

1 Introduction
The global demand for crude oil increased radically in the first years of the 21st century due to the dynamism of “Emerging Countries” such as China and India. Oil consumption rose from about 70 million barrels per day in 1995 to over 80 in 2005 and is expected to grow over 90 million barrels per day in 2020. Moreover, the petroleum market is dominated by a demand for high value petroleum products such as middle distillates while the demand for low value products such as fuel oil and residua decreases. At the same time, the quality of crude oil available to refiners is declining and environmental regulations have been introduced in many countries to reduce drastically the sulfur content in transportation fuels. These specifications, beneficial from the environmental point of view, present a major technical and economic challenge for the petroleum refining industry because refiners are required to produce higher quality products from lower quality feedstocks. Hydrotreatments (HDTs) are the most commonly used processes for upgrading heavy fractions to better quality fuels and even if hydrotreatment is very often considered as a mature subject, due to its major importance, progress in this area is still expected and the development of highly active hydrodesulfurization catalysts appears then as a key factor [1-4]. In 2008, in an overview of the recent studies on the preparation, activation and design of sulfide active phases and supports for preparing hydrotreating transition metal sulfide catalysts, Breysse et al. [5] showed that the development of efficient HDS catalysts could be achieved through different directions working for example on the activation procedure or on the synthesis of new supports. Another direction is the development of new preparation methods.
In hydrotreating catalysts, the active phases of traditionally used catalysts consist of well dispersed MoS2 nanocrystallites decorated with Co or Ni promoter atoms. These phases are obtained by sulfidation of an oxidic precursor that is conventionally prepared by incipient wetness impregnation of an alumina support with ammonium heptamolybdate (AHM) and cobalt or nickel nitrate solutions. These solids are then dried and calcined in order to mainly remove the nitrate and ammonium counterions present in these conventional preparations and form an oxidic precursor. Different methods of preparation of the oxidic precursors have been proposed in the literature by using new starting materials based on heteropolyanions (HPAs) instead of AHM and Co(Ni) nitrate. Various types of heteropolyanions have been reported and they generally led to better catalytic performances than their counterparts based on conventional precursors [6]. Numerous polyoxometallate structures have been described and reported by Souchay [7] and Pope [8]. Polyoxometalates or polyanions constitute a family of mineral condensed ions in the form of isopolyanions and heteropolyanions. Isopolyanions MmOyt− (M = Mo or W) are formed during condensation of MO4n− oxoanions occurring due to solution acidification, elimination of water molecules and creation of μ-oxo bonds between metal ions. In the presence of other XO4p− oxoanions (X = P, Si, B,…) metallic atoms are associated around the second oxoanion to form a heteropolyanion. Polyanions are represented by families possessing defined structures, and few that have been used to prepare hydroprocessing catalysts can be listed (Fig. 1): Lindqvist [MxM′6−xO19](2+x)−, Keggin [XM12O40]n−, Anderson [XM6O24H6]n−, Dawson [X2M18O62]n−, Strandberg [X2M5O23n−] or Waugh [Ni9MoO32]6−, where M(M′) is a metallic cation and X is a non-metallic one. These species are generally synthesized in solution and deposited on a support by impregnation methods in order to prepare a supported catalyst. Nevertheless, HPA species can also be formed during the impregnation procedure due to the “interaction” with the support causing for example the extraction of aluminum cations from alumina. The HPA–support interaction is also largely related to the support zero point charge (ZPC) influencing the pH in the support pores and the pH range stability of the HPA. In this review, after a general presentation of the hydrotreating studies dealing with the use of different polyoxotungstate or polyoxomolybdate structures, we will focus on the Anderson and the dimer derivative Anderson structures involved in the preparation of sulphide catalysts for hydroprocessing. Modifications have also been explored to prepare efficient catalysts such as the formation of metallic salts and more recently the use of chelating agents.
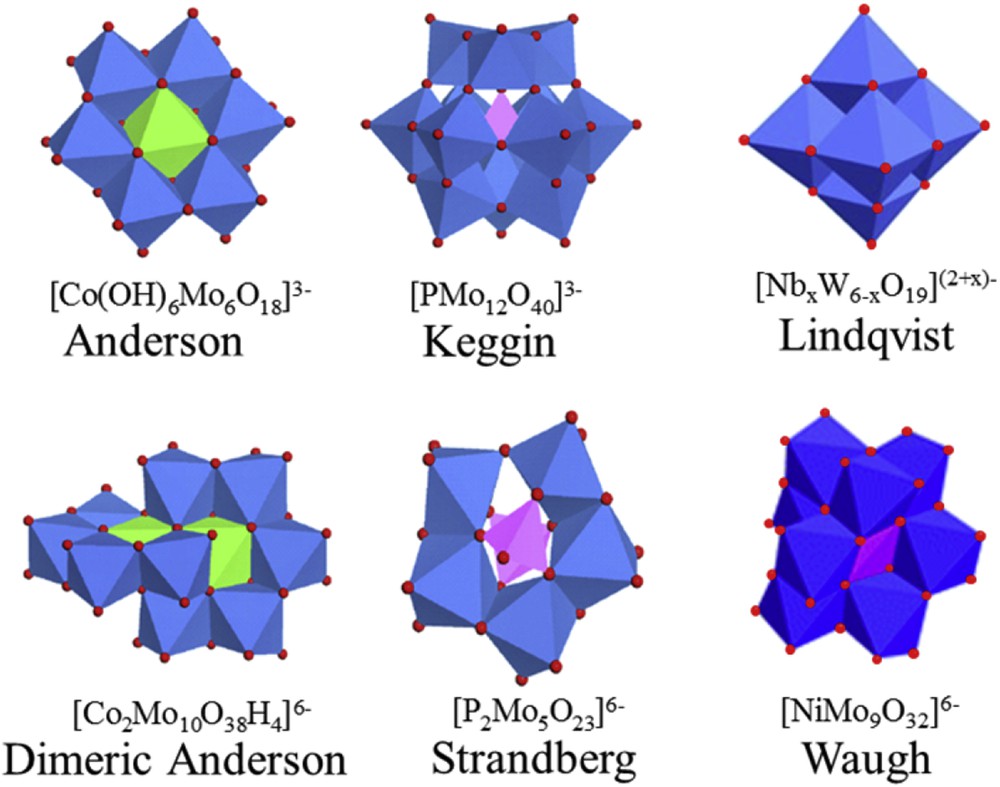
Typical HPA structures used for hydroprocessing catalyst preparation. Color scheme: Mo (blue); O (red); P and Si (pink); Co (green).
