

1 Introduction
Over the past decades, there has been a great interest in the synthesis of luminescent pyrimidine derivatives [1,2]. The pyrimidine ring is a highly π-deficient aromatic heterocycle that can be used as the electron-withdrawing unit in a push-pull structure for intramolecular charge transfer (ICT). Such structures generally exhibit high luminescence properties that can be modulated by the nature of the electron donating group and/or the nature of the π-conjugated bridge [3–5]. Some pyrimidine push-pull derivatives were described as polarity sensors due to their strong emission solvatochromism [6–12] as well as pH sensors due to the basic character of the nitrogen atoms of the heterocycles [9–13]. Some pyrimidine fluorophores have also found applications for the detection of biomolecular species such as proteins and DNA [14–16]. It should be noted also that 4,6-di(arylvinyl)pyrimidines have become well established two-photon absorption chromophores [17] that have found applications in fluorescence microscopy imaging [18–21], 3D data storage [22] and two-photon lithography [23].
However, despite their high emission efficiency in solution, many organic materials are non-luminescent in the solid state, because of the fluorescence quenching caused by intermolecular interactions occurring in the condensed phase. One way to overcome this problem consists of incorporating the fluorophore into a polymer matrix [24–26]. When the fluorophores are covalently bounded to the polymer structure, a better distribution of the dye will be obtained and self-aggregation will be avoided [27–29]. Moreover, a covalent immobilization of fluorescent sensors on carrier matrices improves the practical use because it enables long-term measurements, avoiding leaching and a possible contamination of the sample under investigation [30,31].
The aim of the work described here was to synthesize poly(methylmethacrylate) copolymers doped with (arylvinyl)pyrimidine chromophores. The photophysical properties, including emission solvatochromism and pH sensitivity, are reported and compared to the corresponding free chromophores.
2 Experimental
2.1 General methods
All reagents and solvents were purchased from commercial suppliers (Sigma Aldrich, Acros or Alfa Aesar). Dry THF was obtained by means of the Na-benzophenone ketyl procedure. Methylmetacrylate stabilized with tert-butylcatechol was purified by washing with aqueous 2M NaOH and water, dried over magnesium sulfate, and used immediately. KtBuO was purified by the procedure described by Glinka [32] and kept under nitrogen. In air and moisture–sensitive reactions, all glassware was flame-dried and cooled under nitrogen. Compounds 4 [33] and 8 [34] were synthesized according to a reported procedure. NMR spectra were acquired at room temperature on a Bruker AC-300 spectrometer. Chemical shifts are given in parts per million relative to CDCl3 (1H: 7.26 ppm, 13C: 77.0 ppm). Acidic impurities in CDCl3 were removed by treatment with anhydrous K2CO3. High resolution mass analyses were performed at the “Centre Régional de Mesures Physiques de l’Ouest” (CRMPO, University of Rennes1) using a Bruker MicroTOF-Q II apparatus. Average molar mass (Mn) and molar mass distribution (Mw) values of copolymers were determined by size-exclusion chromatography (SEC) in THF at 30 °C (flow rate 1.0 ml min−1) on a Polymer Laboratories PL50 apparatus equipped with a refractive index detector and a ResiPore 300 × 7.5 mm column. The copolymer samples were dissolved in THF (2 mg mL−1). The elution curve was calibrated with polystyrene standards. UV/vis spectra were recorded with a UVIKON xm SECOMAM spectrometer using standard 1 cm quartz cells. Fluorescence spectra were recorded using Spex FluoroMax-3 Jobin-Yvon Horiba apparatus. Measurements were performed at room temperature with solutions of OD <0.1 to avoid re-absorption of the emitted light, and data were corrected with a blank and from the variations of the detector with the emitted wavelength.
