

1 Introduction
Incorporation of additional coordination sites within the backbone of a ligand coordinated to a catalytic site potentially allows the catalytic activity to be modulated through the complexation of metal cations. Allosteric effects [1] and/or electronic effects [2] may be implicated to explain the modulation of the catalytic activity observed. Porphyrins bearing peripheral coordination site(s) are interesting scaffolds to investigate how it is possible to tune the electronic and catalytic properties of the outer metal(s) thanks to the inner metal (or vice versa) [2–5]. For example, Osuka and co-workers reported the synthesis of metalloporphyrins bearing external N–C–N pincer type PdII complexes, which exhibited markedly different catalytic activities in typical Mizoroki–Heck reactions depending on the inner metals [3]. Although the mechanism of the present Mizoroki–Heck system is unclear, it is known that nearly all Pd precatalysts slowly decompose at high temperatures ( > 120 °C),[6] i.e. temperatures typically used in Mizoroki–Heck reactions, to liberate soluble “ligand-free” palladium colloids or nanoparticles [7]. It has also been shown for other metalloporphyrins bearing external PdII complexes that the porphyrin plays a crucial role in the apparent catalytic activity of the Pd-precatalysts. More exactly, the nature of the inner metal strongly modulates the release of adequate amounts of active Pd0 species in high-temperature Mizoroki–Heck reaction mixtures [2,4,7]. In the course of our studies, we were interested in the synthesis of metalloporphyrins annulated to peripheral N-heterocyclic carbenes (NHCs, Scheme 1) [8]. N-heterocyclic carbenes (NHCs) have become an important class of ligands in the field of catalysis, since they can form very stable bonds with a wide range of transition-metals in low and high oxidation states [9]. Recently, we showed that porphyrin-NHCs scaffolds 1M (Scheme 1, M = Ni, Cu, Zn and H2) provide an interesting way to remote the electronic properties of NHC by varying the inner metal of the porphyrin [10] and to build multiporphyrinic systems to investigate energy transfer processes between chromophores [11]. Here, we present the use of palladium complexes bearing porphyrin-NHCs as precatalysts in the Mizoroki–Heck reaction. We notably demonstrate how the porphyrin core and its inner metal can affect the apparent catalyst activity in the course of the Mizoroki–Heck reaction. Similar to previous reports, mercury poisoning tests and transmission electronic microscopy (TEM) studies suggest that these complexes may readily form Pd nanoparticles at elevated temperature [7].
Structure of the porphyrin-NHC scaffold 1M.
2 Results and discussion
2.1 Electronic properties of porphyrin-NHCs
Imidazolium salts 2M were obtained in good yields starting from meso-tetra(4-tert-butylphenyl)porphyrin, and were characterized by using conventional techniques (Scheme 2) [12]. To evaluate the electron-donating ability of the porphyrin ring, we studied the electrochemical oxidation processes of 2M by cyclic voltammetry (CV) and rotating disk voltammetry (RDV) in CH2Cl2 with Bu4NPF6 as the supporting electrolyte: the first oxidation potentials (EOX1 vs Fc/Fc+) are shifted anodically in the order of 2Zn (+0.36 V) < 2Cu (+0.54 V) < 2H2 (+0.57 V) < 2Ni (+0.64 V). Thus, the electron donating ability of the porphyrin ring increases in the order of 2Ni < 2H2 ≤ 2Cu < 2Zn. To quantify the electron-donor ability of the corresponding carbene ligand, we synthesized the corresponding cis-[(1M)Rh(CO)2Cl] complexes and measured their CO-stretching frequencies to study the influence of the inner metal of the porphyrin on the electronic properties of the outer NHC ligand [10,13]. After obtaining the imidazolium salts of 3M by reacting 2M with AgBF4 in acetonitrile, the rhodium(I) complexes 4M were obtained by the in situ deprotonation of 3M in the presence of [{Rh(COD)Cl}2] (Scheme 2). Then, complexes 5M were obtained by bubbling carbon monoxide through a solution of complexes 4M in CH2Cl2 (Scheme 2). Two CO stretching frequencies were observed by IR spectroscopy confirming the cis geometry of all complexes 5M. Similar average CO stretching vibrations were observed at 2040.5, 2041, 2041 and 2041.5 cm−1, respectively, for 5Zn, 5Cu, 5H2 and 5Ni. These data clearly show that NHCs 1M with M = H2 and M = divalent metal cations such as NiII, CuII and ZnII display very similar electron-donor abilities. It is also noticeable that the σ-donor ability of the NHCs is poorly influenced by the π-electron-donor ability of the porphyrin ring.
Structures of compounds 2M–5M.