2 General issues: HPAs for hydrotreating catalyst preparation
2.1 Keggin structure
Among the different series of heteropolymolybdates, Keggin HPAs (Fig. 1) have been more frequently used as alternatives to classical ammonium heptamolybdate (AHM) and early investigations concerned the heteropolyacids H3PMo12O40 and H4SiMo12O40, and the addition of nickel nitrate allowing us to obtain promoted catalysts efficient for thiophene HDS [9,10]. HPA-based nickel promoted catalysts have also been proved to be efficient in diesel hydrotreating and have shown an enhancement of the HDS and HYD activities compared to their classical counterpart [11]. To avoid the presence of ammonium and nitrate ions in the impregnating solution, the cobalt (nickel) salts of these heteropolyanions such as Co3/2PMo12O40 (with Co/Mo = 0.125) have been prepared [12]. The increase of the Co/Mo ratio has been obtained using cobalt salts of a reduced heteropolyanion PMo12O407− (Co/Mo = 0.29) [13] or of a substituted one Co3PCoMo11O40H6− (Co/Mo = 0.36) [14] allowing us to prepare efficient alumina supported CoMo HDS catalysts. Due to its higher stability when increasing the pH the reduced HPA is preserved after impregnation on alumina while the decomposition of the PCoMo11O407− HPA occurs due to the buffering effect of the alumina support [14]. By a combination of Raman and UV-NIR spectroscopies Bergwerff et al. [15] studied the disintegration of H2PCoMo11O405− entities inside Al2O3 pellets after impregnation that was explained by the reaction between phosphate with alumina resulting in the formation of [Co(H2O)6]2+ and HxMo7O24(6−x)−. Nevertheless, when impregnation was carried out in the presence of citrate and adjusting the phosphate concentration, the formation and distribution of H2PMo11CoO405− can be controlled inside the alumina pellets [15]. The CoMoP/Al2O3 catalyst was prepared from a solution containing MoO3, H3PO4, and Co(OH)2 with an additive impregnation step using an aqueous solution of triethyleneglycol (TEG) and its efficiency in toluene hydrogenation (under HDT conditions) was also attributed to the formation of the Co substituted PCoMo11O407− heteropolyanion at the surface of the alumina [16]. Analysis of the promotion of CoMoP/Al2O3 catalysts prepared from a reduced heteropolyacid H7PMo12O40 and Co was reported in [17]. It was found that the use of Co7/2PMo12O40 as the precursor of CoMoP HDS catalysts led to higher activity than that of a catalyst prepared by co-impregnation with a mixture of AHM–Co(NO3)2–H3PO4. The increased activity of the former catalyst was related to a higher number of Co-promoted molybdenum sites. The amount of Co-promoted sites in the catalyst prepared with Co7/2PMo12O40 depended on the pretreatment applied to the catalyst before its sulfidation. High activity was obtained by avoiding the calcination treatment, and for the calcined catalysts, the activity decreased with the calcination temperature. This was related to the stability of the Keggin structure of Co7/2PMo12O40 that was destroyed at calcination temperatures above 350 °C.
Amorphous silica-alumina (ASA) supported hydrocracking catalysts possessing interesting hydrogenating properties have been successfully developed based on heteropolytungstate nickel salts Ni2SiW12O40 and lacunary Ni4SiW11O39 [18]. Compared to classical H3PMo12O40 and H4SiMo12O40 HPAs, the use of the vanadium substituted heteropolycompound H3[PVMo11O40] led to improved catalytic performances in diesel hydrotreating [11]. The efficiency of MoV sulfide catalysts prepared from H(3+x)PMo(12−x)VxO40 with x = 1, 3 and 6 was also reported for the hydrotreatment of residues requiring to perform hydrodemetalation to remove nickel and vanadium together with hydrotreatment [19]. Trimetallic supported catalysts NiMoW(S)/Al2O3 prepared from heteropolyacids and nickel nitrate have also been investigated and have exhibited higher HDS and HYD activity than the corresponding bimetallic NiMo(S)/Al2O3 catalyst with a MoO3 content equal to the total MoO3 and WO3 content [11,20].
