

1 Introduction
Any heterogeneous catalyst works within a certain range of pressure and temperature, beyond which the catalyst fails to show efficient activity. The most important part of the catalyst development, therefore, is to design a catalyst which shows activity in a broad range of pressure and temperature. Modifications of the catalyst structure and morphology, change of supports, change of active metals are basic parameters which are employed and investigated to broaden the catalysis regime of a catalyst. However, these changes could bring into expensive modifications to catalyst synthesis as well as in the reactor requirements. Without changing its material elements, can we alter the boundary conditions of a heterogeneous catalyst in a simple manner. This seemingly elementary question however turns out to be difficult to answer without addressing the particular reaction system. The reason is the role of reactants imparting influence in the catalyst surface modification (SM). SM of a heterogeneous catalyst can influence its catalytic activity to shift the boundary conditions and this can be used as a handle to tune the activity regime of a catalyst. The elementary steps of heterogeneous surface reactions, such as adsorption and desorption, are simultaneous bond making and bond breaking processes, and they depend on the electronic changes of a surface. If the SM of a solid catalyst can induce electronic changes in the catalyst surface, we believe, that it can also change the activity of the catalyst. The present manuscript shows the shift in activity regime of Pd due to SM in the CO oxidation and reduction of NO by H2.
CO oxidation and NO reduction are main reactions which take place in a three-way catalytic converter (TWC), and Pd is well-known metal used in it. However, like other noble metals, Pd also is ineffective for CO oxidation below 400 K because of strong chemisorption of CO to Pd, and this phenomenon is known as “CO poisoning”. The other aspect is high dissoluting nature of Pd for small species like C, H, and O [1–3]. Especially when oxygen is involved in the reaction, the situation is intricate. The atomic oxygen diffuses into Pd to subsurfaces and/or bulk regions depending on the severity of the reaction conditions. Atomic O diffusion into Pd subsurfaces was observed at and above 500 K by many groups [4–11], and the extent of O diffusion increases linearly up to 900 K, though surface O2 desorption reaches a maximum at approximately 750 K for Pd(111) surfaces. Lundgren et al. [12] recently reported CO adsorption patterns on different Pd surfaces and the nature of Pd during the CO oxidation reaction under in situ conditions. Kinetic and operando studies on NO reduction are also reported by Granger et al. [13]. However, the role of dissolved O in the catalytic activity of Pd has not been explored. Herein we report the role of subsurface oxygen in the catalytic activity of Pd in CO oxidation and selective catalytic reduction of NO by H2.
2 Experimental section
The kinetic experiments were carried out in a home built molecular beam instrument (base pressure 2 × 10−10 Torr). The details about the system and molecular beam generation set up can be found elsewhere [14, 15]. In brief, the molecular beam instrument is a cylindrical chamber equipped with a turbo molecular drag pump, a doser assembly with a microcapillary, a four axis manipulator for sample manipulation, an argon sputter-ion gun, and a quadrupole mass spectrometer (QMS). The QMS was placed out of site of the beam to avoid angular effects from the single crystal as well as direct interaction with the reactants. The effusive molecular beam doser consists of a 13 mm disk multichannel array made up of microcapillary glass tubes of 1 mm in length and 10 μm in diameter each (Collimated Holes Inc), which helps to maintain a very high beam profile. The gaseous reactants and products were detected using the QMS. Pd(111) single crystal (8 mm diameter and 1 mm thick, MaTeck, Germany) and Pd foils (MaTeck) were spot welded on a 0.5 mm thick Ta wire. The samples were resistively heated and the heating was controlled by using a homemade temperature controller fitted with a PID control unit (eurotherm). The temperature was measured using a Cr–Al (K type) thermocouple welded on the periphery of the Pd sample. Pd surfaces were cleaned by Ar+ sputtering in an oxygen environment (total pressure 1.5 × 10−6 Torr) at 900 K followed by flashing at 1100 K. All gases were used with high purity and without any further filtration. In this set up the reactants are mixed first in the gas manifold and the pressure is monitored by using a baratron gauge. The reaction mixture is introduced as a molecular beam through the doser assembly, controlled by a variable leak valve and the leak rate is always kept identical for the whole set of reactions.