2.2 Synthesis of palladium(II) complexes 6M and 7M
To investigate the effects of the inner metal of the porphyrin core on the catalytic activity of a metal anchored at the periphery of the porphyrin, we synthesized palladium complexes containing porphyrin-NHCs 1M as ancillary ligands [8,11]. The reaction of the imidazolium salts 2M with 0.5 equivalent of Pd(OAc)2 in refluxing THF afforded a mixture of two complexes after standard workup and crystallization. No remaining imidazolium salt was observed in the reaction mixtures after two hours. The two complexes formed in the course of the complexation reactions are trans-[PdI2(1M)2] and [{PdI(μ-I)(1M)}2], respectively, labeled 6M and 7M (Scheme 3). They were obtained in a 6M/7M ratio of ∼4:1 and similar results were obtained with M = Ni, Cu, Zn or H2 showing that the inner metal has no influence on the 6M/7M ratio. The two complexes 6M and 7M could be separated by column chromatography on silica gel.
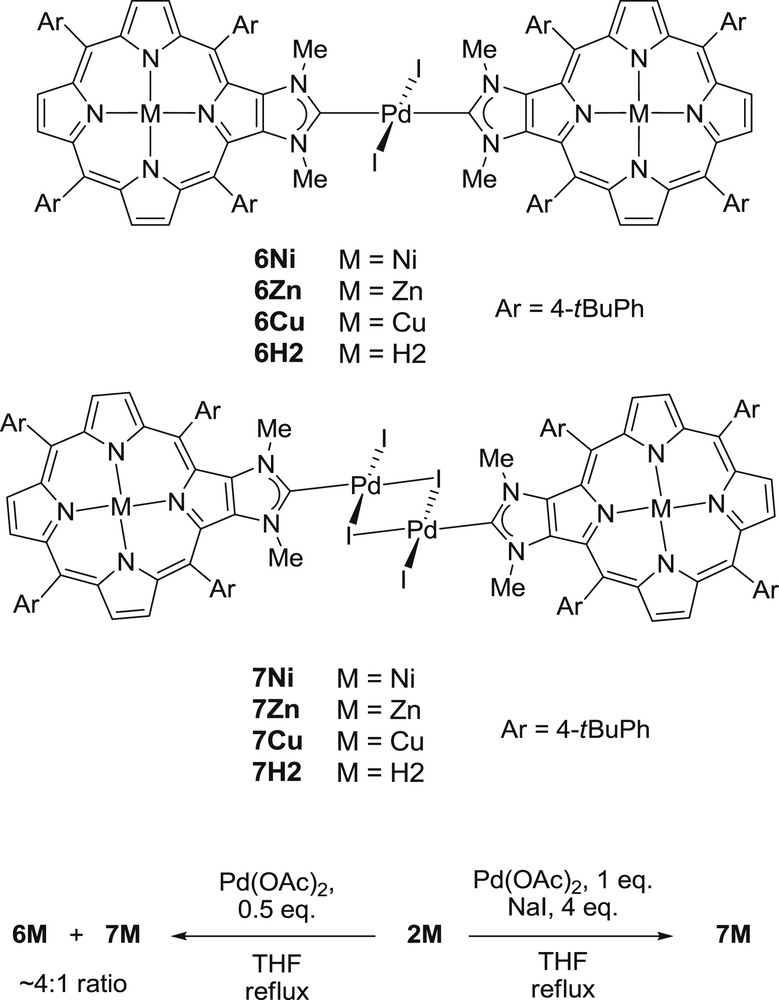
Synthesis of complexes 6M and 7M.
No signal corresponding to the iminium proton could be observed in the 1H NMR spectra of trans-[PdI2(1M)2] complexes, confirming the formation of the NHC–Pd bonds. Their parent mass ion peaks could be observed by ESI-TOF mass spectrometry: for example m/z = 2175.7 for 6H2 (calcd for (C126H135I2N12Pd)+ = 2175.8 [(M+H)+]). 1H NMR spectroscopy also showed the 2-fold symmetry of 6M complexes. The trans geometry around the PdII was only confirmed by X-ray diffraction analysis for 6Ni, because no suitable monocrystals for X-ray diffraction analysis were obtained for other complexes 6M [8]. However, we observed that 6Ni could be also obtained by reacting 6H2 with Ni(acac)2 and this experiment indirectly showed the trans geometry around the PdII of 6H2. Knowing that 6H2 has a PdII center with a trans geometry and that metallation reactions with Zn(OAc)2 and Cu(OAc)2 afforded respectively 6Zn and 6Cu, we presume that PdII centers of 6Zn and 6Cu have a trans geometry too [11]. Herrmann and co-workers showed that cis-[PdI2(NHC)2] complexes could be obtained by reacting N,N′-dimethylimidazolium salts with Pd(OAc)2 in refluxing THF.[14] Cis isomers are usually obtained with small N-substituting groups such as methyl groups, and trans isomers are obtained with bulky N-substituting groups. In our case, the NHC precursors of 1M are also N,N′-dimethylimidazolium salts, but we did not observe the formation of cis-[PdI2(1M)2] isomers, which are presumably energetically unfavored [11].