Simultaneous use of Keggin HPAs and Co(Ni)-chelates for the preparation of HDT catalysts was investigated in several studies [21,22,23,24,25]. Ramírez et al [21] reported that the use of phosphomolybdic acid and nickel citrate as active phase precursors led to more active catalysts in the HDS of 4,6-DMDBT than those conventionally prepared with AHM + Ni(NO3)2 + H3PO4. Recently, the effect of the Ni/W ratio on Ni–CA–PW/Al2O3 hydrotreating catalysts prepared with H3PW12O40 and nickel citrate (Ni–CA) was studied [22]. It was found that with the rise of Ni content in the prepared catalysts the composition and morphological characteristics of the NiWS active phase species as well as their catalytic properties were changed simultaneously. The turnover frequency (TOF) number in all studied reactions increased with the decrease in the average length of the NiWS phase species together with the Ni/W ratio on the (Ni)WS edges. Both NiW reference catalysts prepared using co-impregnating solutions of (NH4)6H2W12O40 and Ni(NO3)2 had active sites with lower TOF values than their counterparts prepared using H3PW12O40 and nickel citrate. The highest TOF values were achieved at a moderate Ni/(Ni + W) ratio equal to 0.24. However, maximal conversions of the reactants were observed over the catalysts with higher Ni/(Ni + W) ratios equal to 0.33–0.45. These results were explained by a better balance between the TOF value of the active sites and their content in Ni–CA–PW/Al2O3 catalysts. Further improvement of the Ni–CA–PW catalysts can be obtained by using carbon-coated alumina (CCA) as the support [23]. Covering the alumina surface by coke is beneficial to tune the metal–support interactions, which causes easier reduction of tungsten. This property allowed the CCA supported catalysts to be better sulfided compared with the Ni–PW/Al2O3 reference.
Using CCA supports instead of alumina for preparation of CoMo catalysts resulted in improving their activities in hydrodeoxygenation of guaiacol and oleic acid and significantly reduced deactivation degrees [24,25]. Enhanced catalytic properties of CoMo/Cx/Al2O3 catalysts were related to lower acidity of CCA supports. Catalyst activities in guaiacol and oleic acid hydrodeoxygenation were maximal at moderate carbon content in the CCA, which was explained by an optimal balance between the cobalt content in CoMoS phase particles and their average length [25].
2.2 Strandberg structure
The heteropolyanion P2Mo5O236− possesses the Strandberg structure (Fig. 1) characterized by a P/Mo ratio of 2/5. It is spontaneously and quantitatively formed in solution containing molybdenum and phosphoric acid with this ratio [14], the protonation (once or twice) depending on the pH solution [26]. Cobalt or nickel species using nitrate, carbonate or hydroxide precursors can be added to P2Mo5O236− containing solution to prepare promoted catalysts. In a large excess of phosphoric acid to dissolve MoO3, the low pH of the solution leads to the formation of the Dawson HPA P2Mo18O326− [12]. By the addition of cobalt carbonate a solution containing P2Mo5O236− and Co2+ without nitrate or ammonium counterions is obtained due to the pH increase [27]. After deposition, P2Mo5O236− species are at least partially retained in the alumina MoP supported catalyst as well as on the alumina [28] or zirconia [29] CoMoP catalyst while it is destroyed after deposition on TiO2 in agreement with the stability of the Strandberg anion up to pH equal to 6. Thiophene HDS catalytic performances were higher using supported catalysts prepared from solutions containing P2Mo5O236− and Co2+ than those of their corresponding counterparts prepared from AHM and cobalt nitrate [28,29]. After calcination, the presence of a well dispersed polymolybdate phase together with the absence of ammonium and nitrate ions explained the better catalytic results.
2.3 Lindqvist structure
Very few studies deal with the preparation of sulfide catalysts using Lindqvist type isopolyanions (Fig. 1). These anions can be prepared in organic media by precipitation forming a tetrabutylammonium salt such as (NBu4)3[VMo5O19] [30] whose solubility is low. Recently a series of sulfide pre-treatment catalysts was prepared using [NbxW6−xO19](2+x)− (x = 1,2,3,4, and 6) in order to introduce acidic properties in WS2 alumina supported catalysts. After the sulfidation step, niobium remained unsulfided and niobium oxide generating Brønsted acidic sites was shown to enhance cyclohexane isomerization under hydrotreatment conditions (H2 pressure and presence of H2S) [31].
2.4 Waugh structure
Liang et al. [32] investigated the use of Waugh-type HPAs for the preparation of NiMo/Al2O3 HDS catalysts. It was shown that the application of (NH4)6[NiMo9O32] and Ni3[NiMo9O32] salts as active metal precursors produced highly dispersed NiMo catalysts for HDS of DBT. These catalysts possessed suitable reducibility, high dispersion of Ni and Mo species, short length and low stacking of MoS2 slabs. Catalysts prepared from Waugh-type HPAs showed higher catalytic performance in HDS of DBT than their corresponding counterparts obtained with AHM and nickel nitrate. The results demonstrated that Waugh-type NiMo HPAs can make efficient precursors to produce hydrotreating catalysts with high performance.
3 Special issues: Anderson structure and its dimeric form
3.1 Synthesis and characterization
3.1.1 Anderson structure
The XMo6O24H6n− structure was first proposed by Anderson in 1937 [33]. It is a flat structure formed with 6 MoO6 octahedra surrounding a X(OH)6 octahedron (Fig. 1). Numerous central atoms can form the Anderson structure, particularly trivalent cations such as Te, Cr, Co, Al, Fe, Rh, and Ga and also divalent metallic cations Me2+ such as Cu, Zn, and Ni. The synthesis of the ammonium salts of Anderson HPAs is performed generally by boiling an aqueous solution of AHM where the Me3+/2+ cation is added, and H2O2 can be added for the formation of Co3+ from Co2+. The synthesis of cobalt salt has also been proposed in the second step by using cationic exchange between NH4+ and Co2+ according to the following equation:
| 3(NH4)+,XMo6O24H63− + [3/2Co2+, PMo12O403−] → 3/2Co2+,XMo6O24H63− + (NH4)3PMo12O40 |
The yellow precipitate (NH4)3PMo12O40 is removed by filtration and the Anderson cobalt salt is obtained after evaporation [34].
