

1 Introduction
Nitrogen-doped carbon nanotubes (N-CNTs) have received an ever growing scientific interest during the last few decades for their high catalytic performance in several relevant catalytic processes compared to the traditional metal and oxide supported catalysts [1–6]. According to the recent literature survey the catalytic processes involving N-CNTs are continuously expanded with time and part of the results obtained is summarized in some recent reviews [7–10]. The atomic dimensions of carbon, nitrogen and oxygen as well as the bond length in aromatic structures do not differ very much which makes the latter two elements very suitable for incorporation into the CNT matrix. However, the C–N bond is shorter as compared to the C–C and C–O bond lengths. Therefore, incorporation of nitrogen into the carbon matrix may distort a perfectly ordered graphitic matrix leading to possible modification of the overall electronic properties of the material and thus, its catalytic activity.
However, the use of N-CNTs also represents some drawbacks due to its nanoscopic dimension which could pose serious problems for its handling and transport and also for its use in industrial catalytic reactors. Indeed, N-CNTs can be used as such in their nanoscopic shape in the Oxygen Reduction Reaction (ORR) process as it only requires a good dispersion of the N-CNTs in an ink medium before processing to make the electrode [11, 12] whereas for the other applications, involving gas–solid or liquid–solid reactions, the N-CNTs should be shaped in a macroscopic form in order to avoid the problem of pressure drop across the catalyst bed and also to facilitate the problem of reactor charging and discharging as well as the catalyst recovery. Chizari et al. [3] reported that N-CNTs supported on silicon carbide foam displays a significant improvement in the desulfurization performance compared to the bulky N-CNTs. The main advantages of the macroscopic shaping using a SiC as a host structure not only reduce the problem of pressure drop but also allow one to control the problem of heat and mass transfer during the reaction by adapting the macroscopic shape to the reaction conditions which significantly enhance the overall catalytic performance [13, 14]. In this aim several research projects have been conducted to develop N-CNT supported on different macroscopic host structures, i.e. Al2O3, SiO2, carbon and SiC, for different catalytic processes. The supports should also be produced in a simple and scalable way in order to avoid the costly development of new infrastructure and to reduce as much as possible the time to market the process. However, supported N-CNTs imply the use of inert supports which reduce the intrinsic catalytic activity per unit weight of the catalyst and thus, it is of interest to develop a new synthesis method to shape the N-CNTs directly without a need for support in order to improve the intrinsic activity.
The aim of the present article deals with the synthesis of the self-sustained nitrogen-doped carbon nanotubes with a controlled macroscopic shaping for subsequent use as metal-free catalyst in a gas-phase reaction. It is worthy to note that such direct macroscopic shaping of N-CNTs as well as their use as metal-free catalysts has not been reported so far in the literature. The synthesis method is based on a coagulation process using alginate as a gelling agent, followed by a thermal step to make entangled carbon nanotubes with a homogeneous size and shape. The as-synthesized N-CNT beads will be evaluated as metal-free catalysts in the gas-phase partial oxidation of hydrogen sulfide into elemental sulfur. The catalytic performance will be compared with that obtained on the Fe2O3 supported on silicon carbide catalysts which is one of the most active desulfurization catalysts [15, 16].
2 Experimental
2.1 N-CNT characteristics
The N-CNTs were synthesized by the catalytic chemical vapor deposition (CCVD) process using Fe/Al2O3 as the growth catalyst and C2H6 and NH3 as carbon and nitrogen sources [17]. The N-CNTs were recovered after a consecutive base and acid treatment to remove the growth catalyst. The as-synthesized composite was first treated with a soda solution (20 wt. %) at 110 °C in order to dissolve the alumina support of the growth catalyst. The sample was thoroughly washed with distilled water until a neutral pH was reached and further treated with a solution of aqua regia at 110 °C followed by a washing process until neutral pH. The resulting bulky N-CNT sample was allowed to dry at 110 °C for overnight.
