerox® process in laser printers and photocopiers.[6] However, despite the profusion of studies performed on triarylamine derivatives, both by academic researchers and industry, this molecular core was only recently described as a structuring precursor that is able to self-assemble in ordered supramolecular architectures with enhanced physical properties. Indeed, we have first shown that triarylamine derivatives decorated with lateral amide groups can self-assemble into fibrillar aggregates upon visible light stimulation in chlorinated solvents, thanks to a combination of various supramolecular interactions (hydrogen bonding, π–π stacking, and van der Waals).[7] Using a complementary set of experimental techniques and theoretical tools, we have further demonstrated that the detailed mechanism of this unique supramolecular polymerization process is very complex but follows four main steps: i) light-induced oxidation of a catalytic amount of triarylamines to their radical cations associated with a chemical reduction of the chlorinated solvent; ii) formation of a nucleus of a minimum number of triarylammonium radicals; iii) growth of columnar primary fibrils thanks to the stacking of neutral triarylamines onto the nucleus; and iv) lateral secondary aggregations of fibrils leading to the formation of larger bundles of fibers.[8] We have also shown that unusual electric conduction takes place in these doped supramolecular polymers, with optical, magnetic, and electronic signatures of metallic materials.[9] Finally, we have exemplified that these triarylamine units can act as supramolecular structuring synthons for more complex structures, as they retain the general ability to self-assemble upon light stimulation in chlorinated solvents even when modified with various chemical units such as terpyridine, gallate, fullerene, or even rotaxane-type moieties.[10]
We now report on the self-assembly of triarylamine molecules conjugated with another organizing unit, namely benzene-1,3,5-tricarboxamide (BTA). Over the last two decades, BTA has been used as a versatile structuring moiety which spontaneously gives rise to columnar supramolecular polymers in a large variety of solvents, displaying various properties (gels and liquid crystals) and functions (sensors, MRI contrast agents, etc.).[11] Here we synthesize a tris(triarylamine) dendrimer incorporating a BTA core as the connecting unit. Its characterization by optical spectroscopy and microscopy techniques is performed in various solvents. Finally, we show the light triggered self-assembly of this molecule in chlorinated solvents.
2 Experimental
2.1 General
All reactions were performed under an atmosphere of argon unless otherwise indicated. All reagents and solvents were purchased at the highest commercial quality and used without further purification unless otherwise noted. Dry solvents were obtained using a double column SolvTech purification system. Yields refer to purified spectroscopically (1H NMR) homogeneous materials. Thin Layer Chromatographies were performed with TLC silica on aluminium foils (Silica Gel/UV254, Aldrich). In most cases, irradiation using a Bioblock VL-4C UV-Lamp (6 W, 254 nm and/or 365 nm) as well as phosphomolybdic acid staining was used for visualization. Ultra Performance Liquid Chromatographies coupled to Mass Spectroscopy (UPLC-MS) were carried out on a Waters Acquity UPLC-SQD apparatus equipped with a PDA detector (190–500 nm, 80 Hz), using a reverse phase column (Waters, BEH C18 1.7 μm, 2.1 mm × 50 mm), and the MassLynx 4.1 – XP software. 1H NMR spectra were recorded on a Bruker Avance 400 spectrometer at 400 MHz and 13C spectra at 100 MHz in CDCl3 or toluene-d8/MeOD (5%) at 25 °C. The spectra were internally referenced to the residual proton solvent signal (CDCl3: 7.26 ppm, toluene-d8/MeOD (5%): 2.08 ppm for 1H spectrum, and CDCl3: 77.16 ppm and toluene-d8/MeOD (5%): 20.43 ppm for 13C spectrum). For 1H NMR assignments, the chemical shifts are given in ppm. Coupling constants J are listed in Hz. The following notation is used for the 1H NMR spectral splitting patterns: singlet (s), doublet (d), triplet (t), multiplet (m), and large (l). For all experiments in chloroform (regular and deuterated), the solvent was freshly filtered on basic alumina prior to use.