2.2 Synthesis
2.2.1 Synthesis of 4-(4,6-dimethyl-pyrimidin-2-yl)-benzaldehyde (2)
A stirred mixture of 2-Chloro-4,6-dimethyl-pyrimidine 1 (284 mg, 2.0 mmol), 4-formylphenylboronic acid (450 mg, 3.0 mmol) and Pd(PPh3)4 (115 mg, 0.1 mmol) in degassed aqueous 2 M sodium carbonate (3.0 mmol, 1.5 mL)/ethanol (1.5 mL)/toluene (15 mL) was heated under reflux for 48 h under a nitrogen atmosphere. The reaction mixture was cooled and filtered, and EtAcO/water 1/1 (20 mL) was added. The organic layer was separated and the aqueous layer was extracted with additional EtAcO (2 × 10 mL). The combined organic extracts were dried over MgSO4 and the solvents evaporated under reduced pressure. The crude product was purified by column chromatography (SiO2, petroleum ether/EtAcO, 7/3). Pale yellow solid. Yield: 65% (275 mg) Mp: 89–90 °C. 1H NMR (300 MHz, CDCl3): δ 2.55 (s, 6H), 6.97 (s, 1H), 7.96 (d, 2H, J = 8.5 Hz), 8.60 (d, 2H, J = 8.5 Hz); 10.1(s, 1H). 13C NMR and JMOD (75 MHz, CDCl3): δ 24.1 (CH3), 118.7 (CH), 128.8 (CH), 129.8 (CH), 137.4 (C), 143.6 (C), 162.8 (C), 167.0 (C), 192.2 (CH). HRMS (ESI/ASAP, TOF) m/z calculated for C13H12N2ONa [M+H]+ 235.0842, found 235.0844.
2.2.2 Synthesis of 4,6-dimethyl-2-(4-vinyl-phenyl)-pyrimidine (3)
Potassium tert-butoxide (241 mg, 2.16 mmol) was added to a solution of methyltriphenylphosphonium iodide (654 mg, 1.62 mmol) in dry THF (20 mL), and the reaction mixture was stirred at room temperature for 15 min. The aldehyde 2 (229 mg, 1.08 mmol) was added and the reaction mixture was stirred at room temperature for 90 min. The reaction was hydrolyzed with water (20 mL), and the THF was removed under vacuum. The aqueous layer was extracted with CH2Cl2 (3 × 20 mL), and the combined organic layers were dried (MgSO4) and evaporated. The crude product was purified by column chromatography (SiO2, CH2Cl2). Colorless oil that crystallizes slowly. Yield: 68% (154 mg) Mp: 56–57 °C. 1H NMR (300 MHz, CDCl3): δ 2.52 (s, 6H), 5.31 (d, 1H, J = 11.4 Hz), 5.85 (d, 1H, J = 17.7 Hz), 6.77 (dd, 1H, J1 = 17.7 Hz, J2 = 11.4 Hz), 6.90 (s, 1H), 7.50 (d, 2H, J = 8.5 Hz), 8.40 (d, 2H, J = 8.5 Hz). 13C NMR and JMOD (75 MHz, CDCl3): δ 24.1 (CH3), 114.7 (C), 117.9 (CH), 126.3 (CH), 128.4 (CH), 136.6 (CH), 137.5 (C), 139.4 (C), 163.8 (C), 166.7 (C). HRMS (ESI/ASAP, TOF) m/z calculated for C14H15N2 [M+H]+ 211.1235, found 211.1236.
2.2.3 Synthesis of copolymer CP
A stirred mixture of methyl methacrylate (2.24 g, 22.4 mmol), styrene derivative 3 (237 mg, 1.12 mmol) and benzoyle peroxide with 25% water (364 mg, 1.13 mmol) in toluene (55 mL) was heated under reflux for 15 h under a nitrogen atmosphere. The reaction mixture was cooled and toluene was evaporated. The crude copolymer was crystallized 2 times from CH2Cl2/n-heptane. White solid Yield: 58% (1.43 g).
2.2.4 Synthesis of copolymers CP1–CP3
A mixture of CP (600 mg) and the corresponding aldehyde (0.67 mmol, 2.5 equiv.) was dissolved in THF (30 mL). KtBuO (120 mg, 1.08 mmol 1.5 equiv. per methyl group) was slowly added at room temperature. The solution which immediately turned brown was then refluxed overnight. After cooling, water was added, THF was evaporated and the mixture was extracted with CH2Cl2, dried over MgSO4 and the solvent removed under vacuum. The crude product crystallized 2 times from CH2Cl2/n-heptane. CP1: yellow solid yield: 76% (410 mg) CP2: yellow solid yield 66% (380 mg) CP3: cream solid yield: 74% (483 mg).