TEM analysis shows the presence of high aspect ratio N-CNTs containing bamboo-like caps along the tube length (Fig. 1A and B) which is in good agreement with the literature results. [18] The as-synthesized N-CNTs also exhibit a clean surface and no trace of carbon nanoparticles was observed which indicates the high selectivity of the synthesis method. The STEM-EELS analysis evidences the presence of nitrogen and carbon homogeneously dispersed within the tube matrix (Fig. 1C). [18] The X-ray photoelectron spectroscopy (XPS) analysis indicates that the purified N-CNTs contain about 0.12 at % of residual iron. The residual iron in the purified N-CNTs may be attributed to iron localized within carbon nanotubes or encapsulated by graphene layers and thus, is not accessible to the acid during the acid treatment. Such residual iron is expected to be not accessible to the reactant during the catalytic process.

(A, B) TEM micrographs with different magnifications of the as-synthesized N-CNTs after acid and base purification steps. (C) STEM-EELS micrograph showing the presence of nitrogen and carbon homogeneously dispersed within the tube wall and arches.
2.2 N-CNT bead synthesis
The N-CNT bead synthesis was carried out as follows: 2 g of purified N-CNTs were dispersed in 100 mL of distilled water through a tip sonicator for 30 min [19]. The N-CNT suspension was kept under vigorous stirring (750 rpm) and 1 wt. % of alginate (with respect to the weight of the water) was slowly added to the suspension. The solution was kept under stirring for an additional 30 min. A drop of the N-CNT-alginate suspension was added to an aqueous CaCl2 solution (3 wt. %). As soon as the drop comes into contact with CaCl2 solution, N-CNT beads with a diameter of about 5 mm were steadily formed. The as-synthesized N-CNT beads were left inside the CaCl2 solution overnight in order to complete the chemical reaction. The beads were washed with distilled water to remove as much as possible the residual salt. The beads were then dried at room temperature for 12 h and dried at 70 °C for 14 h and then, the alginate in the composite was decomposed by heating up to 600 °C under helium flow for 2 h. After the calcinations, the CNT beads were treated in HCl solution (20 vol. %) at room temperature for 2 h in order to remove as much as possible the remaining sodium and calcium species from the material before the catalytic evaluation process.
2.3 Characterization techniques
Scanning electron microscopy (SEM) was carried out on a JEOL 2600F with a resolution of 5 nm. The sample was deposited onto a double face graphite tape in order to avoid the charging effect during the analysis.
TEM analysis was carried out on a JEOL 2100F working at 200 kV accelerated voltage, equipped with a probe corrector for spherical aberrations, and a point-to-point resolution of 0.2 nm. The sample was dispersed by ultrasound in an ethanol solution for 5 min and a drop of the solution was deposited on a copper covered with a holey carbon membrane for observation.
The specific surface area of N-CNTs and the beads, were determined in a Micromeritics sorptometer. The sample was outgassed at 250 °C under vacuum for 8 h in order to desorb moisture and adsorbed species on its surface. Physisorption measurements were carried out using N2 as adsorbent at liquid N2 temperature at relative pressures between 0.06 and 0.99.
Thermogravimetric analyses (TGA) were performed under air (25 mL min−1) on an EXSTAR thermogravimetric analyzer (TG/DTA) Seiko 6200.
The XPS measurements were carried out in an ultrahigh vacuum (UHV) spectrometer equipped with a VSW ClassWA hemispherical electron analyzer. A monochromated Al Kα X-ray source (1486.6 eV) was used as incident radiation and XP spectra were recorded using a pass energy of 20 eV. Survey and high-resolution spectra were recorded in the constant pass energy mode (100 and 20 eV, respectively). The CASA XPS program with a Gaussian–Lorentzian mix function and Shirley background subtraction was employed to deconvolute the XP spectra.
2.4 Selective oxidation of the H2S process
The catalytically selective oxidation of H2S by oxygen (Eq. [1]) was carried out in an all glass tubular reactor working isothermally at atmospheric pressure.
| H2S + 1/2 O2 → 1/n Sn + H2O ΔH = − 222kJ/mol | [1] |
An appropriate amount of catalyst (2 g) was placed on silica wool in a tubular Pyrex reactor (inner diameter: 16 mm) located inside a vertical tubular electrical furnace. The temperature was controlled by using a K-type thermocouple and a Minicor regulator. The gas mixture of the reactant feed including H2S (1 vol. %), O2 (2.5 vol. %), H2O (30 vol. %) and He (balance) was passed downward through the catalyst bed. The gas flow rates were monitored by Brooks 5850 TR mass flow controllers linked to a control unit. The steam in the reactant feed was provided by bubbling a helium flow through a saturator containing water maintained at 85 °C. The gaseous hourly space velocity (GHSV) was fixed at 2400h–1 and the O2/H2S molar ratio was settled to 2.5, unless otherwise stated.