2.2 Synthesis and characterization of compound 3
A solution of N1,N1-bis(4-(benzyloxy)phenyl)benzene-1,4-diamine7 (356 mg, 754 μmol) and 1,3,5-benzenetricarbonyl trichloride (57 mg, 215 μmol) in dry THF (2 mL) was treated with triethylamine (150 μL, 1.08 mmol). The resulting solution was then stirred at room temperature for two days under argon. After removal of the solvents, the crude product was purified by column chromatography (SiO2, toluene → EtOAc/toluene: 5/95). The solid was then washed with cyclohexane and diethyl ether, and further triturated with diethyl ether to yield compound 3 (281 mg, 83%) as a yellow solid. Rf = 0.77 (50/50 EtOAc/cyclohexane); 1H NMR (400 MHz, CDCl3, 298 K): δ = 8.42 (brs, 3H), 8.38 (brs, 3H), 7.41–7.28 (m, 36H), 6.97 (d, 3J = 9.0 Hz, 12H), 6.87 (d, 3J = 11.2 Hz, 6H), 6.84 (d, 3J = 9.0 Hz, 12H), 4.97 (s, 12H); 1H NMR (400 MHz, C7D8/CD3OD(5%), 298 K): δ = 8.63 (s, 3H), 7.74 (d, 3J = 8.9 Hz, 6H), 7.24 (d, 3J = 7.3 Hz, 12H), 7.14 (t, 3J = 7.3 Hz, 12H), 7.07 (s, 6H), 7.06 (d, 3J = 9.0 Hz, 6H), 7.01 (d, 3J = 8.8 Hz, 12H), 6.77 (d, 3J = 8.8 Hz, 12H), 4.70 (s, 12H); 13C NMR (100 MHz, C7D8/CD3OD(5%), 298 K): δ = 165.88, 155.64, 146.39, 142.06, 137.91, 136.42, 132.47, 129.76, 129.28, 128.78, 128.36, 128.09, 127.77, 126.58, 122.61, 122.32, 116.25, 70.49; FT-IR (ATR): υ = 3418, 3278, 3036, 2923, 2860, 1678, 1651, 1634, 1599, 1499, 1453, 1310, 1236, 1221, 1162, 1025, 826 cm−1; ESI-MS: m/z calcd for C105H84N6O9: 1572.63 [M]+, found 1572.66.
2.3 Characterization of self-assemblies
1H NMR spectra before irradiation with white light and after exposure to visible light were recorded on a Bruker Avance 400 spectrometer at 400 MHz. UV–Vis–NIR spectra were recorded using a Cary 5000 scan Varian spectrophotometer with quartz glass cuvettes of 1 mm optical path under ambient conditions. Fluorescence spectra were recorded using a FluoroMax-4 (Horiba Jobin-Yvon) spectrophotometer with quartz glass cuvettes of 1 cm optical path under ambient conditions. Atomic force microscopy (AFM) images were obtained by scanning the samples using a Nanoscope 8 (Bruker) operating in Peak-Force tapping mode. All cyclic voltammograms were recorded with a three electrode setup (ALS Co., Ltd, Japan) on a PGSTAT101 potentiostat (Metrohm, Germany) under an argon atmosphere at room temperature in 0.1 NBu4PF6/C2H2Cl4. Spectroelectrochemistry was performed using a platinum wire mesh transmission with a silver wire pseudoelectrode and a platinum foil counter electrode. For spectroelectrochemistry, UV–Vis spectra were acquired on an Avaspec-2048 with an Avalight-DH-S-Bal light source and a Thorlabs GG420 nm Glass longpass filter.
3 Results and discussion
3.1 Synthesis and characterization of compounds 3 and A
Compound 3 was synthesized in three steps from commercially available reagents in 75% overall yield (Scheme 1). In particular, compound 1 was obtained by using an efficient modified Ullmann one-pot coupling protocol that we reported some years ago.[7] Orthogonal reduction of the nitro group was achieved using tin chloride at a temperature of 80 °C.[12] Further amidation of compound 2 with 1,3,5-benzenetricarbonyl trichloride afforded compound 3 with a very good yield. The 1H NMR spectrum of 3 at a concentration of 10 mM in toluene-d8/methanol-d4 (5%) shows the expected resonance signal of the BTA core (singlet, 8.63 ppm) and all signals of the triarylamine moiety which are in good agreement with previous derivatives already reported by our group. Furthermore, 3 was characterized by 13C NMR, FTIR, and ESI mass spectrometry with all analyzes confirming the presence of the desired compound (see Experimental section). Triarylamine A, which will serve as a reference compound in this study, was synthesized by acetylation of 2 following a previously described protocol.[7]
Synthetic route and chemical structures of compound 3 and reference compound A.