2.2.5 General procedure for the Knoevenagel condensation reaction
A stirred mixture of the corresponding methylpyrimidine derivative (1 mmol) and the appropriate aldehyde (1 mmol, 2 mmol in case of 9) in aqueous sodium hydroxide (5 M, 10 mL) containing Aliquat 336 (44 mg, 0.1 mmol) was heated under reflux for 2 h. The mixture was cooled. The filtrate was filtered off, washed with water and purified by crystallization and/or column chromatography.
2.2.6 4-[2-(4-N,N-Dimethylamino-phenyl)-vinyl]-2-phenyl-pyrimidine(5)
Purified by crystallization from CH2Cl2/n-heptane. Yellow solid. Yield: 62% (188 mg). Mp: 136–137 °C. 1H NMR (300 MHz, CDCl3): δ 3.05 (s, 6H), 6.74 (d, 2H, J = 8.7 Hz), 6.95 (d, 1H, J = 15.9 Hz), 7.14 (d, 1H, J = 5.1 Hz), –7.58–7.51 (m, 5H), 7.96 (d, 1H, J = 15.9 Hz), 8.55–8.52 (m, 2H), 8.69 (d, 1H, J = 5.1 Hz) 13C NMR and JMOD (75 MHz, CDCl3): δ 40.2 (CH3), 112.1 (CH), 116.0 (CH), 121.3 (CH), 123.8 (C), 128.2 (CH), 128.5 (CH), 129.2 (CH), 130.4 (CH), 137.5 (CH), 138.2 (C), 151.2 (C), 157.2 (CH), 163.3 (C), 164.3 (C). HRMS (ESI/ASAP, TOF) m/z calculated for C20H20N3 [M+H]+ 302.1652, found 302.1652.
2.2.7 4-[2-(4-N,N-Diphenylamino-phenyl)-vinyl]-2-phenyl-pyrimidine(6)
Purified by column chromatography (SiO2, petroleum ether/EtAcO, 7/3). Non-crystalline yellow solid. Yield: 62% (265 mg). 1H NMR (300 MHz, CDCl3): δ 7.02 (d, 1H, J = 15.9 Hz), 7.13–7.08 (m, 4H), 7.19–7.11 (m, 5H), 7.35–7.28 (m, 4H), 7.55–7.50 (m, 5H), 7.97 (d, 1H, J = 15.9 Hz), 8.57–8.54 (m, 2H), 8.69 (d, 1H, J = 5.1 Hz) 13C NMR and JMOD (75 MHz, CDCl3): δ 116.0 (CH), 122.3 (CH), 123.7 (CH), 123.9 (CH), 125.1 (CH), 128.2 (CH), 128.5 (CH), 128.7 (CH), 129.2 (C), 129.4 (CH), 130.5 (CH), 136.6 (CH), 138.0 (C), 147.1 (C), 149.0 (C), 157.4 (CH), 162.7 (C), 164.4 (C). HRMS (ESI/ASAP, TOF) m/z calculated for C30H24N3 [M+H]+ 426.1965, found 426.1970.