The reaction was conducted in a continuous mode. The sulfur formed in the reaction was vaporized (because of the high partial pressure of sulfur at the adopted temperatures) and then condensed at the exit of the reactor in a trap maintained at room temperature. The analysis of the inlet and outlet gases was performed online using a Varian CP-3800 gas chromatograph (GC) equipped with a Chrompack CP-SilicaPLOT capillary column and a thermal conductivity detector (TCD), which allowed the detection of O2, H2S, H2O and SO2.
3 Results and discussion
3.1 N-CNT bead characteristics
The macroscopic shape of the as-synthesized N-CNT beads is presented in Fig. 2A. The beads show a high homogeneous diameter ranged at around 4 mm. For comparison the digital photos of the beads, before and after the drying process, are presented in Fig. 2B. According to the results a significant contraction of the N-CNT spheres was observed upon drying due to the van der Walls forces generated between the tubes during the drying process [20, 21]. Digital photos also evidence the large apparent volume reduction of the N-CNTs after the macroscopic shaping compared to the powdered N-CNTs (Fig. 2C) due to the van der Waals forces which confer to the N-CNTs a higher entanglement inside the macroscopic beads. Moreover, the apparent density of the pristine N-CNTs was significantly increased after wetting in the ethanol, which could be due to the loose contact between N-CNTs. Interestingly, the macroscopic shaped N-CNT beads show no apparent density change in the ethanol which confirms the high self-sustained mechanical strength of the beads to maintain its form upon wetting. Similar results have already been reported for the CNTs after the wetting and drying process where the apparent density was increased from 190 kg/m−3, after wetting with water, to 230 kg/m−3 after drying. [19]

(A) Digital photos show large-scale products of the N-CNT beads which are synthesized by a simple gelation method. (B) Comparison of the as-prepared and thermally treated N-CNT beads, showing the large volume contraction of the N-CNT beads before (B1) and after (B2) the drying process. (C) Digital photos showing the apparent volume of the same mass of bulk N-CNTs and N-CNT beads, and the apparent volume changes after wetting in the ethanol solution. (D and E) SEM micrographs of the N-CNT beads with different magnifications. Masquer
(A) Digital photos show large-scale products of the N-CNT beads which are synthesized by a simple gelation method. (B) Comparison of the as-prepared and thermally treated N-CNT beads, showing the large volume contraction of the N-CNT beads before (B1) and ... Lire la suite
SEM analysis evidences the high entanglement of the N-CNTs within the beads along with the presence of large pores contributing to the high accessibility of the gaseous reactants to the active sites (Fig. 2D and E). These meso- and macro-porosities present within the material will be extremely useful for they are further used as a catalyst support, especially in the reactions with high mass transfer like Fischer–Tropsch synthesis [22, 23] or in a high space velocity process such as the partial oxidation of H2S into elemental sulfur studied in the present work. According to the SEM analysis the CNT beads are macroscopic objects with a fully accessible surface connected by voids generated within the entangled carbon nanotubes instead of ink-bottled pores as usually encountered with traditional granular supports. However, it is expected that these pores are somehow less accessible compared to those present in the bulky N-CNTs due to the enhancement of the tube entanglement during the macronization and drying processes and also in good agreement with the increase of the apparent density of the material upon drying.
