

1 Introduction
There is no longer a need to prove the synthetic power of radical chemistry. The latter has indeed shown exceptional reliability in terms of efficiency and stereoselectivity to reach complex and polyfunctional molecular architectures [1]. Nevertheless, rendering the reaction conditions more sustainable, notably the elimination of heavily used tin-based mediators, remains a major challenge [2]. While searching a chemical agent acting as a surrogate to tributyltin hydride has been the object of intense investigations [2], the single electron transfer (SET) approach [3] has also emerged as an efficient solution toward greener radical chemistry, featuring also fascinating developments in photocatalysis [4]. Notably, the oxidation of electron rich centers has been widely accepted as a powerful method for the generation of carbon-centered radicals. For example, α-carbonyl radicals have been generated by employing metal salts at high oxidation states [5] or by organocatalysis with SOMO activation from neutral carbonyl substrates [6]. Alternatively, oxidation of enolates or related carbanions has been reported using a variety of reagents [7]. Such processes have been incorporated in ionic/radical domino reactions [8] to access carbo- and heterocycles. Notably, Dulcère and Rodriguez reported a pioneering ionic/radical tandem reaction involving conjugate addition onto nitroalkenes followed by cyclization of the radical formed by oxidation of the resulting carbanion onto an alkene [9]. In the same line, Jahn has widely developed the Michael additions of nucleophiles onto α,β-unsaturated esters and the oxidation-cyclization of the resulting enolates [10]. Finally, quite recently, the photocatalytic oxidation of enolates has opened new perspectives for this type of transformation [11].
Our contribution to the field of ionic/radical tandem reactions consists of the conjugate additions of anionic reagents to alkylidene bis-sulfoxides followed by transformation of the resulting carbanion intermediate into a radical species through oxidation with iron (III) [12]. The outstanding diastereoselectivity observed in the addition of nucleophiles such as alkoxides, amines, organometallic species or enolates to alkylidene bis-sulfoxide 1 led us to consider its subsequent oxidation into radical species for further synthetic transformations [13]. We envisioned that a SET from carbanion 2 would give the corresponding radical 3 which could be further involved in radical reactions (Scheme 1). As the bis-sulfoxide moiety can be easily converted into a carbonyl or a methylene group, the intermediate bis-sulfoxide radical could be considered as a chiral equivalent of an acyl or a methylene radical 4 and 5.

Anionic/radical tandem reactions from alkylidene bis-sulfoxides.
In this context, investigations toward the generation and behavior of bis-sulfinyl radical intermediates have appeared to us of high interest for the development of sustainable and stereoselective radical transformations.
2 Results and discussion
Our study started by exploring the possibility to generate a bis-sulfinyl radical by SET from the corresponding carbanion with iron(III) salt (Scheme 2). Deprotonation of the (Ss, Ss) bis-(p-tolylsulfinyl) methane 6 with n-BuLi at −40 °C was followed by the simultaneous addition of ferrocenium hexafluorophosphate and TEMPO. After usual work up, adduct 7 was isolated in 34% yield, presumably resulting from the trapping by TEMPO of the intermediate radical [12].

Radical trapping from the oxidation of methylene bis-sulfoxide carbanions.
Having demonstrated that we can turn a bis-sulfoxide carbanion into a radical, we examined the application of this process in tandem reactions according to Scheme 1. Cinnamyl and allyl alcohols were used in this sequence (Table 1). After deprotonation by butyllithium, the resulting lithium alkoxides (8a or 8b) were added to the alkylidene bis-sulfoxides (1a, 1b or 1c) at −40 °C. Then, after 1 h, a mixture of the oxidant (3 equiv.) and TEMPO (2 equiv.) was introduced into the reaction vessel [12]. While Fe(acac)3, FeCl3 or Cu(II) ethylhexanoate was ineffective to give conversion, the use of ferrocenium tetrafluoroborate resulted in the formation of cyclization products. Thus, substituted tetrahydrofuran derivatives 9aa–ca were obtained in 41–59% yields and only one stereoisomer was detected when allyl alcohol was used as the nucleophile. The trans stereochemistry of 9aa was confirmed by the presence of a NOE interaction between the benzylic proton and the methylene attached to the alkoxyamine. These findings suggested a complete diastereoselectivity of the Michael addition process [13] as well as of the radical cyclization step. When cinnamyl alcohol was used, 9ab was isolated as a mixture of epimers at the benzylic stereocenter connected to the alkoxyamine, suggesting a fast homolytic scission – recombination at that center [10f].