3.2 Characterization of the self-assembly of compound 3
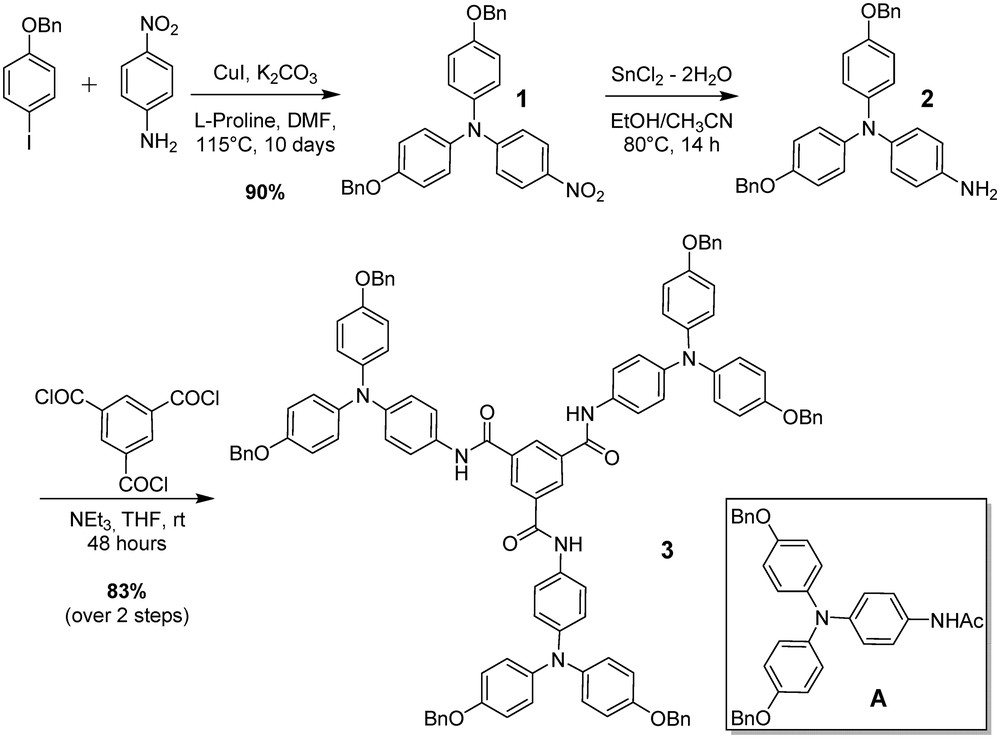
In order to evaluate the influence of the BTA core on the photophysical properties of compound 3, we investigated its absorption and emission properties in various solvents (Fig. 1, Table 1). Compound 3 features a maximum absorption at ∼300 nm (in all solvents) and a broad shoulder peak whose maximum absorption wavelengths vary between 349 and 363 nm (depending on the polarity of the solvent). Compared with the UV–Vis absorption reference spectra of N-(4-(bis(4-(benzyloxy)phenyl)amino)phenyl)acetamide A,[7] the main absorption at ∼300 nm clearly arise from the triarylamine core whereas the shoulder at higher wavelength has been already observed in supramolecular stacks of triarylamines.[7,9b] When excited at 400 nm, fluorescence experiments display a) broad structureless fluorescence spectra with a maximum intensity located between 528 and 543 nm, b) large Stokes shifts (>160 nm) and c) no mirror image relationship between the absorption and fluorescence spectra, indicating a probable aggregation induced fluorescence (AIE) phenomenon as already described for stacks of triarylamines.[9b,10b] All these features suggest that in all solvents tested, the triarylamine units are forming stacks with the help of the BTA core. Indeed, for compound A, no UV shoulder neither fluorescence can be observed in the same conditions of solvent and concentration (Fig. 1, dotted lines).
Absorption (full line) and aggregation induced emission (dashed line; excitation wavelength 400 nm) spectra of compound 3 in CHCl3 (black), THF (red), toluene/5%MeOH (blue). Absorption spectra of N-(4-(bis(4-(benzyloxy)phenyl)amino)phenyl)acetamide A in the different solvents are represented in dotted lines.