To avoid the formation of a mixture between trans-[PdI2(1M)2] and [{PdI(μ-I)(1M)}2] complexes, we used the classical conditions for the synthesis of dimeric mono-coordinated [{PdI(μ-I)(NHC)}2] complexes, i.e. addition of 1 equiv of Pd(OAc)2 and NaI in excess to a solution of 2M in THF (Scheme 3). Following this procedure, complexes 7M were obtained in good yields (83% yield for 7H2, see reference [11]). Mass spectrometry analyses were performed for these complexes. The parent mass ion peak of 7H2 could be observed at m/z = 2537.5 (calcd for (C126H135I4N12Pd2)+ = 2537.52 [(M+H)+]) in its ESI-TOF mass spectrum, but parent mass ion peaks of the complexes including metallopoporphyrins 7M (M = Ni, Cu, Zn) could not be detected and only fragments were observed. The 1H NMR spectra of the complexes 7M are similar to those of the trans isomers 6M except some signals for the protons of the two meso aryl groups of 7M which were broad and split at room temperature and sharper at higher temperature. Variable temperature 1H NMR experiments confirmed the dynamic nature of this phenomenon.[11] This is consistent with the perpendicular orientation of each porphyrin mean plane with respect to the Pd2(μ-I)2 bridge in complexes 7M, the slower rotation of the meso aryl groups at room temperature, and thus, different chemical environments for the protons close to the Pd2(μ-I)2 unit. Energy-dispersive X-ray (EDX) analyses appeared also to be an interesting tool to characterize complexes 6M and 7M (M = Ni, Cu or Zn) since different M/Pd/I chemical compositions were expected for these two families (Table 1). EDX analyses of complexes 7M showed chemical compositions of M/Pd/I in ca. 1:1:2 ratios in accordance with the [{PdI(μ-I)(1M)}2] structures proposed for these complexes. Chemical compositions of M/Pd/I in ca. 2:1:2 ratios were observed for 6M confirming that one palladium cation was coordinated by two 1M ligands and two iodides.
Energy-dispersive X-ray (EDX) analyses of 6M and 7M.a
| % atomicb | |||
| M | Pd | I | |
| 6Nic | 34.9 (3.0) | 22.0 (1.7) | 43.1 (2.1) |
| 6Cud | 40.8 (8.2) | 20.0 (3.1) | 39.2 (5.8) |
| 6Zne | 38.9 (1.7) | 21.3 (1.1) | 39.8 (0.6) |
| 7Nic | 24.9 (1.4) | 26.7 (1.2) | 49.3 (2.5) |
| 7Cud | 27.8 (3.6) | 26.2 (1.3) | 46.0 (3.8) |
| 7Zne | 26.0 (1.9) | 26.9 (1.2) | 47.1 (0.8) |
a Mean normalized values obtained for four different spectra. Values indicated in parentheses correspond to the standard deviations.
b Theoretical % atomic values calculated for 6M and 7M (M = Ni, Cu or Zn) are, respectively, M/Pd/I 40:20:40 and 25:25:50.
c Normalized for Ni, Pd and I elements.
d Normalized for Cu, Pd and I elements.
e Normalized for Zn, Pd and I elements.
2.3 Mizoroki–Heck coupling reactions with 7M used as precatalysts
Knowing that complexes 6M were only poorly soluble in DMF, we used complexes 7M as precatalysts for the Mizoroki–Heck coupling reaction between iodobenzene and styrene. The catalytic experiments were conducted in DMF at 120 °C with 1 equiv. of iodobenzene, 1.1 equiv. of styrene, 2 equiv. of diisopropylethylamine, and 0.04 mol% of the precatalyst 7M, i.e. 0.08 mol% of [Pd]. The reaction progress was monitored by gas chromatography and 1H NMR spectroscopy. We have previously shown that 1M ligands with M = Ni, Cu, Zn and H2 display very similar electron-donor abilities. They also exhibit identical steric properties, since no modification has been brought to the organic backbone. Knowing that, it was reasonable to anticipate that reaction profiles obtained for Mizoroki–Heck reactions catalyzed by the four 7M precatalysts should be similar.
Reaction profiles (i.e. conversion of iodobenzene vs. time) at one hour intervals are displayed in Fig. 1. 1H NMR studies confirmed that styrene was converted to trans-stilbene. At 0.04 mol % of the precatalyst 7M, conversions of 80–90% were reached by 7Zn, 7Ni and 7Cu after 10 h. No significant induction period was observed for 7Cu, the most active pre-catalyst in the early stage of the reaction. Sigmoidal curves could be observed for the conversion of iodobenzene for 7Ni and 7Zn. These induction times are commonly observed for high-temperature Mizoroki–Heck reactions using NHC-PdII precatalysts, and it was proposed that during this period, catalytically active Pd0 species are formed and enter catalytic cycles. UV-visible spectroscopy was performed in the course of the Mizoroki–Heck reactions catalyzed by complexes 7M, and the case of 7Zn is represented in Fig. 2a as an example. Data obtained indicate that metalloporphyrins did not decompose in the course of the reactions and, more importantly, remained metallated. For example, the Soret band of 7Zn was only slightly red-shifted (∼1–2 nm) and Q bands remained at similar positions (Fig. 2a).