The ambient pressure experiments were carried out in a custom built near-ambient pressure photoelectron spectrometer (NAPPES) (Prevac, Poland). In this set up, XPS can be performed up to 1 mbar and UVPES up to 0.3 mbar with an analyzer cone aperture of 0.8 mm. Full details about the system is available in Refs. [16,17]. The analysis chamber equips a differentially pumped high pressure electron analyzer (VG Scienta, R3000 HP), a monochromatic Al Kα X-ray source (VG Scienta, MX 650), a dual anode (Al Kα, Mg Kα) source, and a helium UV discharge lamp. The reactant gases are introduced into the analysis chamber through a variable leak valve. The samples can be cleaned by ion sputtering in the preparation chamber, which is connected to the main analysis chamber, and is separated by using a manual gate valve.
3 Results and discussion
3.1 Surface modification
The clean Pd(111) surface was modified with oxygen in the subsurface layers by direct dosing of O2 beam for 10–110 min at 900 K with an oxygen flux of 0.5 ML s−1. CO titration was carried out at 600 K to remove any surface oxygen, after the above surface modification. A significant subsurface population starts from a dosing period of 10 min and then gradually improves until a threshold/saturation coverage of 0.45 ML in subsurface layers [11]. Pd surfaces with 50 min of O2 dosing shows a significantly higher activity compared to the virgin Pd surfaces. The results reported in the present communication is after surface modification by dosing O2 beam (flux = 0. ML s−1) at 900 K for 50 min. Fig. 1 shows a typical surface modification experiment in the molecular beam set up for 20 min of dosing with labelled oxygen (18O2). All relevant mass numbers (12, 18, 32, 36, 28, 44 and 46 amu) were followed during the course of the reaction. The CO titration, followed by oxygen dosing, does not produce any significant isotopic CO2 (C18O16O) as evidenced from the 46 amu signal. CO + O2 reaction carried out, after the above titration, exclusively produces CO2. After the above reaction, temperature programmed desorption (TPD) was carried out with a heating rate of 5 K/sec (not shown). 18O2 desorption was seen at higher temperatures around 1000–1100 K. A similar observation was recorded by Leisenberger et al. [18] from TPD spectroscopy, where they found chemisorbed oxygen desorbs around 750 K, whereas the subsurface oxygen desorbs at higher temperatures (>1000 K). Two main points can be summarized from the experiment: (a) the subsurface oxygen does not diffuse out to the surface or is consumed during the CO titration, which emphasizes its non-participation in the reaction. (b) The subsurface oxygen is stable atleast up to 900 K. Any reaction carried out on the above surface below 900 K is unlikely to affect the subsurface oxygen population.