2.2.8 4-[2-(6-Methoxy-naphthalen-2-yl)-vinyl]-2-phenyl-pyrimidine (7)
Purified by column chromatography (SiO2, petroleum ether/EtAcO, 1/1), pale yellow solid. Yield: 89% (300 mg). Mp: 142–143 °C. 1H NMR (300 MHz, CDCl3): δ 3.96 (s, 3H), 7.24–7.17 (m, 4H), 7.56–7.54 (m, 4H), 7.82–7.79 (m, 3H), 7.98 (s, 1H), 8.18 (d, 1H, J = 15.9 Hz), 8.59–8.56 (m, 2H), 8.77 (d, 1H, J = 5.1 Hz) 13C NMR and JMOD (75 MHz, CDCl3): δ 55.4 (CH3), 106.0 (CH), 116.3 (CH), 119.3 (CH), 124.3 (CH), 125.4 (CH), 127.4 (CH), 128.3 (CH), 128.5 (CH), 128.8 (CH), 128.9 (C), 129.5 (CH), 130.6 (CH), 131.2 (C), 135.2 (C), 137.1 (CH), 138.0 (C), 157.6 (CH), 158.5 (C), 162.6 (C), 164.4 (C). HRMS (ESI/ASAP, TOF) m/z calculated for C23H19N2O [M+H]+ 339.1492, found 339.1490.
2.2.9 4,6-Bis-[2-(4-N,N-dimethylamino-phenyl)-vinyl]-2-phenyl-pyrimidine (9)
Purified by crystallization from CH2Cl2/n-heptane. Yellow solid. Yield: 86% (384 mg). Mp: 197–198 °C. 1H NMR (300 MHz, CDCl3): δ 2.96 (s, 12H), 6.65 (d, 4H, J = 8.7 Hz), 6.87 (d, 2H, J = 15.9 Hz), 7.02 (s, 1H), 7.49–7.42 (m, 7H), 7.88 (d, 1H, J = 15.9 Hz), 8.55–8.52 (m, 2H). 13C NMR and JMOD (75 MHz, CDCl3): δ 40.2 (CH3), 112.1 (CH), 113.0 (CH), 122.0 (CH), 124.3 (C), 128.3 (CH), 128.4 (CH), 129.0 (CH), 130.1 (CH), 136.5 (CH), 138.8 (C), 151.0 (C), 163.4 (CH), 164.0 (C). HRMS (ESI/ASAP, TOF) m/z calculated for C30H31N4 [M+H]+ 447.2548, found 447.2552.
3 Results and discussion
The copolymers CP1–CP3 were obtained in 4 steps from commercially available 2-chloro-4,6-dimethylpyrimidine 1 (Scheme 1). The first step consists of a Suzuki cross-coupling reaction with 4-formylphenylboronic acid. The aldehyde 2, obtained with a 65% yield, is then involved in a Wittig reaction with methyltriphenylphosphonium iodide to obtain the styrene derivative 3 with a 68% yield. A copolymerization of methyl methacrylate with 10 w% of 3 is then carried out to yield copolymer CP. The last step consists of the functionalization of pyrimidine fragments by the Knoevenagel condensation reaction with a series of aldehydes.

(i) 4-Formylboronic acid, Pd(PPh3)4, Aq Na2CO3/EtOH/Toluene, Δ, 48 h, 65%. (ii) Methyltriphenylphosphonium iodide, tBuOK, THF, rt, 2 h, 68%. (iii) Methylmethacrylate, benzoyl peroxide, Toluene, Δ, 15 h, 58%. (iv) Corresponding aldehyde, tBuOK, THF, 15 h, 65–76%.
It proved to be difficult to determine the chemical structures of the copolymers CP1-3. Because of the low concentrations of the pyrimidine monomer used (<10 w%), NMR is not a sufficiently sensitive technique, and the signal corresponding to the pyrimidine fragments was not easily observed. IR spectra did not provide any information about the structure, and the only characteristic peak observed was that corresponding to the ester function of the methyl methacrylate moiety (1732 cm−1). UV–vis and fluorescence spectra of dichloromethane solutions of the copolymers permit to indicate the presence of arylvinylpyrimidine chromophores (Table 1). Surprisingly, whereas it is generally difficult to isolate the monocondensation products from 4,6-dimethylpyrimidine [9], comparison of absorption spectra of the copolymers CP1-3 with those of the corresponding 4-arylvinylpyrimidine [11] and 4,6-diarylvinylpyrimidine derivatives [9] seems to indicate that only the monocondensation products of dimethylpyrimidine fragments are observed in our case. To confirm our hypothesis, and to verify that the phenyl ring in position 2 of the pyrimidine does not modify the photophysical properties of the pyrimidine dyes, the chromophores 5–7 and 9 were synthesized from 2-phenylpyrimidine derivatives 4 and 8 by Knoevenagel condensation of the corresponding aldehyde in boiling aqueous 5M NaOH solution in the presence Aliquat 336 as the charge transfer catalyst [35] (Schemes 2–3).