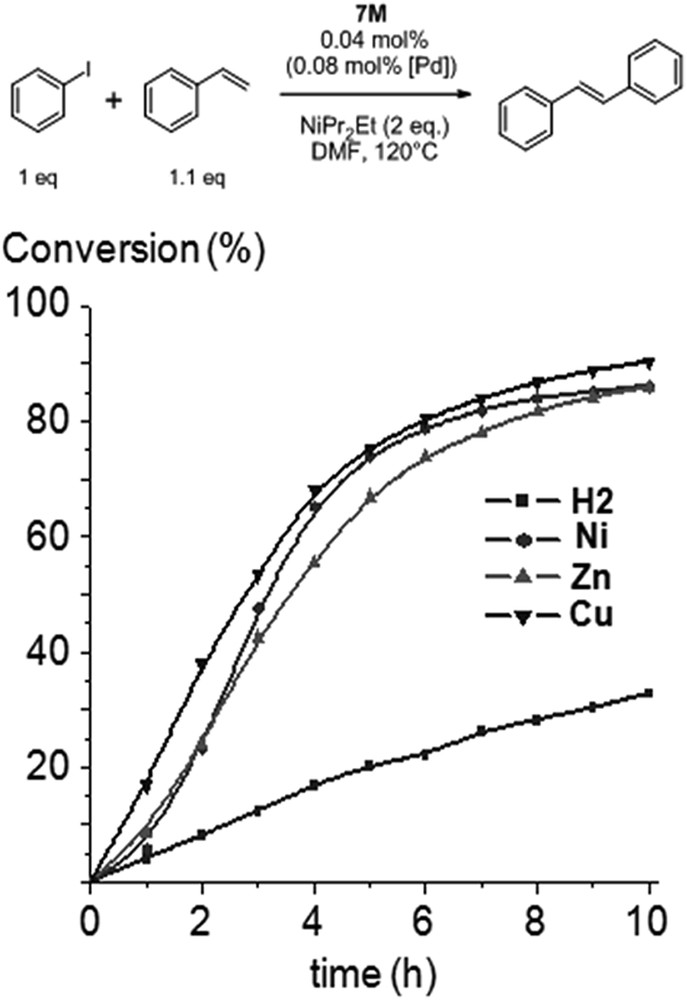
Time-profile of the Mizoroki–Heck reaction with 7M used as precatalysts: conversion of iodobenzene vs. reaction time.
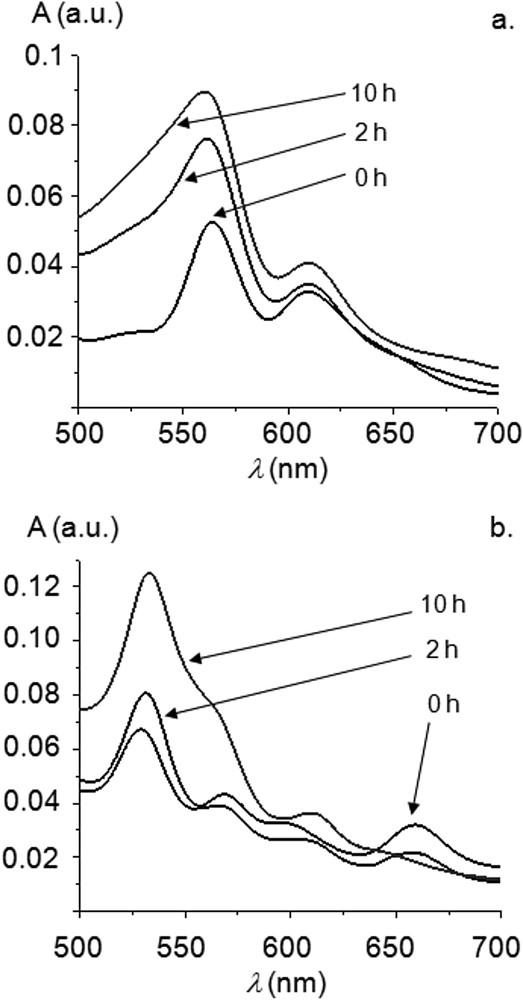
(a) Evolution of the Q bands during Mizoroki–Heck reactions (Soret band normalized at A = 1) catalyzed by 7Zn. (b) Evolution of the Q bands during Mizoroki–Heck reactions (Soret band normalized at A = 1) catalyzed by 7H2.
Surprisingly, the precatalyst 7H2 appeared to be significantly less active and a conversion of only 30% was reached after 10 h (Fig. 1). To ascertain if the incorporation of the inner metal in the porphyrin core was able to increase catalytic activity, the Mizoroki–Heck reaction catalyzed by 7H2 was performed and an excess of zinc(II) acetate monohydrate was added to the reaction mixture after two hours (Fig. 3a). We observed that the catalytic activity increased immediately and this could be correlated with the metallation of the inner core of the porphyrin by ZnII by analyzing aliquots of the reaction mixture using UV-visible spectroscopy. For the Mizoroki–Heck reaction catalyzed by 7H2, the four Q bands of the free-base porphyrin were observed before the addition of Zn(OAc)2 (Fig. 3b, dotted line). However, the fourth Q band at low energy completely vanished 30 min after the addition of Zn(OAc)2 as a consequence of the metallation of the inner core of the porphyrin with ZnII (Fig. 3b, dashed line). This experiment confirmed that the increased catalytic activity observed after the addition of Zn(OAc)2 could be correlated with the metallation of the inner core of the porphyrin with ZnII. This begs the question, “why is the precatalyst 7H2 less active compared to 7M (M = Ni, Cu, Zn) despite the fact that the corresponding NHCs 1M exhibit identical steric and quite similar electronic properties?”