3.1.2 Dimer structure
Tsigdinos showed that monomeric Anderson species CoIIIMo6O24H63− can be converted entirely to dimeric ones if activated charcoal or Raney nickel is added to the aforementioned boiling solution [35]. The ammonium salt formula (NH4)6Co2Mo10O38H4·7H2O has been later established by Evans and Showell [36]. The dimeric anion is obtained by the association of two monomers from which one MoO2 has been removed and that are oriented so that the two CoO6 octahedra could share an edge (Fig. 1). It appeared more interesting for hydrotreating applications to prepare the cobalt salt of the dimer that was first obtained by cationic exchange as already performed for the Anderson ammonium salt [37]. This preparation method of Co3Co2Mo10O38H4 in several steps was later replaced by a very simple procedure consisting of mixing of MoO3, H2O2 and cobalt carbonate in a stoichiometric amount to obtain quantitatively Co3Co2Mo10O38H4 (named after Co2Mo10Co) [38]. By varying parameters such as the Co/Mo ratio, molybdenum concentration, and pH, the stability of the decamolybdate heteropolyanion was recently studied by UV-visible and Raman spectroscopies [39].
3.2 Anderson XMo6O24n−-based catalysts
3.2.1 AlMo6
In the first studies rationalizing the interaction of molybdenum entities with the support, alumina was not taken into account and the nature of the adsorbed species was considered to depend on the Mo loading and on the alumina adsorption surface sites. By increasing the Mo loading, Raman spectroscopy identified the presence of isolated tetrahedral entities (MoO42−) of the polymolybdate phase and of MoO3 microcrystallite [40,41,42,43].
More recent results have shown through Raman, NMR and Mo-Kedge EXAFS spectroscopic studies that upon impregnation of alumina with AHM solution the Mo7O246− isopolyanion is decomposed while extraction of aluminum atoms of the support occurs to form the stable Anderson heteropolyanion AlMo6O24H63− (AlMo6) [44,45]. This aluminum-based Anderson species observed after the maturation step is maintained after drying. On conventional promoted catalysts the formation of AlMo6 entities is also observed after maturation and drying [28]. Whatever the preparation procedure (AHM- or MoO3-based impregnating solutions), after calcination and transfer in air a well-dispersed alumina supported catalyst presents a Raman spectra characteristic of well-dispersed AlMo6 entities [28]. At low Mo loading AlMo6 is not observed, the Raman spectrum being characteristic of MoO42− entities, while increasing the Mo content (4 at Mo∙nm−2) leads to the precipitation of AlMo6O24H6(NH4)3 presenting well-resolved Raman peaks. Taking into account the low solubility of AlMo6 ammonium salt, the corresponding Co salt AlMo6O24H6(Co)3/2 was prepared by cationic exchange in solution. However its solubility remained low (0.18 molMo.L−1) leading to its decomposition in MoO42− species after deposition on alumina due to an increase in pH in the alumina pores by a buffering effect [28].
3.2.2 CoMo6/NiW6 ammonium salts
As early as 1989, Maitra et al. [46] proposed the use of Anderson heteropolyanionic solutions (ammonium salts or free acids) for the impregnation of alumina in order to prepare hydrotreating catalysts. In the anions of general formula [H6XY6O24]x−, where X is Co or Ni and Y is Mo or W, both catalytically active metals are in the same complex and should thus be deposited in the same position in the pellet. The authors showed that by tuning the pH, they could also be used to prepare catalysts with controlled profiles of impregnation throughout the support pellet diameter (shell-like or uniform profiles). As the solubility of the anions is higher than that of conventional impregnating agents, they also could prepare catalysts with higher concentrations of metals (tungsten particularly). The efficiency of solids was evaluated in hydrotreating of a model feedstock, containing thiophene, quinoline, dibenzofuran and phenanthrene. The most performant solid was a NiW6 catalyst, with a W content of 11.8 wt% and presenting a uniform distribution of both metals. It showed good HDS and HYD activities, that surpass that of a commercial NiW sample (W = 19 wt%), despite a lower content of metal, especially of the promoter (respective conversions of 87 and 90% for HDS and 20% for both in HYD, at 633 K). The low promoter/metal ratio appeared however unfavorable to HDN activity, with a conversion of 5% compared to 9% for the commercial catalyst.
Cabello et al. [47] prepared CoMo catalysts by impregnation of an alumina support with a (NH4)3[CoMo6O24H6]·7H2O Anderson phase without calcination. They found that the catalytic activity in thiophene HDS of the most efficient CoMo6/Al2O3 catalyst of the series was similar to that obtained for the Co–Mo/Al2O3 conventional catalyst at an equivalent molybdenum content, despite its Co/Co + Mo ratio of 0.14, considerably lower than that of the conventional catalyst (0.25). The promoting action of cobalt seems to be more effective in the CoMo6/Al2O3 catalysts. The planar molecular structure of the anion and the adsorptive interaction with the support are factors that make easier not only the coverage of the support surface (monolayer of CoMo6) but also the availability of metallic sites for promotion. The absence of calcination also prevents the formation of the inactive CoAl2O4 spinel oxide, allowing a maximum of the Co species to be available for promotion.
Close results were obtained by Nikulshin et al. [48,49,50,51,52,53] when (NH4)4[Ni(OH)6Mo6O18] salt was used as a precursor. The obtained NiMo6(S)/Al2O3 catalyst had higher activity in thiophene HDS [53], DBT HDS [52] and diesel hydrotreating [48,49,50,51,52] compared with conventional references.