UV–Vis and photoluminescence data for copolymers CP1–CP3 and compounds 5–9.
| Compound | UV–Vis in CH2Cl2 λmax (nm) (ɛ, mM−1 cm−1) | PL in CH2Cl2 λmax (nm) (ΦF) | PL powder λmax (nm) | PL thin film λmax (nm) |
| CP1 | 394 | 525 (0.14) | 518 | 503 |
| CP2 | 402 | 522 (0.55) | 518 | 499 |
| CP3 | 360 | 450 (0.11) | 466 | 456 |
| 5 | 398 (30.4) | 493 (0.066) | 536 | |
| 6 | 404 (27.2) | 528 (0.75) | 527 | |
| 7 | 351 (31.4) | 448 (0.068) | 465 | |
| 9 | 428 (44.9) | 534 (0.58) | 530 |

(i) Corresponding aldehyde, aliquat 336, NaOH 5 M, Δ, 2 h, 62–89%.
(i) 4-N,N-dimethylaminobenzaldehyde, aliquat 336, NaOH 5 M, Δ, 2 h, 86%.
The photophysical data of the compounds 5–7 and 9 indicated the negligible influence of the phenyl substituent of the pyrimidine ring on the absorption spectra confirming the structure proposed for the copolymer CP1-3. The weight percentage of chromophore fragments was estimated by UV–vis on the assumption of a similar molar absorptivity for the free dye and the linked dye (Table 2). The w% of chromophores was comprised to be between 2.5 and 7.1 for macromolecule CP1-3.
Data for synthesized copolymers.
| Copolymer | Mwa (g mol−1) | Polydispersitya | Weight % of chromophoreb |
| CP1 | 2034 | 1.61 | 5.4 |
| CP2 | 2007 | 1.53 | 2.5 |
| CP3 | 2996 | 1.79 | 7.1 |
a Molecular weight (Mw) and polydispersity were determined by SEC using polystyrene as the standard.
b Determined by UV–Vis measurement.
Size exclusion chromatography analyses were also performed to determine the molecular weights and polydispersities of CP1-3. The synthesized copolymers showed relatively low molecular weights ranging from 2007–2996 g.mol−1 and polydispersities ranging between 1.53 and 1.79 (Table 2).
CP1-3 was dissolved in 1,1,2-trichloroethane at concentrations of 170 g·L−1, 280 g·L−1 and 190 g·L−1, respectively. The solutions were then spin-coated on silicon wafers at a speed of 2000 rpm for 10 s. The resulting films were subsequently baked at 100 °C for 20 min yielding film thicknesses of 500–800 nm (measured with a Veeco Dektak 150+ Surface Profiler).
The fluorescence properties of the copolymers CP1-3 were examined in dichloromethane solution, as well as in powder and thin film states (Table 1). In the case of CP2 and CP3, the macromolecules in dichloromethane solution show similar emission spectra compared to the corresponding free chromophores (respectively 6 and 7). In the case of dimethylaminophenyl substituted copolymer CP1, a red shift of the emission spectra is observed in comparison with free chromophore 5. The copolymers exhibit fluorescence quantum yield in the same range compared to the corresponding free chromophores. The highest quantum yield (0.55) is obtained, as expected, for diphenylamino substituted copolymer CP2. The copolymers CP1-3 are also luminescent either in powder and thin film states. For each copolymer, the emission wavelength maxima are slightly blue-shifted in the thin film state compared to those observed in the powder state and in dichloromethane solution (except for CP3).