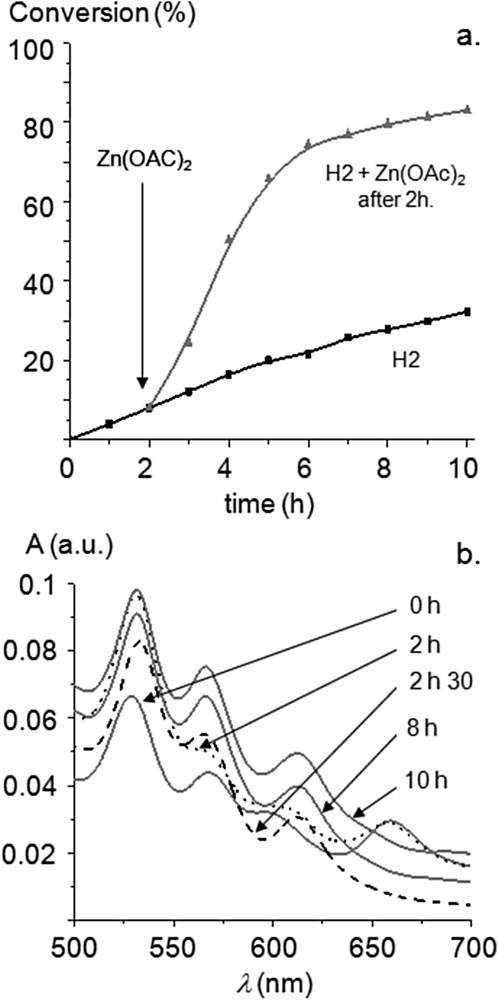
(a) Time-profile of the Mizoroki–Heck reaction with 7H2 used as the precatalyst: zinc(II) acetate monohydrate added (200 equiv vs 7H2) after two hours. (b) Evolution of the Q bands during Mizoroki–Heck reactions (Soret band normalized A = 1) catalyzed by 7H2 + 200 equiv of Zn(OAc)2 after 2 h.
To answer this question, UV-visible spectroscopy was performed in the course of the reaction catalyzed by 7H2 and significant changes occurred in the Q band regions, notably, the Q band at ∼660 nm which slowly vanished during the course of the reaction (Fig. 2b). Knowing that several studies reported that NHC-Pd complexes decomposed in the course of high-temperature Mizoroki–Heck reactions,[6,7] we attributed this spectral evolution to the metallation of the free-base porphyrin cores with leached palladium issued from the decomposition of precatalyst 7H2. To verify this hypothesis, we added an excess of trifluoroacetic acid to aliquots of the reaction mixture at different times (Fig. 4). When TFA is added to aliquots of the reaction mixture after 10 min, a change of color from purple to green occurred due to the protonation of the free-base porphyrin derivatives. This was further confirmed by UV-visible spectroscopy: the Soret band was bathochromically shifted from 429 to 455 nm and one intense Q band appeared at 676 nm (Fig. 4a,b) [10a]. When TFA was added to aliquots of the reaction mixture after four hours, two kinds of porphyrin derivatives could be observed (Fig. 4c): protonated porphyrin derivatives (Soret band and Q band, respectively, at 455 and 676 nm) and PdII-porphyrin derivatives (Soret band at 425 nm; TFA alone is not able to remove PdII from the inner core of the porphyrin). According to these data, the progressive metallation of the inner core of porphyrin during the reaction with PdII was obvious. We assume that all Mizoroki–Heck reactions catalyzed by 7M (M = H2, Ni, Cu, Zn) release palladium in a similar manner, because they exhibit identical steric and quite similar electronic properties.
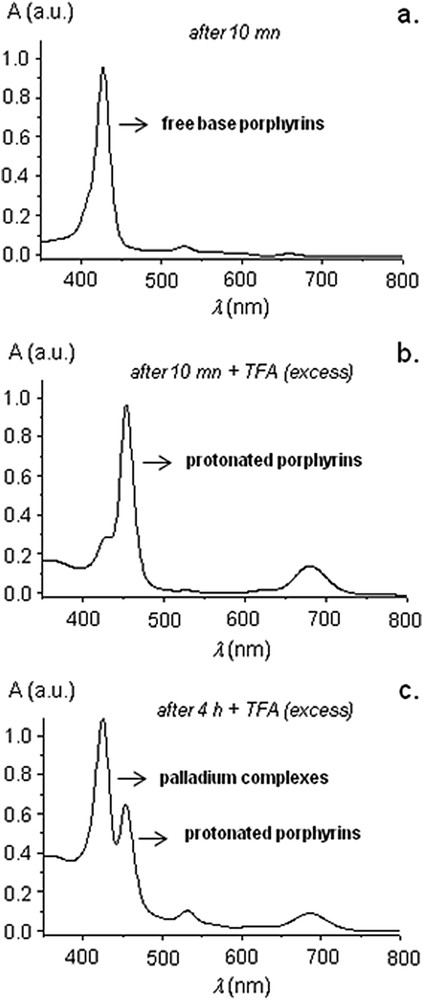
UV-visible spectra of the reaction mixture during Mizoroki–Heck reaction catalyzed by 7H2. (a) After 10 min. (b) After 10 min + TFA. (c) After 4 h + TFA.