These results appeared promising concerning the efficiency of Anderson-based hydrotreating catalysts compared to conventional ones despite the low Co/Mo ratio, and thus encouraged studies aiming to increase the metal/promoter ratio.
3.2.3 Influence of the Co(Ni)/Mo ratio on the CoMo6/NiMo6-based catalyst
Cabello et al. varied the Co/Mo ratio in CoMo6/Al2O3 catalysts by co-impregnation of alumina with CoMo6 and (NH4)6[TeMo6O24]·7H2O (TeMo6), the Mo amount being kept around 8% [54]. The Anderson phase of Te was chosen because of its good solubility and because no promoting effect of Te was observed in hydrotreatment. Both Anderson structures were adsorbed on the alumina during the equilibrium impregnation of the support. Increasing the Co/Mo ratio from 0 to 0.15 by varying the quantity of TeMo6 added to CoMo6 led to an increase in the HDS of thiophene and HYD of cyclohexene, the higher conversions being obtained for a Co/Mo ratio of 0.15, corresponding to that of the pure CoMo6-based catalyst.
Palcheva et al. thus considered to increase the Co/Mo ratio above that imposed by the stoichiometry of the salt, up to a Co/Mo ratio of 0.68, by the addition of variable amounts of cobalt preliminary loaded on the alumina support [55]. On a CoMo catalyst with 12 wt% Mo, an optimum in both thiophene and 1-benzothiophene HDS is obtained for a preliminary incorporation of 1.3 wt% Co in the support, corresponding to an optimum Co/Mo ratio equal to 0.27. The authors assumed that cobalt species present on the support are exchanged during impregnation with the ammonium ions of the heteropolyoxomolybdate and occupy the counterion state, being thus available to form an additional CoMoS phase during sulfidation. This was supported by the absence of cobalt aluminate in the catalyst. When still increasing the Co content, the extra-added cobalt is not in the decoration position and participates in the formation of other cobalt compounds, leading to lower efficiency in HDS reactions.
Alumina previously modified with nickel, cobalt and boron was also used for the preparation of NiMo catalysts by impregnation with Anderson NiMo6 heteropolyoxomolybdate [56]. Nickel incorporation into the alumina prior to loading led to the most efficient catalyst (Ni/(Mo + Ni) = 0.24) and resulted in an activity for 1-benzothiophene hydrodesulfurization that was nearly twice as much as that observed for the NiMo6 deposited on alumina and B modified alumina. The better performance of the NiMo6/Al–Ni catalyst was related to the formation of a larger amount of active sites, due to better dispersion of the molybdenum phase as well as to a higher sulfidation degree of nickel and molybdenum as determined by XPS.
3.2.4 RhMo6
Another metal other than Co and Ni was also considered in XMo6 Anderson-type heteropolyoxomolybdate: (NH4)3[RhMo6O24H6]·7H2O was used by Cabello et al. to prepare RhMo6/Al2O3 catalysts [57]. The HDS activity of the RhMo6/Al2O3 catalyst was found similar to that observed for CoMo6/Al2O3 and Co–Mo/Al2O3 systems with equivalent Mo loading (thiophene conversion around 70%). However, its HYD ability was considerably higher than that of the CoMo6 and conventional CoMo catalysts (cyclohexane hydrogenation conversion of 85, 30 and 15% respectively), and also higher than that of the commercial Rh/Al2O3 catalyst (65%). The presence of Rh was found to induce the Mo(VI)-Mo(IV) reduction at lower temperature.
3.2.5 XMo6 (X = Cr, Mn, Fe, Ni, Co, Cu, Zn, and Ga)-based catalysts
Catalysts XMo6(S)/Al2O3 and Ni3–XMo6(S)/Al2O3 were also prepared from heteropolycompounds of Anderson-type, where X–Co, Ni, Cr, Mn, Fe, Cu, Zn, and Ga [58,59]. The precursors and synthesized catalysts were characterized by FTIR, XRD, EXAFS, and HRTEM techniques. The catalytic performances were evaluated in thiophene HDS, benzene HYD and hydrotreating of diesel. The catalytic activities in HDS and HYD and the heats of thiophene adsorption were found to be related to the nature of the heteroatom proving its role in the formation and behavior of the HYD and HDS active sites. It was found that heteropolycompounds are effective precursors of a multilayered active phase of hydrotreating catalysts. The heteroatom X in Anderson-type HPAs in the catalysts promotes the optimization of the electron density on the anti-bonding d-orbital of Mo in the active mixed sulphide phase and, thereby, facilitates productivities of the active sites both in HDS and in HYD reactions.
3.2.6 Al13−xMo6 composite
Munoz et al. [60] have developed an advanced inorganic composite material obtained by direct combination of Al13 Keggin isopolycation [AlO4Al12(OH)24(H2O)12]7+ with the Anderson heteropolyanions AlMo6, CrMo6 and CoMo6. These composites, consisting of an ordered distribution of the Anderson heteropolyanion with an Al13 network, could be of interest as hydrotreatment oxidic precursors.
3.2.7 CoMo6 cobalt salt
Cobalt salt of the CoMo6 heteropolyanion was prepared by Lamonier et al. [61], the advantages compared to the ammonium salt being a much higher solubility and an increased Co/Mo ratio (0.41 instead of 0.17). The performance of the catalysts prepared from this salt was found slightly higher than that of a reference catalyst prepared from conventional precursors, with the same metal loading (8 wt% MoO3) but with a Co/Mo ratio of 0.5.