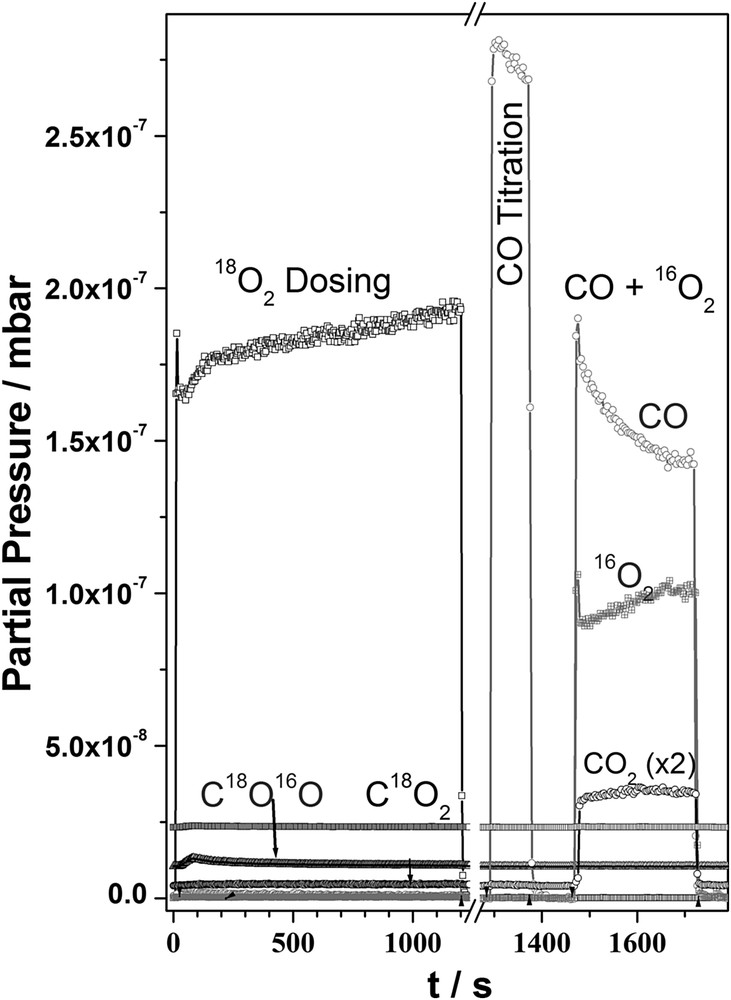
Surface modification of Pd(111) by dosing labelled 18O2 in a molecular beam setup. 18O2 was dosed at 900 K for 20 min, followed by CO titration at 600 K to remove any surface adsorbed oxygen. A 1:1 CO + 16O2 molecular beam was dosed on SM-Pd to show that subsurface oxygen does not diffuse to form C18O2 or CO18O. Labelled CO2 traces are vertically shifted to avoid the overlap with other traces.
3.2 NO + H2 + O2 reactions on SM-Pd
We carried out the NO + H2 + O2 reaction on surface modified Pd (represented as SM-Pd) and the results were compared with that of virgin Pd surfaces (Fig. 2). The kinetic experiments were measured with NO + H2 + O2 (1:1:1) from 325 K to 700 K. All the measurements were carried out in an identical manner mentioned in our earlier publications [19, 20]. The sample was kept at an initial temperature and then a beam of reactants NO + H2 + O2 was turned on; the reaction reaches to the steady state (SS) within a very short period (about 60s), and the steady state rate of formation was determined by deliberately closing and opening the shutter (i.e. blocking and releasing the beam to interact with the sample). Once the SS rate of the reaction has been measured for a particular temperature, the temperature of the sample was increased by 50 K. This process was repeated up to 700 K. Fig. 2 shows the comparison of the rate of formation of all the products N2, N2O, NH3 and H2O for 1:1:1 NO:H2:O2 beam composition on the virgin Pd(111) surface and on SM-Pd(111) at different temperatures. The result shows a higher rate of formation of N2 for SM-Pd(111) compared to virgin Pd(111) surfaces. Not only product enhancement, it is also worth mentioning that the onset temperature for SM-Pd(111) shifts to 325 K. Whereas the reaction starts at 450 K on virgin surfaces and reaches the maximum at 550–600 K (Fig. 2). Indeed the decomposition temperature of NO on Pd(111) surfaces is known to occur above 300 K, but nitrogen desorption occurs above 400 K [14,21].

A comparison of NO + H2 + O2 reactions performed on (a) virgin Pd(111), and (b) surface-modified Pd(111) between 700 and 325 K. All the products (N2, H2O, NH3, and N2O) formation and response to beam oscillation are shown. The value of the y-axis range remains the same on both panels.
Fig. 3 shows the rate of formation of all the products at various temperatures for the 1:1:1 reaction on SM-Pd(111). The results were compared with virgin Pd surfaces. It is very appealing for lean de-NOx at colder conditions with a large amount of oxygen content (33%) in the beam. The formation of N2 and H2O can be seen up to 325 K, while no NH3 could be observed below 450 K. A shift in rate maximum towards low temperatures, at least by 50 K, was observed for all the products on SM-Pd(111), compared to virgin surfaces. Further the rate maximum also broadens to large temperature range highlighting the influence of surface modification on the fundamental aspects of adsorption, dissociation of reactants and product formation. These experiments are repeated multiple times to confirm the low-temperature activity, due to the fact that subsurface oxygen does not diffuse out or take part in the reaction. Otherwise, a sustainable and stable product (N2 or H2O) formation would not have been observed.