The specific surface area (SSA) of the pristine N-CNTs and the N-CNT beads measured by means of the BET method is displayed in Fig. 3 along with the pore size distribution. According to the results, the SSA of the N-CNT beads (237 m2/g) slightly increased compared to that of the bulky N-CNTs (205 m2/g). Such SSA increase could also be due to the formation of some residual carbon phase on the N-CNTs surface consecutively to the decomposition of the alginate precursor. As can be found in Fig. 3A, the N-CNT beads presents the type IV isotherm in the relative pressure region between 0.35 and 1.0, which confirms the meso-/macroporous network in the as-prepared microsphere. It is also worthy to note that the pore volume was increased significantly after the macroscopic shaping process. The pore volume of CNT beads is amounted to 1.01 cm3/g, and in the mean time only 0.58 cm3/g is obtained from the pristine N-CNTs, which could be attributed to the entangled network formed after N-CNTs were shaped by the presence of alginate (Fig. 3B). The N-CNT beads display a significantly lower pressure drop compared to that measured on the bulky N-CNTs [19] and confirm the advantage of the macronization process developed in this study.
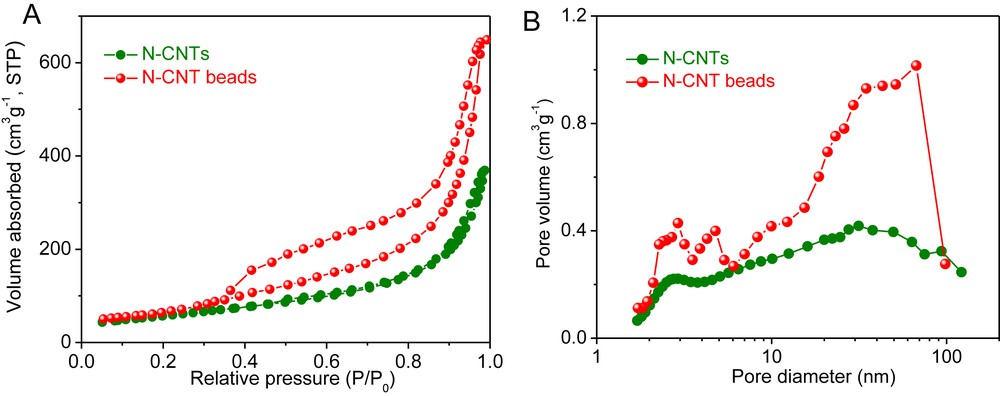
(A) Nitrogen adsorption–desorption isotherm and (B) pore size distribution of the pristine N-CNTs and the N-CNT beads.
Thermal gravimetric analysis of the pristine N-CNTs and N-CNT beads was carried out whose results are presented in Fig. 4. The unshaped N-CNTs display a single and relatively large combustion peak centered at around 578 °C (Fig. 4A) while the N-CNT beads show several combustion peaks (Fig. 4B). The low combustion peak at around 430 °C in the macroscopic sample could be attributed to be originated from the carbon phase formed from the carbonization of the alginate while the two other peaks could be attributed to the combustion of the N-CNTs with different entanglements inside the macroscopic beads.
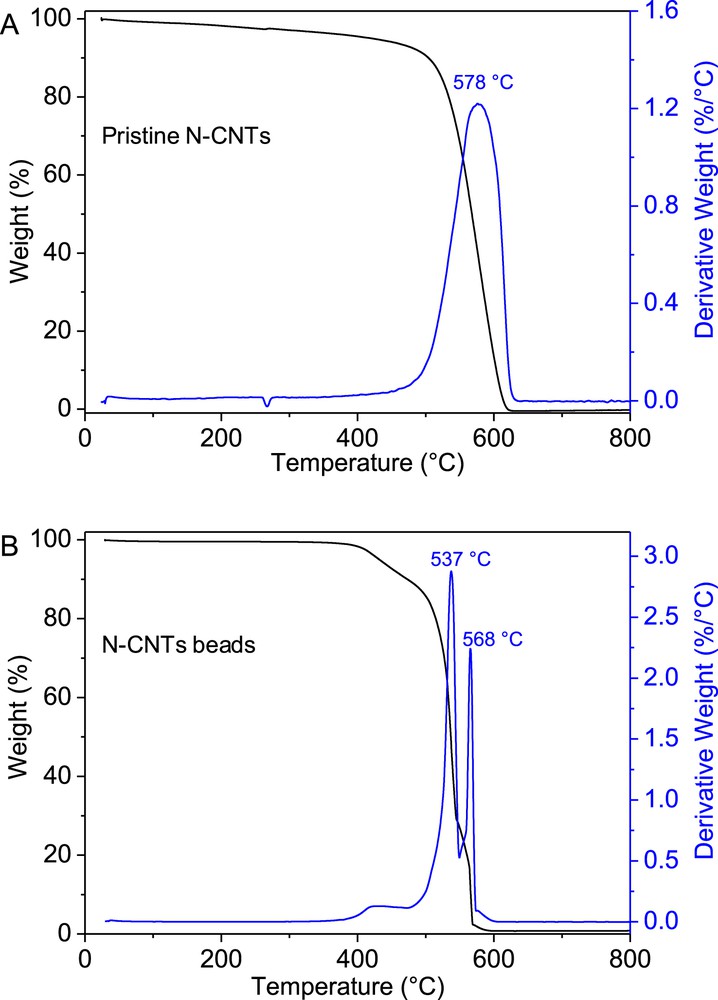
TGA spectra of the pristine N-CNTs and the N-CNT beads.