Synthesis of bis-sulfinyl tetrahydrofurans 9.
| Alkylidene 1 | Alkoxide 8 | Yield | |
| 1 | 1a, R1 = Ph | 8a, R2 = H | 9aa, 59% |
| 2 | 1a, R1 = Ph | 8b, R2 = Ph | 9ab, 48%a |
| 3 | 1b, R1 = i-Pr | 8a, R2 = H | 9ba, 41% |
| 4 | 1c, R1 = 2-(PhO)–C6H4– | 8a, R2 = H | 9ca, 56% |
a 1.1:1 ratio of diastereoisomers was isolated.
Stereochemical studies were then conducted in order to confirm the enantiomeric purity of the cycloadducts and to understand the factors that govern the trans stereoselectivity of the radical cyclization. Benzylidene bis-sulfones 10a,b, analogs of 1a,b, were engaged in the tandem ionic/radical reaction (Scheme 3). Cycloadducts 11a,b were isolated respectively in 64 and 74% yields and as trans single diastereomers as determined by NOESY on 11b and derivatization of 11a (see below). This observation suggests that the diastereoselective formation of the trans isomers 9 is independent of the stereogenicity of the sulfoxides. It is reasonable to hypothesize that bis-sulfoxides 9 and bis-sulfones 11 derivatives originate from pseudo-chair radical intermediates 12 in which the R substituent occupies a pseudo-equatorial position to minimize steric interactions (Scheme 3) giving radicals 13. Overall the stereocenter between the oxygen and the bis-sulfoxide moiety methylene created in the Michael addition of the alkoxide is controlled by the bis-sulfinyl auxiliary while the second stereocenter created by the radical cyclization is controlled through a pseudo-chair transition state [14].
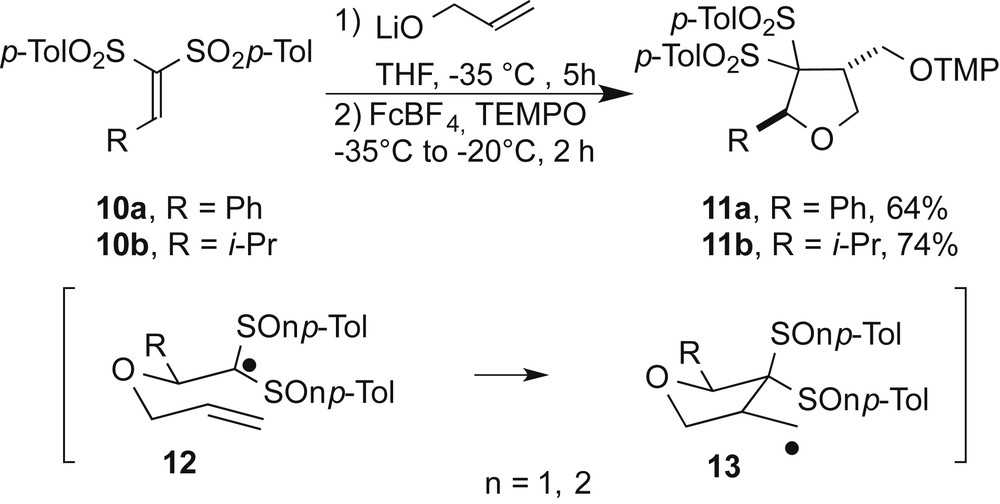
Synthesis of bis-sulfonyl tetrahydrofurans.
Adduct 9aa was engaged in different oxidation reactions to transform the sulfoxide groups into sulfones and obtain enantioenriched 11a. This could allow the comparison with the racemic sample 11a obtained through the bis-sulfone route. However, all attempts failed to offer the desired compound, so we investigated other derivatizations. The carbon-sulfur bond is known to be reduced under electron transfer conditions by metallic complexes. Thus, both compounds 11a and 9aa were reacted with 7 equiv. of the SmI2/HMPA complex in the presence of tert-butanol (7 equiv.) (Scheme 4).