Steady state rate of the formation of (top panel) N2, and H2O, and (bottom panel) N2O, and NH3. Solid and open symbols for the rates obtained on SM and virgin Pd(111) surfaces, respectively.
3.3 CO + O2 reactions on SM-Pd
Our earlier reports on CO oxidation [10, 11] highlighted that subsurface diffusion of oxygen in palladium led to an altered catalytic activity toward CO oxidation, and the modified surfaces show CO oxidation activity even at 900 K, whereas virgin Pd(111) shows no significant activity at and above 700 K [22]. We investigated the impact of SM- Pd toward low-temperature CO oxidation. The results obtained are very promising. The SS rate of CO oxidation is presented in Fig. 4 for the CO:O2 ratio of 7:1 to 1:7 from temperature 300–525 K for virgin and SM-Pd(111) surfaces. CO oxidation activity can be observed from 400 K and above on virgin Pd(111) surfaces, which is in good correlation with literature reports [11,22]; whereas CO oxidation activity can be observed from 300 K and above for any ratio of CO:O2. This observation demonstrates the capability of SM-Pd in ambient temperature CO oxidation aspect. For virgin surfaces the activity maxima can be seen toward high temperatures (around 475–550 K). With increase in SM, the onset of the reaction shifted toward lower temperature. Oxygen rich beams show higher shifts in the rate maxima. The 1:1 composition on SM-Pd shows maxima at 400–450 K, whereas the 1:2 beams shows the maxima around 350–400 K. However, with oxygen increment, the overall rate of the reaction decreases due to the decrease in the overall CO content. It is also to be noted that the 1:1 beam composition shows a significantly higher rate of CO2 production than 1:2; we attribute this deviation from the Langmuir–Hinshelwood mechanism to significantly higher sticking coefficient for CO than O2 [11,22].
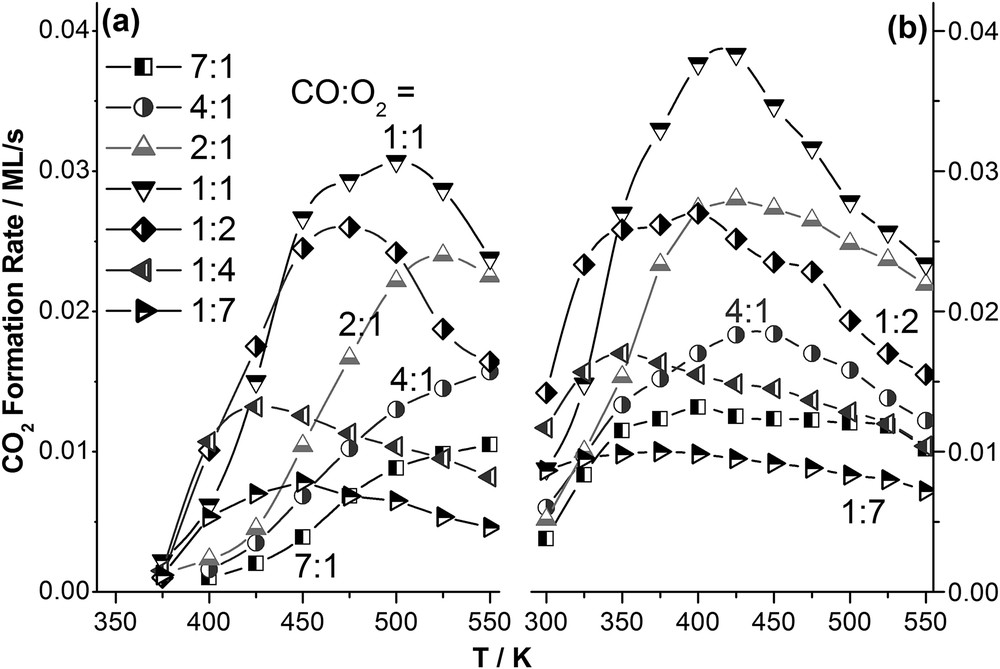
The SS rate of the formation of CO2 on (a) Pd(111), and on (b) SM-Pd(111) surfaces. The CO:O2 composition varied between 7:1 to 1:7.