To identify more accurately the porphyrin derivatives generated in the reaction mixtures, mass spectrometry (ESI-TOF MS, positive mode) was performed after 10 h (See Supporting Information, Figs. S1 and S2). For the Mizoroki–Heck reaction catalyzed by 7Ni, we identified NiII complexes 2Ni and 8Ni (Scheme 4). We presume that compounds 8M with a phenyl ring at the C(2’) position could be obtained in the course of the Mizoroki–Heck reaction through a reductive elimination of PdII affording Pd0 species which are catalytically active (Scheme 4). For the Mizoroki–Heck reaction catalyzed by 7H2, we identified by mass spectrometry the Pd complexes 2Pd (imidazolium salt 2M with M = Pd) and 8Pd, and the corresponding free-base 2H2 and 8H2. Thus, it is quite obvious that in the case of the Mizoroki–Heck reaction catalyzed by 7H2, Pd leached in the course of the reaction can be trapped by the porphyrin cores. This phenomenon is expected to dramatically change the concentration of free palladium in the reaction mixture (reduction of the concentration was expected in the present case) compared to reactions catalyzed by 7M with M = Cu, Ni and Zn. It also explains why a significant increase of the catalytic activity was observed upon metallation of the free-base porphyrin with zinc(II) in the course of the Mizoroki–Heck reaction, since leached palladium could not be trapped anymore by the porphyrin cores.
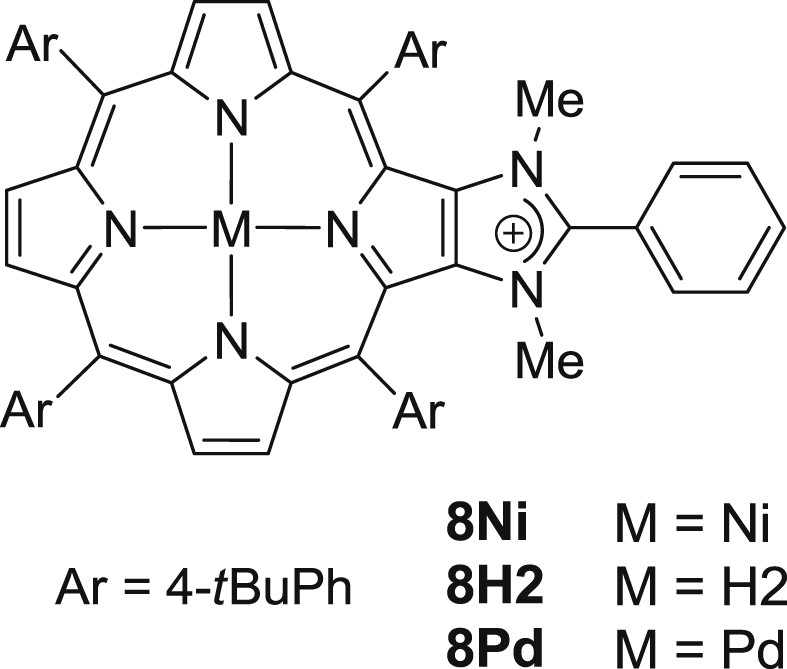
Proposed structures of compounds 8M.
Knowing that the previously reported Pd-NHC precatalysts used in C–C coupling reactions such as Heck reactions decomposed to Pd nanoparticles [7], we therefore attempted to detect the presence of heterogeneous Pd. First, it should be outlined that the formation of black palladium was not observed during the reaction. Then, we performed mercury poisoning experiments by exposing reaction mixtures to an excess of Hg and the subsequent catalytic activity was completely inhibited. This strongly suggests that heterogeneous Pd nanoparticles are involved in the catalytic reactions. Samples of the reaction mixtures were also analyzed by TEM and, in some cases, the formation of nanoparticles was clearly evidenced. A representative TEM image is presented in Fig. 5 and shows the presence of nanoparticles after the Mizoroki–Heck reaction catalyzed by 7H2, confirming the results obtained from Hg poisoning experiments.