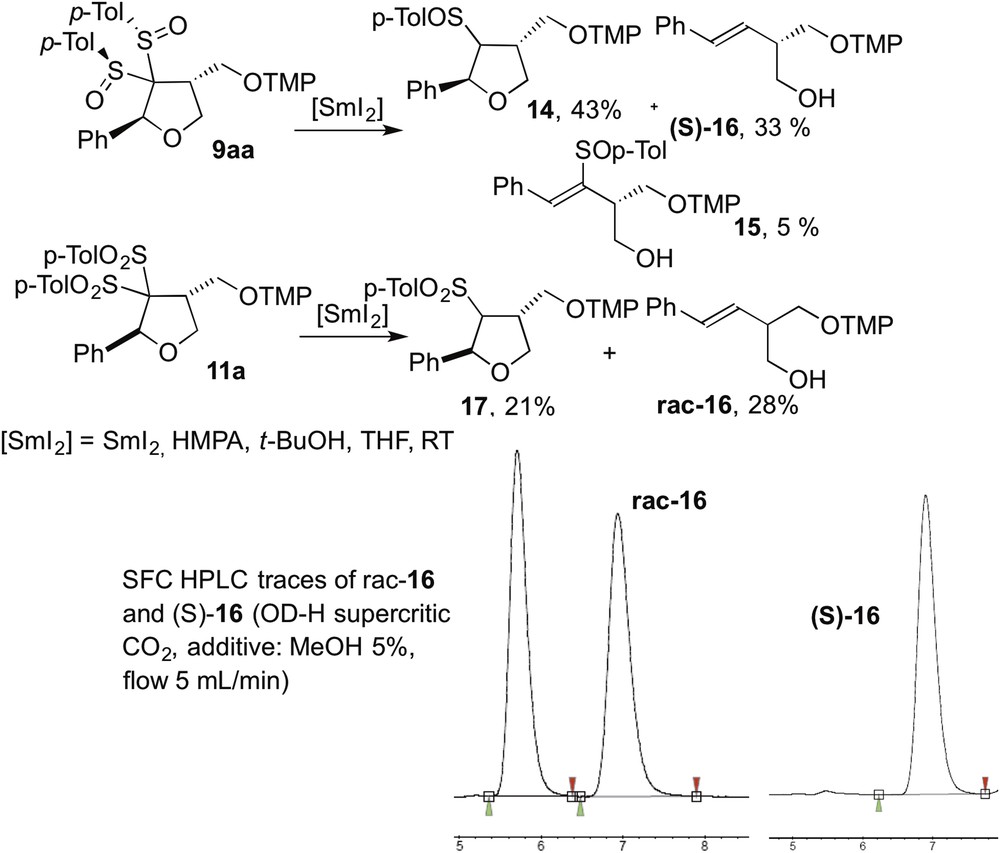
Determination of the enantiopurity of bis-sulfinyl tetrahydrofuran 9ba.
Under these conditions, bis-sulfoxide 9aa provided sulfoxide 14 as the major product, resulting from a single reductive cleavage of one carbon-sulfur bond. Presumably, over-reduction of the radical intermediate provides a carbanionic entity which β-eliminates affording ring-opened vinylsulfoxide 15 in 5% yield. Alternatively, the carbon-sulfur bond of 14 was reduced by a second electron transfer process and a similar ring-opening occurred to generate the interesting styryl derivative 16 in 33% yield. It should be noted that we cannot exclude that 15 can also be reduced into 16. A similar behavior was pointed out with the bis-sulfone analog 11a and a mixture of 16 and 17 [15] was obtained in 28% and 21% yields, respectively. Compound 16 isolated from the two reactions was used to determine the enantiomeric excess of the stereogenic center formed by the 5-exo-trig radical cyclization. The tandem process from the alkylidene bis-sulfone 10a gave 16 as a racemate whereas the enantioenriched (S)-16 was obtained from the bis-sulfoxide analog 1a. Chiral HPLC analyses showed that the enantiomeric excess of (S)-16 is 98% which confirmed the high stereoselectivity of the sequence (Scheme 4).