Photophysical characteristics of compound 3 in different solvents.
| Solvent | Absorption λCT (nm) | Emission λem (nm) | Stokes shift (nm) | ɛmax (×105) | |
| CHCl3 | 0.259[13] | 363 | 528 | 165 | 8.8 |
| Toluene/MeOH (5%) | n.d. | 358 | 538 | 180 | 6.7 |
| THF | 0.207[13] | 349 | 543 | 194 | 6.0 |
Interestingly, optical microscopy from a drop-cast solution of 3 in toluene/MeOH (5%) revealed the formation of monodimensional single fibers, with lengths of several hundreds of microns after evaporation of the solvent (Fig. 2a,b). Imaging these objects by atomic force microscopy (AFM) indicates the presence of a rigid ribbon-like structure with a width of ∼200 nm (Fig. 2c). A closer look at the internal part of these ribbons shows the presence of thinner fibrils (with a width < 5 nm, Fig. 2d) which have a unidirectional alignment that accounts for the piling of compound 3 thanks to intermolecular hydrogen bonding at the BTA/triarylamine junction. Similar observations were made by optical microscopy in the other solvents.[14]

a, b) Optical microscopy images of compound 3 from a 3 mM solution in toluene/MeOH (5%); c, d) AFM images of compound 3 from a 1.5 mM solution in toluene/MeOH (5%).
3.3 Light-induced oxidation process for compound 3
Based on the knowledge of triarylamine-amide compounds, we then studied the light-induced oxidation of compound 3 in chlorinated solutions (chloroform or tetrachloroethane) by different spectroscopic experiments (Fig. 3). When a solution of compound 3 in deuterated chloroform (∼5 mM) was kept in the dark, all the expected set of proton resonance signals were observed by NMR spectroscopy (Fig. 3a, i). However, after light irradiation using a 20 W lamp (power density: 0.07 W cm−2), the disappearance of proton resonance signals corresponding to the triarylamine core, the BTA unit and the CH2 from the benzyl unit was observed, indicating the partial oxidation of compound 3 in triarylammonium radicals (Fig. 3a, ii). This conclusion is supported by a number of our previous studies in which the characteristic disappearance of the signals is explained by the combination of i) a strong anisotropy of the triarylamine stacks and ii) a paramagnetic contribution of the delocalized radical in the supramolecular polymer.[7,10] UV–Vis–NIR measurements were also performed to monitor the stabilization of triarylammonium radicals upon light irradiation, owing to their characteristic broad absorption band centered at ∼785 nm which is known as another signature of the self-assembly process (Fig. 3b).[7,8,10] Indeed, upon irradiation with white light, compound 3 displayed a strong increase of the 785 nm absorption band, in correlation with its expected propensity to stack triarylamine moieties in charge transfer complexes. This experiment revealed that the maximum absorption at 785 nm is reached after 8 h of irradiation (Fig. 3c), which is slower compared to other triarylamine-amide compounds.[7] This difference could be explained by the presence of the self-assembly before the irradiation with light. Indeed, it was shown previously that when stacked, the triarylamine molecules can only oxidize at the extremities of the self-assemblies before diffusing into the fibers, thus reducing the effective molarity and the kinetics of the process.[8a]

a) Typical 1H NMR spectra of 3 obtained immediately after purification (i) and after 2 h of exposure to a 20 W power halogen lamp (ii), [3] ≈ 5 mM in CDCl3. The red lines indicate the disappearance of the characteristic triarylamine and BTA signals upon light irradiation; b) UV–Vis–NIR spectra obtained as a function of time of irradiation for an initial 0.25 mM solution of compound 3 in chloroform; c) Irradiation kinetic experiment plotted at different wavelengths for a 0.25 mM solution of compound 3; d) Optical microscopy image from a drop-cast solution of compound 3 irradiated for 2 h with a 20 W power halogen lamp in CHCl3 and obtained after evaporation of the solvent.