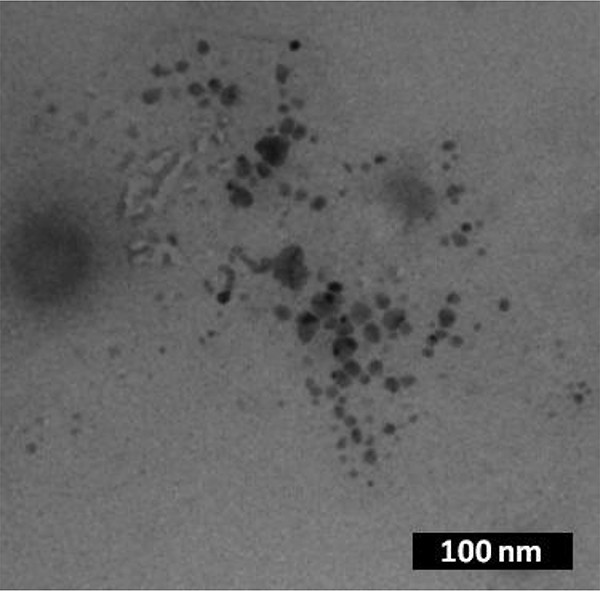
Transmission electronic microscopy image of the reaction mixture after the Mizoroki–Heck reaction catalyzed by 7H2.
3 Conclusion
N-heterocyclic carbene ligands 1M fused to porphyrins across two neighboring β-pyrrolic positions were used to synthesize two kinds of palladium complexes, i.e. trans-[PdI2(1M)2] and [{PdI(μ-I)(1M}2] complexes, respectively, labeled 6M and 7M. Complexes 7M were then used as precatalysts for Mizoroki–Heck reactions. All NHC ligands 1M displayed similar steric and electronic properties according to IR data obtained with cis-[(1M)Rh(CO)2Cl] complexes. Upon the use of complexes 7M in the Mizoroki–Heck reaction of iodobenzene with styrene, precatalysts containing metalloporphyrin, i.e. 7M with M = Ni, Cu, Zn showed quite similar catalytic activities, although some differences were observed in the course of the formation of the catalytically active species. However, the precatalyst 7H2 containing free-base porphyrins exhibited a significantly lower activity; ∼30% of conversion after 10 h compared to 80–90% observed with 7M with M = Ni, Cu and Zn. Although comparisons with other porphyrin-based precatalysts reported by Klein Gebbink [2], Osuka [3] and Matano [4] are not easy to make (palladium complexes are on meso positions and ligands are different), it is clear that precatalysts containing free-base porphyrins showed the lowest activity (excluding Klein Gebbink's MnIII complex for which the reduction of Mn also occurred in the course of the Heck reaction). With porphyrin-NHC ligands 1M and corresponding 7M precatalysts, we showed that Pd leaching occurred in the course of the reaction, and that leached Pd can be trapped by the porphyrin in the case of 7H2. This dramatically decreased the concentration of free Pd available for the catalytic cycle. Moreover, poisoning experiments with Hg and TEM analyses also showed that Pd nanoparticles may play a leading role in the catalytic process in all cases. Indeed, steric and electronic properties are important factors to take into account when investigating the catalytic properties of porphyrin-based catalysts, but other important factors should also be taken into account: the metallation of the free-base porphyrin, i.e. incorporation of the Pd in the inner core of the porphyrin, is one of them. Other phenomena should also be taken into consideration, such as possible aggregation of the catalysts (commonly observed with porphyrins) or possible coordination of additional ligands on the inner metal of the porphyrin. This is actually under investigation in our group.
4 Experimental
4.1 General
The synthesis of imidazolium salts 2M and 3M is described in references [8] and [12]. The synthesis of RhI complexes 4M and 5M is described in reference [10]. Reactions were performed under argon using oven-dried glassware. Dry THF was obtained by distilling over CaH2, and then Na/benzophenone. Preparative separations were performed by silica gel flash column chromatography (Baeckeroot-Labo 60M). Chemicals were obtained from Alfa-Aesar, Sigma-Aldrich and Acros and used as received. Reactions were monitored by thin-layer chromatography using Merck© TLC Silica gel 60 F254 plates. NMR spectra were recorded in CDCl3 on either a Bruker 300 MHz or a 200 MHz Fourier-transform spectrometer. Chemical shifts are reported in ppm referenced to the CHCl3 solvent residual peak at 7.26 ppm for 1H. Abbreviations for 1H NMR spectra used are as follows: s, singlet; d, doublet. UV-visible spectra were recorded on a Perkin Elmer Lambda 35 spectrophotometer in quartz cells. Infrared spectra were obtained in CH2Cl2 solutions in a CaF2 cell with a Nicolet Avatar 320 FT-IR spectrometer. ESI mass spectra were recorded on a Q-Tof Waters 2001 MS spectrometer. MALDI-TOF mass spectra were recorded on a Bruker Ultraflex III MS spectrometer using an anthracene-1,8,9-triol matrix and PEG as the internal reference. EDX microanalyses were performed on a FEI Quanta DEG 200 environmental microscope.
4.2 Synthesis of PdII complexes 6M and 7M
4.2.1 Synthesis of complex 6H2
The synthesis and characterization data of complex 6H2 are reported in reference [11].
4.2.2 Synthesis of complex 6Ni
The synthesis and characterization data of complex 6Ni are reported in reference [8].
4.2.3 Synthesis of complex 6Zn
The synthesis and characterization data of complex 6Zn were reported in reference [11].