We next investigated the potential of this process for the creation of nitrogen-containing heterocycles (Table 2). For this purpose, allylic alcohols were replaced by N-Boc allylamines. Lithium salts 18a and 18b were generated by deprotonation of the corresponding allylcarbamate with n-BuLi at −40 °C. At that temperature, the alkylidene bis-sulfoxide was added to the reaction mixture and the conjugate addition progress was monitored by TLC. After a complete disappearance of the starting material, the ferrocenium salt was added and the temperature was increased slowly to −20 °C. After the work-up, the corresponding dihydropyrrole compounds 19a–f were obtained in moderate to good yields. Presumably, as before, SET occurs followed by a 5-exo-trig cyclization and TEMPO trapping, leading to the formation of bis-sulfinyl pyrrolidine derivatives. The latter undergoes steric strain release by a regioselective sulfinic acid β-elimination [16].
Synthesis of sulfinyl dihydropyrroles 19.
| Entry | Alkylidenes 1 | Allylcarbamates 18 | Pyrrolidines 19 |
| 1 | 1a, R1 = Ph | 18a, R2 = H | 19aa, 55% |
| 2 | 1a, R1 = Ph | 18b, R2 = Ph | 19ab, 56% |
| 3 | 1b, R1 = i-Pr | 18a, R2 = H | 19ba, 58% |
| 4 | 1e, R1 = n-Pr | 18a, R2 = H | 19ea, 73% |
| 5 | 1f, R1 = n-C12H25 | 18a, R2 = H | 19fa, 71% |
The N-deprotonated allylcarbamate 18a gave similar results to benzylidene (1a, entry 1) and isopropylidene (1b, entry 3) acceptors, as a single diastereomer was isolated in 55% and 58% yields, respectively. When the N-deprotonated cinnamylcarbamate was used (entry 2), a comparable yield was obtained and a single diastereoisomer was isolated after purification. The latter readily epimerized at the benzylic center bearing the TEMPO residue so that equimolar mixtures of diastereoisomers were observed after 1 day standing in CDCl3 [10f]. The sequence has been exemplified with different bis-sulfoxide derivatives and the lithiated carbamate 18a and in each case, a single diastereoisomer was formed.
The influence of the steric demand of the alkylidene substituent was confirmed with the use of two linear alkyl chains as substituents of the alkylidene. The propyl (1e) and the dodecyl (1f) alkylidene derivatives were engaged in the tandem sequence allowing the formation of the corresponding cyclic products in better yields (Table 2, entries 4 and 5) compared to the isopropyl group.
The following findings show some limitations of this ionic/radical tandem process when dealing with unsubstituted alkylidenes. Addition of the lithium salt of allyl alcohol or allyl carbamate followed by the introduction of the ferrocenium salt and TEMPO into the unsubstituted bis-sulfoxide alkylidene 1g did not lead to the expected cyclized compounds but rather to a complex mixture of the Michael addition product and the sulfonyl and sulfinyl compounds 21–23 (Scheme 5).
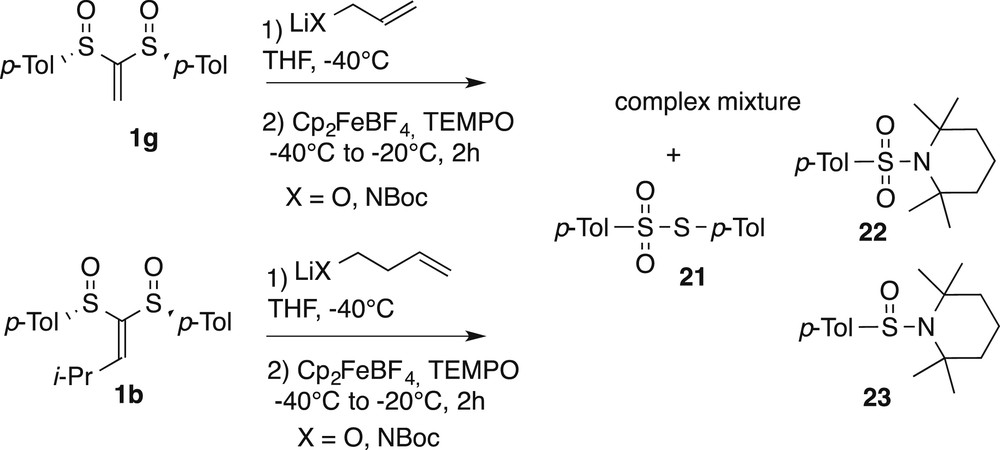
Unsuccessful radical cyclizations.