3.3 Dimer Anderson Co2Mo10O38H46−-based catalysts
3.3.1 Ammonium salt
The CoMo catalyst supported on alumina was prepared using (NH4)6[Co2Mo10O38H4]·7H2O salt [62] by impregnation with excess of solution under continuous stirring for 24 h. After separation by centrifugation and drying at 80 °C, a catalyst with a Mo loading equal to 9.8 MoO3 wt%, and the Co/Mo ratio equal to 0.2 corresponding to the (NH4)6[Co2Mo10O38H4] stoichiometry was obtained. Co2Mo10-based solid catalytic performances were compared to those of a CoMo6-based solid obtained from the ammonium salt with the same preparation procedure and of a conventional catalyst (12.8 MoO3 wt%, Co/Mo = 0.3) prepared from AHM and cobalt nitrate. The catalytic performance of sulfide catalysts has been evaluated both in thiophene HDS and cyclohexene hydrogenation. Even with lower metal content, better catalytic performances were obtained using heteropolymetallate catalytic systems. The authors attributed these good results to a good adsorptive interaction of heteropolyanions with alumina and a uniform distribution of active sites. Thiophene HDS and cyclohexene hydrogenation conversions were higher on the Co2Mo10-based catalyst. Moreover, conversions expressed per Co atom in HPA-based systems were nearly twice that of the conventional catalyst suggesting a good synergetic effect in the sulfide phase arising from the preparation procedure with HPA avoiding the calcination step.
For preparing alumina supported catalysts [62] the use of Co2Mo10 ammonium salt (Co/Mo = 0.2) with low solubility (0.69 molMo.L−1) led to an 8 MoO3 wt% catalyst whose HDS catalytic performance is compared to that of a reference catalyst (10 MoO3 wt%, Co/Mo = 0.3) and a CoMo6Co based catalyst (8 MoO3 wt%). The conversion of the Co2Mo10(NH4)6/Al2O3 catalyst (12.5%) is almost similar to that of the reference catalyst with a higher Co/Mo ratio and lower than that of CoMo6Co/Al2O3 with a Co/Mo ratio equal to 0.42 (24%). Cobalt nitrate has been added to the initial Co2Mo10(NH4)6 impregnating solution to increase the Co/Mo ratio. The thiophene conversion of such Co–Co2Mo10(NH4)6-based catalysts increased with the Co/Mo ratio and for a Co/Mo ratio of 0.5 it became higher than that of the reference catalyst with the same metal loadings. Nevertheless, this conversion was still below that obtained for the Co2Mo10Co/Al2O3. It has been assumed to be due to the presence of foreign counterions (NO3− and NH4+) in the impregnating solutions, their presence hindering a good interaction between Co and Mo.
3.3.2 Cobalt salt
The cobalt salt of the dimer Co2Mo10O39H46− is an interesting starting material to introduce cobalt and molybdenum in the same entity, without foreign counterions and with a Co/Mo ratio equal to 0.5 corresponding to the optimum Co/Mo ratio for conventional HDS catalysts [62]. Its optimum Co/Mo ratio together with a better promoting effect explained that the Co2Mo10Co-based catalyst was more efficient than the CoMo6Co catalyst with the same Mo loading (8 MoO3 wt%). The preservation of Co2Mo10Co on alumina after the drying step was demonstrated through a XAS analysis at the Co Kedge; it prevented the formation of bulk oxides after calcination and allowed a direct cobalt–molybdenum interaction in the HPA cobalt salt resulting in a better promoting effect at the sulfide state. With the same Co/Mo ratio and varying the Mo loading between 6 and 16 (MoO3 wt%), better catalytic performance of the HPA catalyst compared to the reference one was also attributed at high Mo content to the better dispersion of the (Co)MoS2 active phase.
Alumina, titania and zirconia supported CoMo catalysts have been prepared using three different impregnating solutions. Two of them contained well defined Co2Mo10O39H46− and P2Mo5O236− heteropolyanions, respectively, and cobalt cations, the third one was the conventional impregnating solution prepared from AHM and cobalt nitrate [29]. In the three cases, the Co/Mo ratio was set at 0.5 with various Mo content. HDS catalytic results showed that whatever the support Co2Mo10 cobalt salt appeared as the starting material leading to the best thiophene HDS catalytic performances. The best activities were obtained at various Mo loadings for each support but corresponded to a similar Mo density (3.4 Mo atoms.nm2) taking into account the different specific surface area of alumina, titania and zirconia. Impregnation with conventional solution containing ammonium and nitrate counterions led to poorly dispersed alumina and zirconia supported solids whereas molybdenum entities appeared well dispersed on titania whatever the preparation route. In this case, starting from conventional or P2Mo5O236− solutions led to the same catalytic results. The efficiency of the Co2Mo10Co precursor is then related to the formation of well-dispersed solids after calcination allowing to maximize the Mo sulfidation and also to the better promoting effect due to the initial Co-Mo proximity in the heteropolycompound molecular structure.
More recently Costa et al. [16] have investigated the use of additive-impregnated dried catalysts on a series of various CoMo solids prepared from a conventional route, Co2Mo10Co, and a solution containing P2Mo5O236− and Co2+ (Co/Mo = 4). The additive impregnation step was performed on the dried solid by incipient wetness impregnation with an aqueous solution of triethyleneglycol (TEG) with a TEG/Mo molar ratio close to 0.75. Additive-impregnated catalysts calcined or dried additive free catalysts performances were evaluated in toluene hydrogenation and cyclohexane isomerization under sulfo-reductive conditions (H2S partial pressure of 0.215 MPa and hydrogen partial pressure of 3.69 MPa). The CoMoS active phase was characterized by XPS and TEM. The “effective” amount of the CoMoS phase is increased on calcined samples, this amount still increased for dried additive-impregnated catalysts prepared from HPA. It was shown that the formation of the “CoMoS” phase is required but not sufficient to explain catalytic performances and that the number of Co-Mo mixed sites determined from DFT calculations that correlated with the catalytic activity and the use of additive was to produce a higher number of mixed sites rather than to create different active sites.