The nitrogen species present on the surface of the N-CNTs were analyzed by XPS and the results are presented in Fig. 5. The N1s XPS spectrum shows the presence of four types of nitrogen species, i.e. pyridinic (NP), pyrrolic (NPY), quaternary (NQ) and oxidized N (NO), which is in good agreement with the literature data. [5, 24, 25]
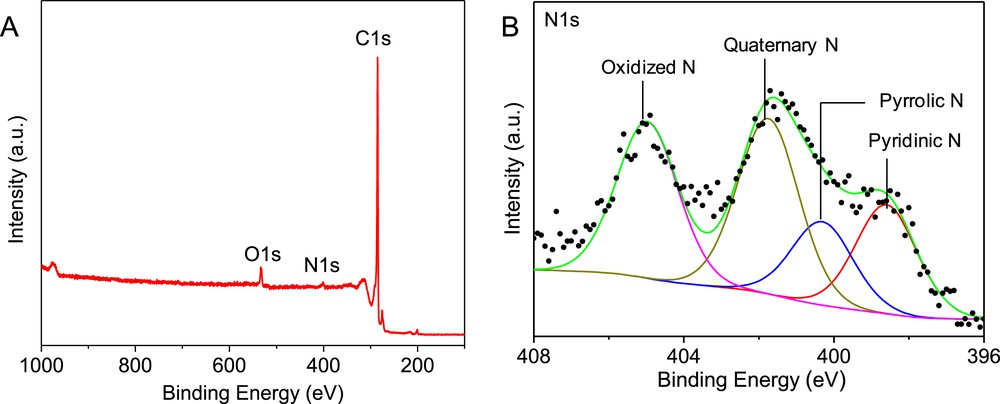
(A) XPS survey scan and (B) high-resolution N1s spectrum of the N-CNTs showing the presence of several nitrogen species.
3.2 Catalytic performance
The desulfurization activity, expressed in terms of H2S conversion, obtained on the N-CNT beads as a function of the reaction temperature is presented in Fig. 6. Both the N-CNTs and N-CNT beads display a high desulfurization activity at 210 °C. The N-CNT beads display a slightly lower desulfurization activity compared to that of the pristine N-CNTs. The lower desulfurization activity of the N-CNT beads at 210 °C could be attributed to the higher entanglement of the nanotubes within the beads leading to a lower effective surface area compared to the bulk N-CNTs. Indeed, the higher entanglement of the N-CNTs inside the beads could induce some problem of diffusion compared to the bulk N-CNTs. The problem of diffusion could be tackled by adding pore formers, i.e. rice powder, polystyrene beads, inside the N-CNTs and alginate mixture before processing. These pore formers will be removed during the thermal treatment step leaving behind macropores which will improve the accessibility of the reactants to the active sites. The high desulfurization activity of the N-CNT-based catalysts could be attributed to the even dispersion of the N–C sites on the surface of the nanotubes which provide high active site density along with the high effective surface area due to the nanoscopic dimension of the tube. The high stability of the catalysts as a function of time on stream is attributed to the direct insertion of the nitrogen atoms inside the carbon matrix which allows one to avoid deactivation through active phase sintering or chemical modification as encountered with the traditional metal and oxide supported catalysts.