The isolation of products 21–23 suggests that SET has taken place, following a favorable conjugate addition on these substrates. But the corresponding radical instead of undergoing cyclization would decompose via fragmentation, liberating a p-Tol-sulfinyl radical, as evidenced in a previous study [17]. It is indeed documented that compound 21 arises from the coupling of two sulfinyl radicals [18], and that sulfonyl piperidine 22 [19] is a rearranged coupling product of a sulfinyl radical with TEMPO and similarly 23 a rearranged coupling product between a thiyl radical and TEMPO [17]. The radical cyclization would presumably not occur because of the lack of substituent on the connecting chain between the radical center and the vinyl group [20], a phenomenon related to the Thorpe-Ingold effect [21]. We also targeted six membered rings of tetrahydropyrans and piperidines. However, the slower 6 exo-trig cyclization was ineffective and again a complex mixture was obtained in which compounds 21–23 were detected (Scheme 6).
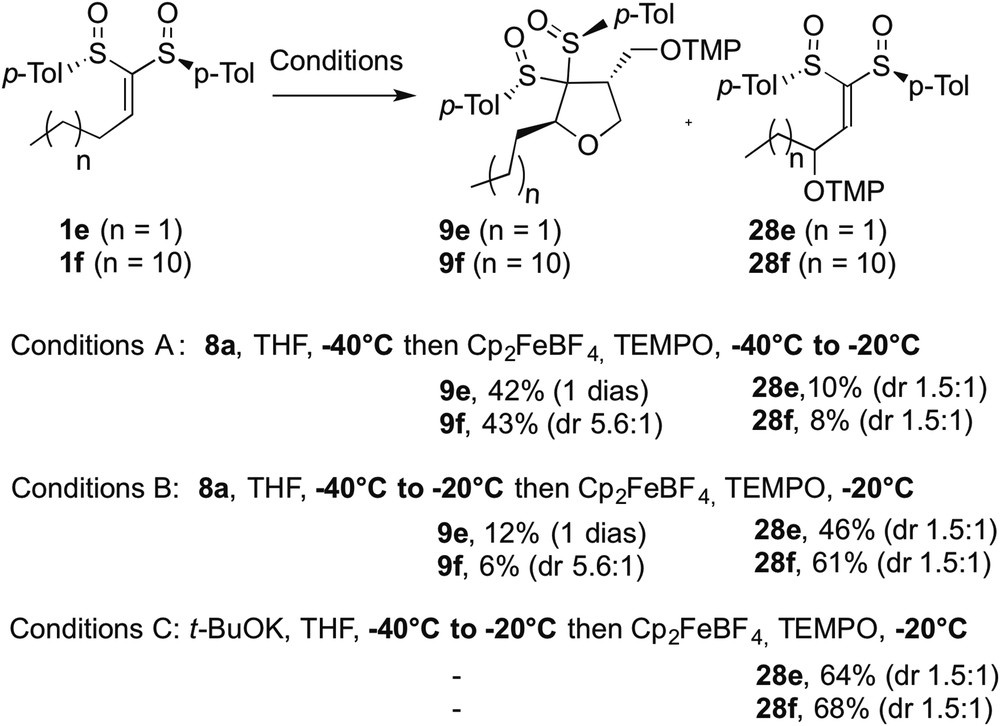
Competition between the formation of tetrahydrofurans 9e,f and allylic oxidation bis-sulfoxides 28e,f.
We have reported that ester enolates are efficient nucleophiles for the conjugate addition onto alkylidene bis-sulfoxides [13]. In this case, a C–C bond is formed instead of a carbon-heteroatom bond and could lead to the formation of carbocyclic derivatives after radical cyclization. The use of bulky tert-butyl or 2,6-dimethylphenyl substituents on the ester is important to avoid the Claisen condensation, transforming the substrate into the corresponding ketoester. Moreover, E enolates could be formed predominantly from these esters and high diastereoselectivities could be obtained from their addition onto alkylidene bis-sulfoxides. Usually higher diastereoisomeric excesses are obtained with the more hindered 2,6-dimethylphenyl [22]. Therefore, the corresponding aryl pentenoate 24 was engaged in the tandem process (Table 3). The enolate was generated by deprotonation of 24 with LDA and a diastereoselective conjugate addition at −78 °C in THF led to the formation of the anti adduct due to the formation of the E enolate.