Fig. 2 illustrates the efficiency of the Anderson HPA and its dimer cobalt salts to prepare HDS catalysts. These catalysts present higher activity in thiophene HDS than references based on NiMo6HPA [63] or CoMo6HPA [64] and on Keggin Co–PMo12 HPA. It is clearly due to the increase in the Co/Mo ratio reached in the dimeric cobalt salt that allowed after sulfidation the best promoting effect. When compared to a completely different system based on the formation of nanometric bimetallic sulfides from Anderson-based emulsions, the Co2Mo10Co catalyst is still the most efficient.
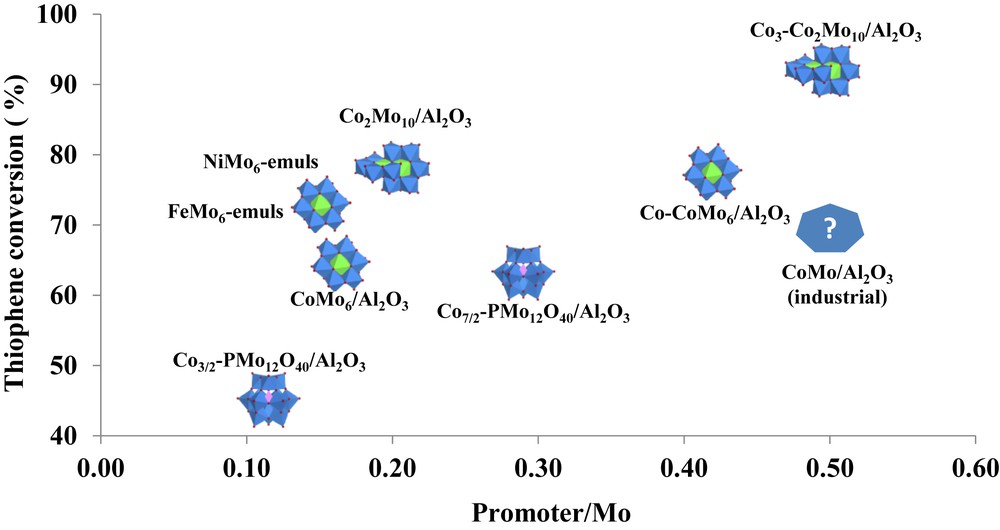
Thiophene HDS over catalysts based on HPA of different types and promoter/Mo ratios. Adapted using data [12, 29, 58, 59, 61, 62, and 65]
More precisely the effective Co content in CoMoS phase species as well as the Co/Mo ratio in the slab edges calculated from XPS were higher when Co2Mo10HPA and cobalt salts of Со2Мо10HPA were used for catalyst preparation compared with CoMo6HPA [64] (Fig. 3). Catalyst activities in HDS of DBT and 4,6-DMDBT increased with increasing (Co/Mo)edge ratio.
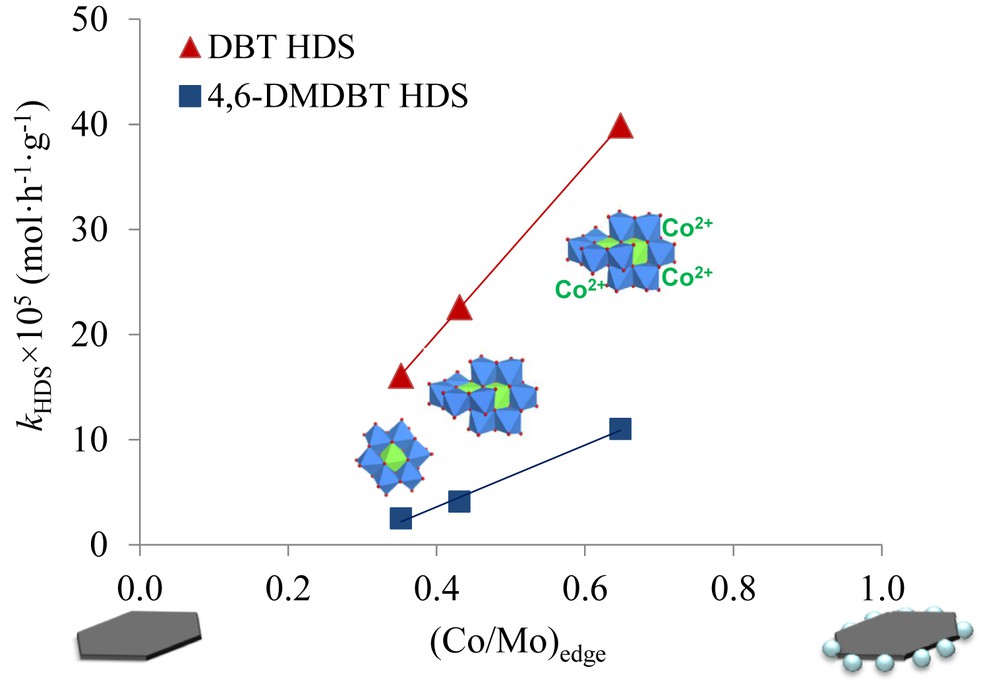
Dependence of rate constants in HDS of DBT and 4,6-DMDBT on the promotion ratio of MoS2 slab edges and the (Co/Mo)edge of catalysts prepared from CoMo6HPA, Co2Mo10HPA and Co3[Co2Mo10HPA]. Adapted using data from [64]
3.3.3 Co2Mo10HPA and Co-chelates
The effect of Co/Mo ratio on the CoMo/Al2O3 hydrotreating catalysts prepared by simultaneously using Со2Мо10HPA and cobalt citrate was investigated [66]. It was established that the Co/Mo ratio in the catalysts significantly influences the structure and morphology of CoMoS-II species and catalytic activity examined in the model reaction of 4,6-DMDBT HDS, in hydrotreating of straight-run gas oil (SRGO) and a mixture of 80 vol. % SRGO with 20 vol. % light coker gas oil (LCGO). Catalysts with λ = Co/(Co + Mo) = 0.360 had maximal activities in 4,6-DMDBT HDS and diesel HDT and showed the best stability. Increase in the Co/Mo ratio led to simultaneous increase of both average length (from 3.0 to 4.0 nm) and stacking number (from 1.4 to 1.6) of the CoMoS multilayer active phase of the catalysts. It was found that activity of catalysts decreased with λ from 0.35 to 0.45. Analysis of textural properties of the catalysts, active phase morphology, their phase composition and catalytic activities allowed us to suppose that with the increase in Co/Mo ratio blocking of the mesopores by active component sulfides and by coke species with the formation of new micropores would take place. The activity of the most efficient CoMo/Al2O3 catalyst was competitive with the activity of well-known commercial catalysts for ultra-low sulfur diesel in hydrotreating of SRGO or a mixture of SRGO with LCGO [66].