To investigate the sustainability of the activity due to SM, the reaction was carried out for a long period (1 h) at isothermal conditions, T = 325 K. Fig. 5 shows the SS production of CO2 at 325 K with CO: O2 (1:x, x = 2, 4, 7) beam compositions on SM-Pd(111) for a period of 1 h. During the course of the reaction, multiple shutter operation was performed to determine the SS rate of the reaction at 325 K. A clear CO and O2 uptake (not shown) and CO2 formation underscores the fact that the reaction is sustainable at 325 K. If the effect is transient and the subsurface oxygen would have participated in the reaction, we believe that this continuous sustainable CO2 formation would not have been observed. The rate of CO2 formation is the same as given in Fig. 4 for all CO:O2 compositions.

CO + O2 reaction with different compositions, (a) 1:2, (b) 1:4, and (c) 1:7) carried out on SM-Pd(111) at 325 K for 60 min.
3.4 Ambient pressure photoelectron spectroscopic study of SM-Pd
Virgin Pd surfaces do not show activity for NO reduction below 400 K, even though NO dissociation occurs but nitrogen desorption requires 400 K or above [14,21]. On the other hand, for CO oxidation reactions activity below 400 K is hindered because of the “CO poisoning” of Pd metal. To investigate the role of SM-Pd in low-temperature catalytic activity, photoelectron spectroscopy studies were carried out under near-ambient pressure conditions. Before exploring the CO + O2 reaction system, we tried to understand the Pd-oxygen (gas-solid) interaction at different pressure regimes. Palladium is known to form mainly three kinds of oxides- surface oxides, Pd5O4 which are metastable, subsurface oxide, and bulk oxide. To sort out the stability aspects of subsurface oxide, we chose a wide pressure and temperature regime for SM. We also matched molecular beam SM conditions by keeping a similar oxygen partial pressure and Pd surface temperature in the ambient pressure XPS set up to confirm subsurface oxide formation. O2 interaction with Pd(111) surfaces are presented in Fig. 6 under molecular beam conditions as well as under near-ambient pressure (0.07 mbar) conditions. The UV photoelectron spectrum at ultrahigh vacuum (UHV) conditions shows a Pd 4d doublet with strong Fermi level intensity, which is a characteristic of the clean Pd-metal. Upon introduction of oxygen, a feature at around 5.7–6 eV (dashed line in Fig. 6) appears when heated up to 650–900 K at 1 × 10−5 mbar pressure. Fermi level intensity decreases and a clear broadening of the Pd 4d levels could be observed. The work function increases from 6 eV for virgin Pd(111) surfaces to 6.4 eV for SM-Pd surfaces demonstrating the change in surface nature. On prolonged exposure of oxygen under this condition, the feature remains the same (not shown). The system was evacuated after oxygen exposure and further spectra were recorded. The feature at 5.8 eV remains stable on evacuation even up to 900 K heating, and the valence band features are restored back similar to the metallic state. This experiment clearly indicates that the feature cannot be due to metastable surface oxides or bulk oxides. The increase in oxygen pressure broadens the feature at 5.7–6 eV, and a prolonged exposure (150 min) at 0.07 mbar and 675 K develops a feature at 6.4 eV (dotted line in Fig. 6), which we assign to PdO-like surface oxide. XPS spectra also confirm a distinct peak at 336.5 eV (see Supplementary data, Fig. S1). We carried out CO dosing to check the stability of the feature between 5.7 and 6.5 eV. After oxygen dosing at 0.07 mbar, the system was evacuated and 0.07 mbar CO was introduced at 675 K. Interestingly, the feature at 6 eV shows no sign of disappearance. Neither heating in the presence of CO nor heating under UHV conditions up to 900 K changes the intensity of the above feature. We attribute this feature to be subsurface oxide, and hence Pd surfaces with this kind of attribute were represented as SM-Pd and they showed ambient temperature catalytic activities for CO oxidation and NO + H2 + O2 reaction.