Synthesis of sulfinyl cyclopentane derivatives 25–27.
| Alkylidene | Yield | 25:26:27 |
| 1a, R = Ph | 81% | 1.8:1:1.6a |
| 1b, R = i-Pr | 89% | 0:1.5:1 |
a 0:1:1.7 after 3 days in CDCl3.
The subsequent addition of TEMPO and ferrocenium initiated the radical process of the tandem reaction to afford the bis-sulfinyl cyclopentane 25a as a single diastereoisomer in the mixture with the two sulfinyl cyclopentenes 26a and 27a in 81% yield and a respective ratio of 1.8:1:1.6. Cyclopentenyl derivatives 26a and 27a originate from the bis-sulfinyl compound 25a following two different modes of β-elimination of sulfinic acid [16]. The mixture ratio was modified after three days in CDCl3. A complete disappearance of 25a was observed and the composition was stabilized as a 1:1.7 mixture of 26a and 27a. All our attempts to crystallize 25a were unsuccessful due to the fast elimination processes.
We gained insight into the chemical structure and the relative stereochemistry of the reaction products by an X-ray crystal structure analysis of 27a (Fig. 1) [23]. The ester and the CH2OTMP substituents show a cis relative relationship which is in agreement with the enolate addition model and the stereoselectivity of the 5-exo cyclization.
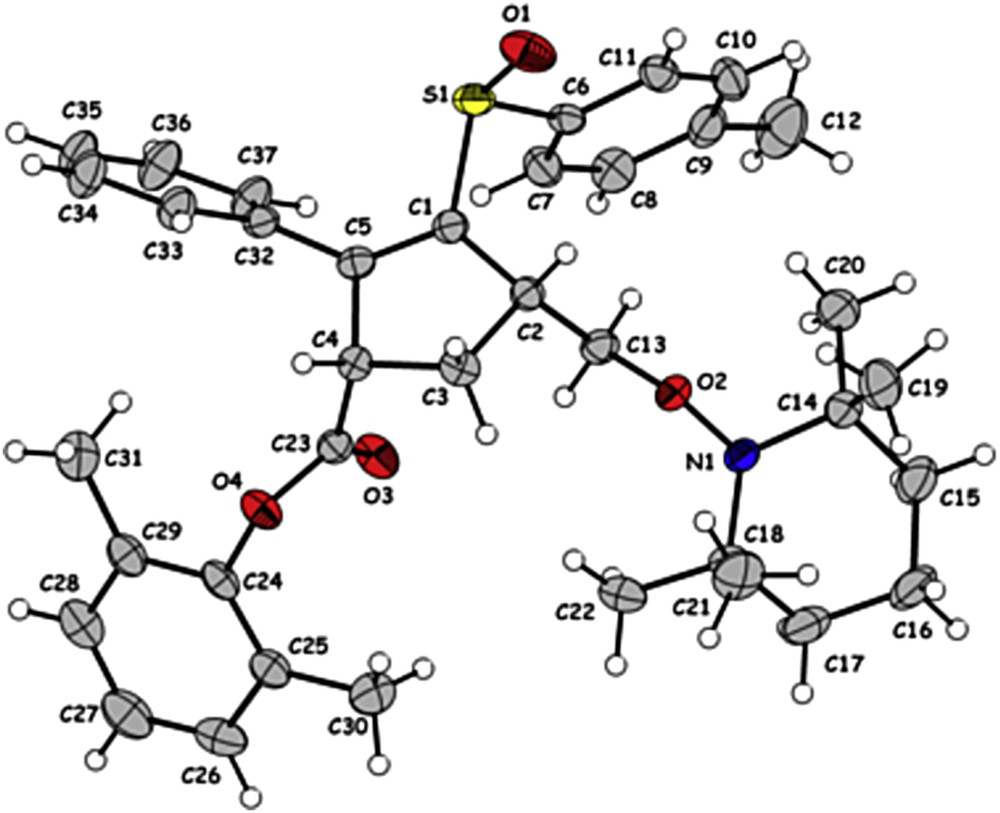
X-ray crystal structure of 27a.