In previous studies, we have described the important emission solvatochromism of push-pull pyrimidine chromophores [4,9–12, 16]: a bathochromic shift of the emission band is observed with increasing solvent polarity as predicted by using the Dimroth-Reichardt polarity parameter (ET(30)) [36] whereas the absorption wavelength is not significantly shifted. Copolymers CP2 and CP3 exhibit the same characteristics (Table 3, Fig. 1). In the case of CP1, a positive emission solvatochromism is observed from Toluene (ET(30) = 33.9) to Acetone (ET(30) = 40.7). A decrease of emission maxima is observed for a more polar solvent (MeCN and DMSO); this might be explained by an encapsulation of the chromophore fragments into the polymer structure which is not observed in the case of CP2 and CP3 that bear more voluminous dye units.
Emission solvatochromism of copolymers CP1–CP3 in aprotic solvents.
| Copolymer | λem (nm) | |||||
| Toluene ET(30) = 33.9 | THF ET(30) = 37.4 | CH2Cl2 ET(30) = 40.7 | Acetone ET(30) = 42.2 | MeCN ET(30) = 45.6 | DMSO ET(30) = 45.1 | |
| CP1 | 483 | 510 | 525 | 543 | 517 | 526 |
| CP2 | 476 | 502 | 522 | 533 | 543 | 546 |
| CP3 | 423 | 438 | 450 | 458 | 465 | 466 |
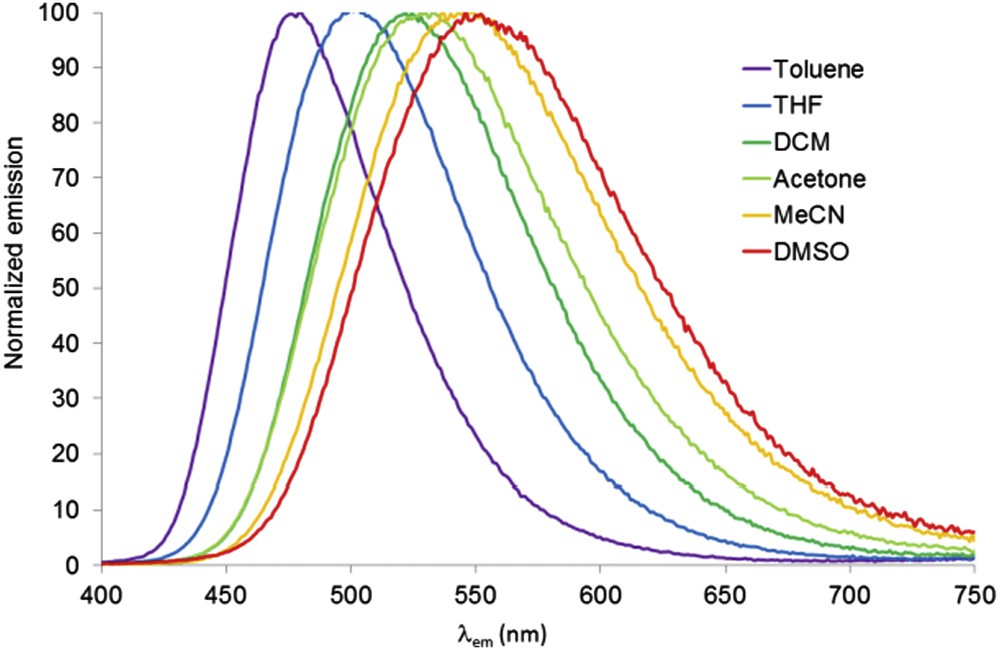
Normalized emission of copolymer CP2 in various solvents.
In previous studies [9–12], we also demonstrated the ability of arylvinylpyrimidines to act as colorimetric and luminescent pH sensors due to the basic character of the nitrogens atoms of the pyrimidine ring. This is also the case of CP1–CP3. A dichloromethane solution of the copolymers CP1–CP3 underwent a significant color change upon protonation due to an increased charge transfer from the donors to the pyrimidinium moiety (Table 4). This color change is fully reversible by neutralization with a base such as Et3N. The changes observed in the UV–Vis spectra of CP1 are illustrated in Fig. 2. The increase in the concentration of TFA led to the progressive attenuation of the absorption band for the neutral forms and the appearance of a new, more intense red-shifted band corresponding to the protonated species. As far as the fluorescence response is concerned, in the case of amino substituted chromophores, the emission is partially (CP1) or totally (CP2) quenched. However, in the case of methoxy-substituted chromophores (CP3), an increase of the luminescence intensity is observed upon protonation (Fig. 3). The emission is also red-shifted due to an increase in the charge transfer into the chromophore fragments (see Fig. 4).