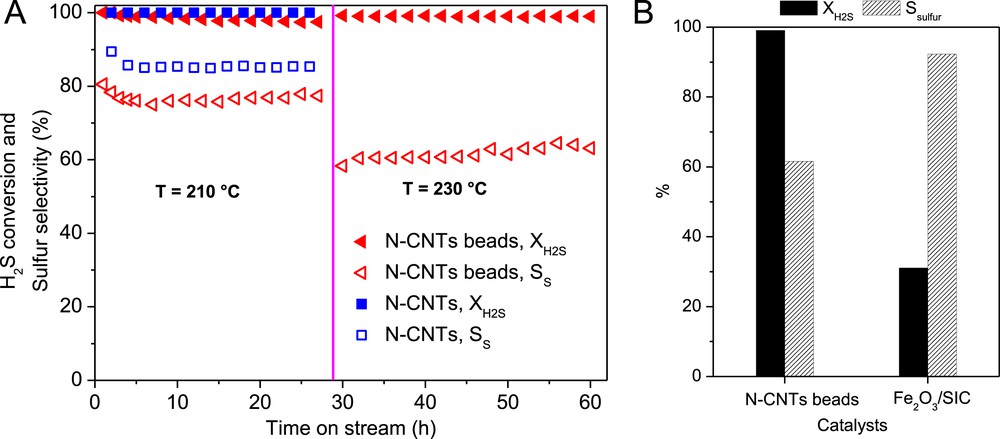
(A) Desulfurization performance, H2S conversion and sulfur selectivity, of the N-CNT bead catalyst. (B) The steady-state catalytic desulfurization performance over N-CNT beads and Fe2O3/SiC catalyst at 230 °C. Reaction conditions: [H2S] = 1 vol. %, [O2] = 2.5 vol. %, H2S-to-O2 molar ratio = 2.5, [H2O] = 30 vol. %, GHSV (STP) = 2400 h−1.
The sulfur selectivity on the N-CNT beads is amounted to about 76% at 210 °C for the whole test which is lower compared to that of the bulky N-CNTs, i.e. 83%. Such relatively lower sulfur selectivity could be attributed to the longer residence time of the formed sulfur inside the catalyst macroscopic body which favors the secondary reaction between the sulfur and excess oxygen to form SO2. Indeed, in the bead form it is expected that higher entanglement is generated within the N-CNT structure leading to a longer residence time which could favor the secondary reaction between the formed sulfur and excess oxygen to yield SO2.
Increasing the reaction temperature from 210 to 230 °C leads to an increase of the desulfurization activity of the N-CNT beads with, however, a net loss regarding the sulfur selectivity. Additional work is underway to investigate the influence of the reactant gaseous space velocity on the desulfurization performance of this macroscopic catalyst. The desulfurization performance of the N-CNT beads was also compared with that of the industrial-like Fe2O3/SiC catalyst and the results are presented in Fig. 6B. The N-CNT beads display a very high desulfurization activity compared to the Fe2O3/SiC catalyst under similar reaction conditions with a lower sulfur selectivity due to the problem of diffusion discussed above.
In order to evaluate the influence of the O2-to-H2S molar ratio on the N-CNT beads experiments were carried out at 230 °C with different O2/H2S ratios and the results are displayed in Fig. 7. According to the catalytic results the decrease of the O2-to-H2S molar ratio leads to a significant improvement of the sulfur selectivity. Decreasing the O2-to-H2S ratio, O2/H2S from 2.5 to 0.6, a drop of the H2S conversion was observed, 92% instead of 100%, while the sulfur selectivity was increased from 60 to 80%. It is worthy to note that during the whole test lasting for more than several dozen hours no deactivation was observed on the catalyst which confirms again the high stability of this metal-free catalyst.