When the phenyl group of 1a was replaced by an isopropyl, the eliminations occurred more rapidly to afford a mixture of 26b and 27b in 89% yield in a 1.5:1 ratio (inverse to the 26a:27b ratio), while the bis-sulfinyl compound was not detected in the reaction mixture.
In studying the scope of the tetrahydrofuran synthesis (see above), a very interesting temperature effect was evidenced with primary substituted alkylidene bis-sulfoxide (e.g., 1e and 1f) and with lithium salt of allylic alcohol 8a as a nucleophile. In a first series, lithium alkoxide 8a was reacted with propylidene and dodecylidene bis-sulfoxides 1e and 1f at −40 °C and the conjugate addition was monitored by TLC (conditions A). After the disappearance of the starting material, the ferrocenium salt and TEMPO were added at −40 °C and the temperature was increased to −20 °C. These conditions allowed the formation of the cyclic derivatives 9e and 9f as the major products in 42% and 43% yields, respectively; 9e was obtained as a single diastereoisomer whereas a 5.6:1 mixture was recorded for 9f. Interestingly, allylic oxidation products 28e and 28f were isolated in 10% and 8% and as a 1.5:1 mixture of diastereoisomers. We then discovered that a slight modification of the temperature events led to a dramatic modification of the reaction outcome. When conditions B were applied, corresponding to the addition of the oxidant at −20 °C instead of −40 °C, the proportions of 9e/9f and 28e/28f were reversed affording mainly the oxidation compounds. Thus, 28e was formed in 46% and a better yield of 61% was found for 28f. The minor cyclic products 9e and 9f were not obtained at higher yields than 12%, with similar diastereoselectivity as recorded under conditions A (Scheme 6).
To rationalize the formation of 28e and 28f, we proposed the mechanism shown in Scheme 7. At low temperature, the conjugate addition of 8a to 1 affords the carbanionic intermediate 29. The subsequent oxidation of this anion into the corresponding radical 30 followed by a radical cyclization and a spin-trapping generate the cyclic compound 9. If the temperature is increased, a retro-Michael addition would occur regenerating the allylic alkoxide that can react as a base to form allylic carbanion 31 and allylic alcohol in equilibrium with 1 and 8a. This equilibrium can be shifted by a single electron oxidation converting 31 into 32. Then, the so-formed allylic radical 32 reacts with TEMPO to give allylic oxidation compound 28. To support this mechanism, the lithium salt of the allylic alcohol was replaced by potassium tert-butoxide, proving to be unreactive toward the conjugate addition onto alkylidene bis-sulfoxides (conditions C, Scheme 7). tert-Butoxide only acted as a base and under the same temperature conditions as conditions B, products 28e and 28f were isolated in 64% and 68% yields, respectively.
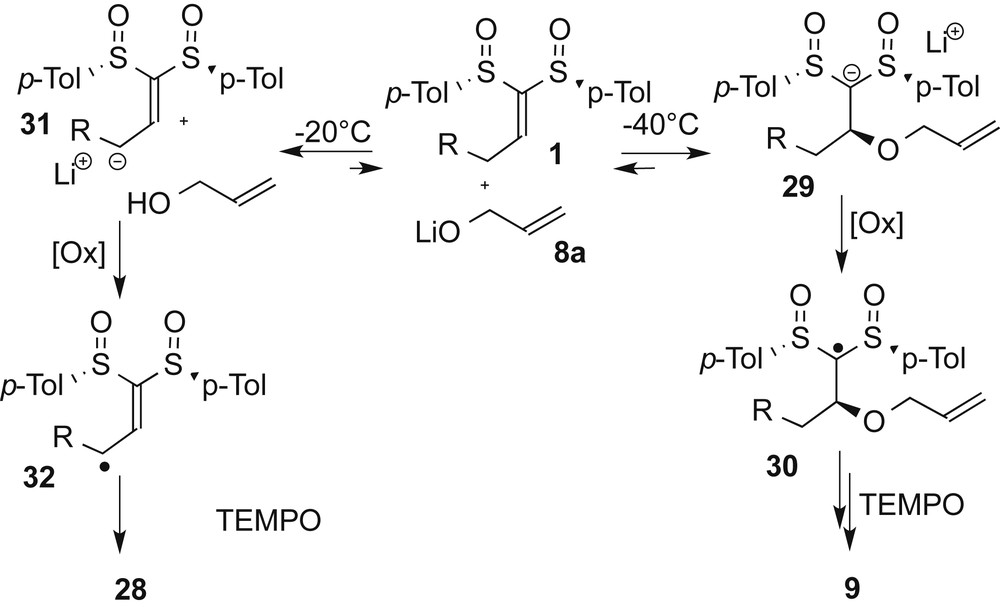
Divergent pathways for the synthesis of products 9 and 28.