Optical properties of copolymers CP1–CP3 with and without the addition of TFA.
| Copolymer | CH2Cl2 | TFA 10−2M in CH2Cl2 | ||
| λabs (nm) | λem (nm) (ΦF) | λabs (nm) | λem (nm) (ΦF) | |
| CP1 | 394 | 525 (0.14) | 515 | 603 (0.063) |
| CP2 | 402 | 522 (0.55) | 515 | – |
| CP3 | 360 | 450 (0.11) | 435 | 565 (0.27) |
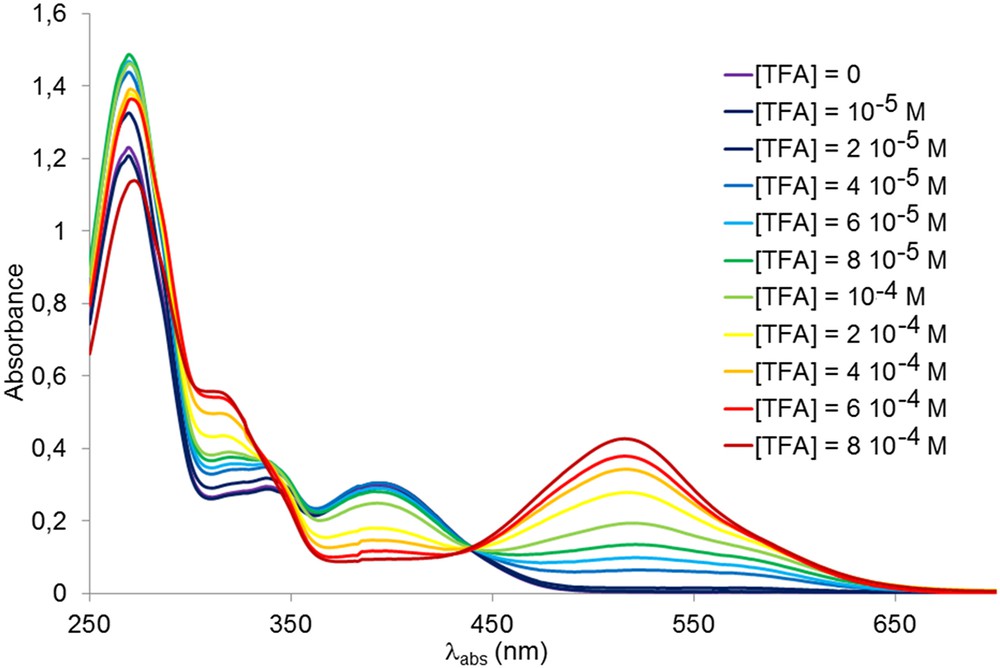
Change in the UV/Vis spectrum of CP1 (6.2 10−2 g L−1 in CH2Cl2) upon the addition of TFA.

Change in the emission spectrum of CP3 (5.1 10−3 g L−1 in CH2Cl2) upon the addition of TFA.

Color change of CH2Cl2 solution of CP1 in the presence of TFA (10−2 M).
To conclude, we have efficiently synthesized a series of polymethyl methacrylate based copolymers bearing arylvinylpyrimidine chromophores. The macromolecules display similar photophysical properties in solution compared to the free chromophores. The thin film of these copolymers was obtained and also exhibits strong luminescence. Due to their sensibility to polarity and pH, these materials have potential for sensing applications. Investigations in this direction are currently being carried out in our laboratories.
Acknowledgments
Morgane Prince, Morgan Lecoublet, Jean Rabouan, and Christophe Nogues, students at IUT Lannion, are acknowledged for preliminary synthesis and optical measurements. Guislaine Barouti is acknowledged for SEC experiments.