The genesis of catalysts prepared with the use of Co2Mo10HPA and cobalt citrate during their sulfidation processes and deactivation in diesel HDT was investigated [67]. The sulfidation stage was studied for two procedures: gas phase sulfidation by H2S/H2 and liquid phase treatment by a mixture of dimethyldisulfide in diesel at various temperatures and durations. It was found that gas phase sulfidation led to the formation of the CoMoS active phase with higher cobalt content compared to liquid sulfidation of the catalyst and the initial activity of the gas phase treated catalysts in diesel HDT was also higher than that of catalysts subjected to liquid sulfidation. Catalytic examination after accelerated deactivation conditions showed that the liquid phase sulfided sample was more resistant to deactivation. This occurred probably due to stabilization of active phase particles by coke formed during liquid phase sulfidation. This effect prevents the loss of Co atoms from the CoMoS phase.
The effects of the composition and morphology of the active phase in CoMo/Al2O3 catalysts prepared using Co2Mo10HPA and chelating agents (Chels) simultaneously on their catalytic properties in HDS and HYD reactions were investigated [59]. It was found that CoMoS phase species were formed more selectively when using Co2Mo10HPA and Co-chelate complexes simultaneously during HDT catalyst preparation. Adding the Co-chelate significantly changed the active phase morphology due to the different interactions between the precursors and carrier surfaces in each catalyst. The presence of abundant NH4+ cations on the catalyst surfaces synthesized using N-containing Chels may partially destroy the Co2Mo10HPA, change the dispersion of the (Co)MoS particles and decrease their stacking number.
The catalytic activities of the Co3[Chel]-Co2Mo10/Al2O3 samples depended strongly on the Chel used during catalyst preparation. The HDS and HYD activities of the catalysts in HDT of a mixture containing thiophene and n-hexene-1 were higher than those of non-chelate-containing materials. The citric acid (CA)-containing catalyst showed the highest HDS and HYD activities and the best HDS/HYD selectivity factor. Only the CA- and tartaric acid (TA)-containing catalysts had a higher activity in HDS of DBT and 4,6-DMDT and the HDT of diesel relative to the activity of the catalyst without a chelate. In contrast, the HDS activity of the nitrilotriacetic acid- and ethylenediaminetetraacetic acid-containing samples was lower than that of the Co3–Co2Mo10/Al2O3. A detailed analysis of the catalyst morphology accounting for a second structural factor (effective Co content in a multi-slab CoMoS species ()) revealed that only CA- and TA-containing catalysts had higher promoter ratios in the CoMoS slabs and edges than Cо3–Со2Мо10/Al2O3, leading to higher activities during the HDS of DBT and 4,6-DMDBT and the HDT of diesel.
The Co2Mo10/Al2O3 catalyst exhibited much higher TOF values than the industrial catalysts and the laboratory samples supported on alumina and activated carbon or different types of alumina using different Co precursors and preparation methods [64]. Further increases in the catalyst activity will most likely be achieved by using supports with a higher surface area than that of alumina.
4 Conclusion
The development of new preparation methods based on HPA catalysts led to efficient catalytic systems due to better sulfidation allowed by a better dispersion of the oxidic precursor even at high metallic loadings. The increase of the Co/Mo ratio in the heteropolycompounds up to 0.5 was possible due to the formation of dimer cobalt salt. The association of Co and Mo at the molecular level in the same heteropolycompound preserved after drying during the catalyst preparation ensured the efficiency of the promoting effect with a higher quantity of the CoMoS active phase. This HPA-based system was further improved by the simultaneous use of HPAs and chelating agents. The high catalytic performances were more precisely related to the (Co/Mo) ratio at the edge of the MoS2 slabs.
Acknowledgments
P.A. Nikulshin and A.V. Mozhaev thank the Ministry of Education and Science of Russian Federation (project 10.1516.2014/K) and Russian Foundation for Basic Research (projects 15-03-01845, 15-33-20511) for financial support of this work. French authors would like to thank IFPEN that provided financial support for PhD thesis.