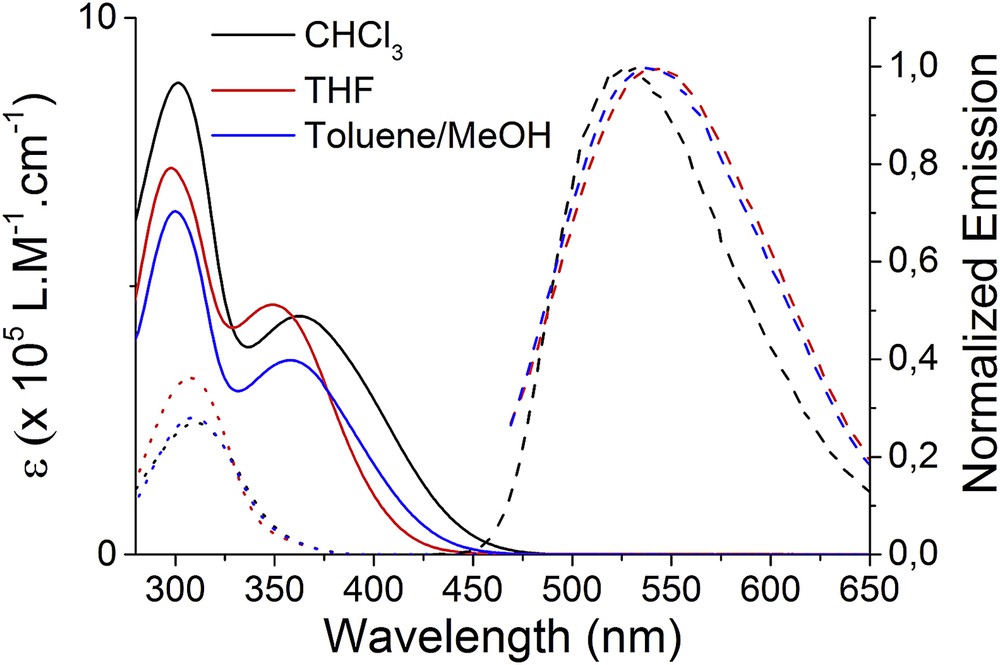
We then investigated the electrochemical properties of compound 3 by cyclic voltammetry (CV), which was performed in solution with all potentials referenced to the ferrocene/ferrocenium couple. The cyclic voltammogram was recorded at a scan rate of 100 mV s−1 in tetrachloroethane (TCE) and features two chemically reversible oxidation waves (Fig. 4a). The first one can be associated with the formation of radical cation species on the three triarylamine units (E1/2 = 0.32 V) while the second one can be attributed to the corresponding dications (E1/2 = 1.31 V). Spectroelectrochemical data were then monitored by applying increasing potentials to a solution of compound 3 in TCE (containing 0.1 M of nBu4NPF6). For potentials above the first oxidation potential, a clear signature corresponding to the presence of radical cations was recorded with the appearance of new absorption bands at 645 and 756 nm, as observed when previously irradiated with light (Fig. 4b). For potentials above the second oxidation potential, no clear signature of the dication was observed, but this species has already been reported to be not stable.[15] Finally, we checked by optical microscopy the structures of the self-assemblies obtained after oxidation, and confirmed the presence of thin fibers even when doped in the form of their radical cations (Fig. 3d).
a) Cyclic voltammogram for the oxidation of compound 3 (0.5 mM in tetrachloroethane with 0.1 M TBAPF6) showing two reversible oxidation peaks; b) UV/Vis/NIR spectra recorded during oxidation at 0.024 V (black) and 1.252 V (red).
4 Conclusions
We have reported the synthesis of a new dendritic molecule combining a benzenetricarboxamide core and three well-designed triarylamine arms. Its characterization by optical spectroscopies has been performed in different organic solvents and demonstrates an aggregated state thanks to the presence of the BTA unit. Micrometric crystalline fibers formed by the hierarchical self-assembly of rigid fibers with a high aspect ratio have been observed by optical and atomic force microscopies. The catalytic oxidation of triarylamine units in self-assemblies of 3 has also been studied. It can be performed in chlorinated solvents upon simple visible light stimulation and leads to the migration of charges within the doped fibers, a characteristic already observed on triarylamine derivatives decorated with lateral amide groups. Overall, this work enlarges the scope of chemical modifications that can be made on triarylamine-amide cores to produce ordered supramolecular architectures. The formation of such monodimensional doped self-assemblies by simple light irradiation is particularly appealing for further use as optoelectronic materials.
Acknowledgements
This work has been supported by the European Union (FP7/2007-2013, ERC n°257099, ITN Read n° GA-211-289723), the ANR (Dynablocks and STANWs ANR-11-BS08-0006) and the international center for Frontier Research in Chemistry. We also would like to thank CNRS, UdS, Labex CSC and IUF for additional financial support.