4.2.4 Synthesis of complex 6Cu
The complex 6Cu was synthesized following the synthetic procedure described for the synthesis of complex 6Zn in reference [11]. UV-visible (CHCl3): λmax (ε): 429 (546,000), 548 (39,000), and 583 nm (10,000 L.mol−1·cm−1). MALDI-TOF+ MS: calculated for C126H128N12Ni2PdI+: 2167.7, found: 2167.7 ([M−I]+).
4.2.5 Synthesis of complex 7H2
The synthesis and characterization data of complex 7H2 are reported in reference [11].
4.2.6 Synthesis of complex 7Ni
The complex 7Ni was synthesized following the synthetic procedure described for the synthesis of complex 7H2 in reference [11]. 1H NMR (200 MHz, 25 °C, CDCl3): δ 8.74 (s, 4H, Hpyrrole), 8.70 (s, 8H, Hpyrrole), 8.10 (d broad, = 8.2 Hz, 8H, HAr ortho), 7.99 (d, = 8.2 Hz, 8H, HAr ortho), 7.84 (d, = 8.2 Hz, 8H, HAr meta), 7.75 (d, = 8.2 Hz, 8H, HAr meta), 3.41 (s, 12H, HNMe), 1.64 (s, 36H, HtBu), and 1.59 (s, 36H, HtBu) ppm. 13C{1H} NMR (50.3 MHz, 25 °C, CDCl3): δ 167.3 (carbene), 153.1, 151.3, 144.7, 143.8, 143.3, 142.0, 138.0, 137.5, 135.4, 134.3, 133.2, 132.8, 132.7, 126.6, 125.5, 124.5, 120.8, 116.0, 38.8, 35.5, 35.3, 32.1, 32.0, 31.9, 30.1, and 1.4 ppm. UV-visible (CHCl3): λmax (ε): 428 (542,000), 540 (36,500), 575 nm (sh 12,000 L.mol−1.cm−1). MALDI-TOF+ MS: calculated for C126H128I3N12Ni2Pd2+: 2517.43, found: 2517.4 ([M−I]+).
4.2.7 Synthesis of complex 7Zn
The complex 7Zn was synthesized following the synthetic procedure described for the synthesis of complex 7H2 in reference [11]. 1H NMR (200 MHz, 25 °C, CDCl3): δ 8.98 (s, 4H, Hpyrrole), 8.97 (d, = 5.0 Hz, 4H, Hpyrrole), 8.86 (d, = 5.0 Hz, 4H, Hpyrrole), 8.41 (d broad, = 8.2 Hz, 4H, HAr ortho), 8.31 (d broad, = 8.2 Hz, 4H, HAr ortho), 8.19 (d, = 8.2 Hz, 8H, HAr ortho), 7.92 (d, = 8.2 Hz, 8H, HAr meta), 7.81 (d, = 8.2 Hz, 8H, HAr meta), 3.64 (s, 12H, HNMe), 1.73 (s, 36H, HtBu), and 1.66 (s, 36H, HtBu) ppm. UV-visible (CHCl3): λmax (ε): 433 (546,500), 561 (35,500), 605 nm (14,300 L.mol−1·cm−1). MALDI-TOF+ MS: calculated for C63H65N6Zn+: 969.46, found : 969.47 ([M-Pd2-I4−1Zn]+).
4.2.8 Synthesis of complex 7Cu
The complex 7Cu was synthesized following the synthetic procedure described for the synthesis of complex 7H2 in reference [11]. UV-visible (CHCl3): λmax (ε): 426 (595,500), 548 (44,600), and 584 nm (sh 10,900 L.mol−1.cm−1). MALDI-TOF+ MS: calculated for C126H128I4N12NaCu2Pd2+: 2677.35, found: 2677.3 ([M+Na]+); calculated for C63H64I2N6CuPd+: 1327.17, found: 1327.1 ([M-Pd-I2-1Cu]+).
4.3 Heck coupling reactions between iodobenzene and styrene
4.3.1 Materials
Conversion percentages were determined by GC analyses performed on a Varian 3900 instrument (FID, HP5, Tinj = 250 °C, Tdet = 275 °C, oven 75–250 °C, 20 °C.min−1) with ethylbenzoate as internal GC standard according to the first sample (t = 0). The GC column was a SLB-5 MS column, length 30 m, diameter 0.25 mm. Samples for UV-visible spectroscopy (30 μL) were diluted in dichloromethane (2.97 mL).
4.3.2 Typical procedure
In a typical run, the precatalyst 7M (1.7 μmol) was weighed accurately and charged in a Schlenk tube fitted with a rubber septum. The Schlenk tube was evacuated and refilled with argon. Then, anhydrous DMF (8.6 mL), ethylbenzoate (400 μL), diisopropylethylamine (1.4 mL, 8.00 mmol), styrene (460 μL, 4.50 mmol) and phenyl iodide (450 μL, 4.00 mmol) were added via a syringe. The reaction flask was placed into a preheated oil bath at 120 °C and the progress of the reaction was monitored by GC every hour. Coupling reactions were tested at least three times.
Acknowledgments
The authors are grateful to the CNRS and the Agence Nationale de la Recherche (ANR) for financial support of the research project ANR-09-JCJC-0089-01.