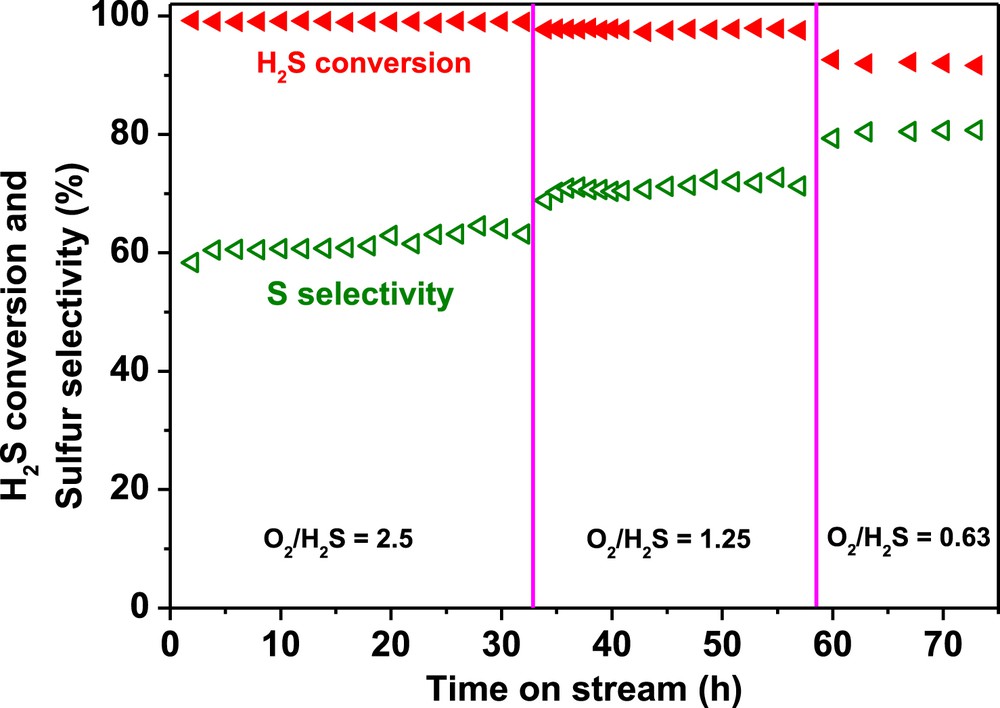
Desulfurization performance, H2S conversion and sulfur selectivity, of the N-CNT bead catalyst as a function of the O2-to-H2S molar ratio. Reaction conditions: [H2S] = 1 vol. %, [O2] = 0.5 to 2.5 vol. %, [H2O] = 30 vol. %, reaction temperature = 230 °C, GHSV (STP) = 2400 h−1.
The desulfurization results obtained above indicate that some optimization process is necessary in order to increase the porosity inside the N-CNT beads leading to a higher accessibility of the active sites to the reactants and also to a better escape of the intermediate sulfur product from the catalyst body in order to reduce as much as possible the secondary reaction to form SO2.
4 Conclusion
In summary, we have developed an easy method for preparing a macroscopic N-CNT composite with a controlled shape and a fully accessible surface area which can be efficiently used as a metal-free catalyst in the selective oxidation of H2S. The material also displays a high specific surface area, i.e. 237 m2 g−1 along with a fully accessible surface thanks to the presence of a meso- and macro-porous network. The diffusion of the reactant inside the catalyst matrix and the evacuation of the products are also favored by the nanoscopic shape of the carbon nanotubes which significantly reduces the diffusion length. The N-CNT composite displays an extremely high desulfurization performance compared to the Fe2O3/SiC catalyst, and especially in a high space velocity operating process. The high desulfurization performance was attributed to the high exposure of the active site towards the reactant while the high stability was due to the direct insertion of the nitrogen atoms inside the carbon matrix which prevents sintering of the active phase. It is expected that such self-sustained micronized nitrogen-doped carbon nanotubes can be efficiently used as metal-free catalysts in other relevant catalytic processes in place of traditional supported ones. Work is underway to increase the porosity of the N-CNT beads in order to reduce as much as possible the problem of diffusion.
Acknowledgments
The authors would like to thank the EU for the financial support of this project through the FREECATS project (NMP3-SL-2012-280658). CDV would like to acknowledge the Vietnamese government for the grant during his stay at the ICPEES. The SEM and TEM were carried out at the facilities of the IPCMS (UMR 7504 CNRS) and T. Romero (ICPEES) and Prof. O. Ersen (IPCMS) are gratefully acknowledged for the characterization experiments.