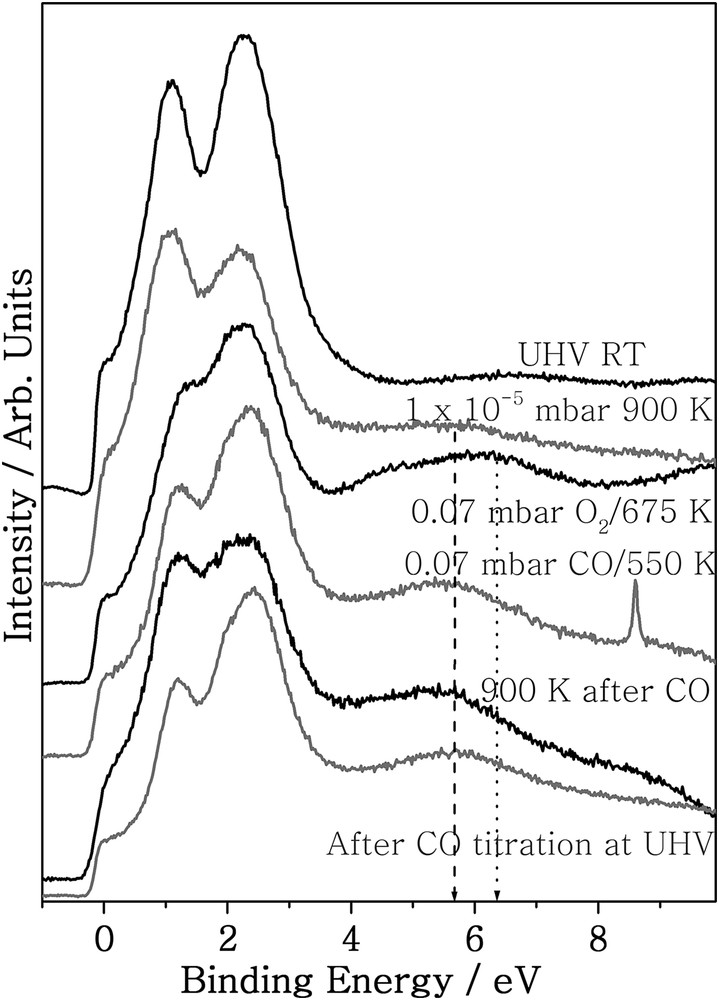
UVPES spectra recorded for Pd–O2 interaction at various temperatures and oxygen pressures are shown. Dashed and dotted lines indicate the formation of subsurface oxide upon Pd-O2 interaction. A sharp molecular vibration feature from CO can be seen at 8.62 eV.