The retro-Michael addition was also illustrated by the reaction of 1h with the benzylic lithium alkoxide, known as a very efficient nucleophile for the conjugate additions onto the alkylidene bis-sulfoxide [13] (Scheme 8). After the TLC monitoring of the reaction has shown a complete conversion of 1h, the oxidant and the spin trap were added at −40 °C and the temperature was increased to −20 °C. Under these conditions, only 28h was isolated in a high yield. The 6-exo-dig cyclization product was not observed and the retro-Michael process was almost exclusive. Moreover, this oxidation showed a high selectivity in favor of the allylic position versus the propargylic one.
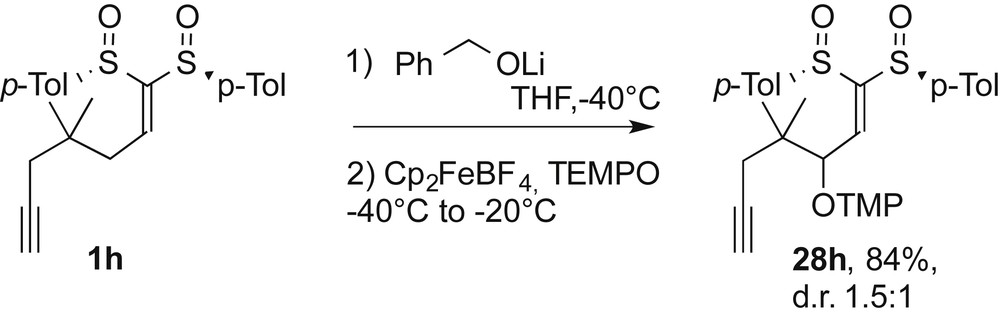
Synthesis of 28h.
Finally, we focused on the use of dienyl-bissulfoxide 1i. Our plan was to oxidize the carbanion resulting from the 1,6-addition of a nucleophile. We first tested this idea with lithium ethoxide. Gratifyingly, [1,6]-conjugate addition of lithium ethoxide onto dienylidene bis-sulfoxide followed by the addition of iron(III) and TEMPO allowed the formation of ethylether 33 in 65% yield with a 1.2 diastereoisomeric ratio (Scheme 9). One can imagine the synthetic potentiality of such transformation in the preparation of 1,2 diol or aminoalcohol. Cuprate reagents could also be engaged in this type of transformation [24], providing good yields of TEMPO adducts 34–35. Further investigations are in progress in order to improve the diastereoselectivity and the versatility of such process.

1,6-addition – oxidation tandem.
3 Conclusion
In conclusion, we herein report the utilization of alkylidene bis-sulfoxide derivatives in ionic/radical tandem reactions. Alkoxides, carbamates and ester enolates could be introduced in a [1,4]-addition. Radical cyclization induced by single electron transfer from iron(III) salts could then respectively lead to enantiopure tetrahydrofurans, dihydropyrroles and cyclopentenes. Almost all of the cyclization compounds were isolated as single diastereomers thanks to highly stereoselective processes. An interesting allylic oxidation process resulting from allylic deprotonation and oxidation or from [1,6]-addition of alkoxides or organocopper reagents followed by oxidation was discovered.
Acknowledgments
The authors thank UPMC, CNRS, IUF (MM & LF), ANR CREDOX (ANR-10-BLAN-0701) & Radicaux Verts (ANR-06-BLAN-0309) for financial support. Special thanks to Lise-Marie Chamoreau for the X-ray analyses of 17 and 27a.