The CO oxidation reaction was followed in-situ by APPES. Fig. 7 shows the He I UVPES spectra measured at 350 K (total pressure: 0.1 mbar) on clean Pd(111) and on SM-Pd with a 2:1 ratio of CO:O2. Deliberately the CO-rich composition was employed to explore the CO-poisoning aspects on virgin and SM-Pd(111) surfaces. The unique aspect of photoelectron spectroscopy under ambient conditions is that the gas–solid interactions can be appreciably understood from the gas phase vibrational features. On SM-Pd surfaces, gas-phase O2 and CO vibrational features appear at 6.4–7.4 and 8.35 eV, respectively. CO2 gas phase features are distinctly observed at 12.4–12.9 eV during the CO + O2 reaction on SM-Pd. To verify this feature reference CO2 gas phase spectra were also recorded and it is in good correspondence with the spectra observed above. The core-level C 1s spectrum recorded under the same NAP reaction conditions exhibits gas-phase CO2 at 291 eV and adsorbed CO at 286 eV (Fig. 7, inset) [7]; this observation unequivocally demonstrates CO2 production at 350 K on SM-Pd(111). We believe that oxygen diffusion into the subsurface layers makes the Pd surface to be marginally oxidized. Due to the oxidized character, there is no back-donation of electron on CO adsorption. This makes the adsorbed CO to be mobile, in comparison to virgin surfaces, and hence facilitates CO oxidation at ambient temperatures. Nonetheless, further studies, such as in-situ IR, are required to confirm this aspect. The above APPES results demonstrate CO oxidation on SM-Pd and are in resonance with the MBI results. However, no CO2 feature was observed on virgin Pd(111) surfaces under similar reaction conditions. It is also to be noted that the intensity of the first Pd 4d feature at 1.2 eV seems to be very susceptible to CO and O2 and the same can be seen in Figs. 6 and 7. Under the CO + O2 environment, the first Pd 4d feature seems to be completely suppressed and we do not know the reason for this. Another interesting change is the shift in the binding energy of molecular vibration features of CO and O2 towards lower energy by 0.25–0.35 eV on SM-Pd(111) compared to the virgin Pd(111) surface. The above shift in energy of vibrational features is not simply due to work function changes. It is also to be underscored that while C–O vibration energy remains the same (200 meV) [23] on virgin and modified surfaces, the electronic state is shifted to lower BE. Shift towards lower binding energy underscores the higher reactivity of the surface due to surface modification. Further, it also reflects the change in surface potential by 0.25–0.35 eV, after surface modification. All these observations highlight the changes observed in catalysis and spectral aspects due to surface modification are in good correspondence. It is worth exploring this aspect for different surfaces as well as different reactions.
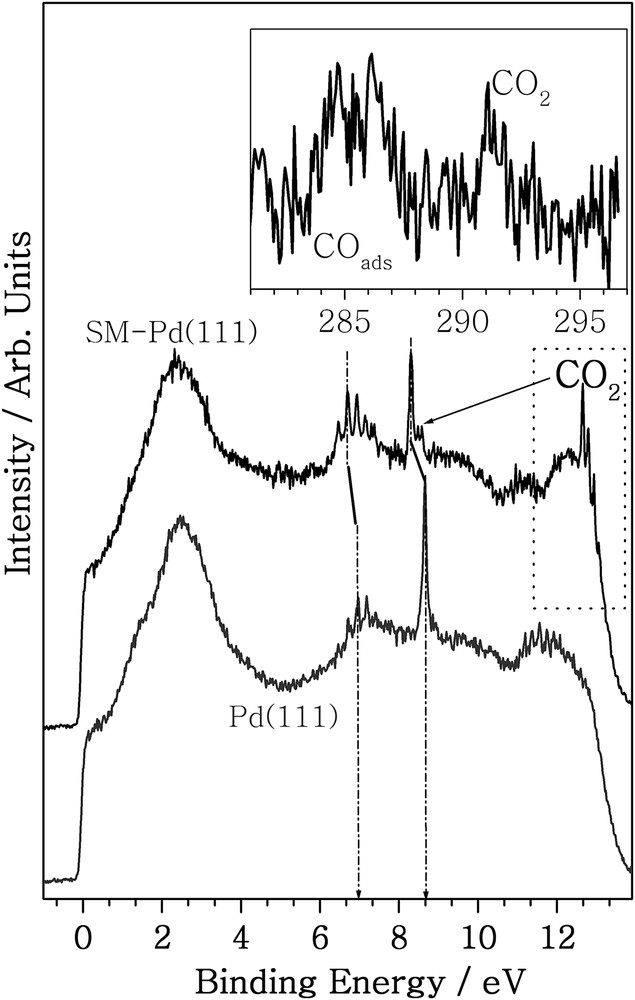
UVPES spectra measured while the CO + O2 (2:1) reaction was carried out on virgin Pd(111) and SM-Pd surfaces at 350 K at a total pressure of 0.1 mbar. Molecular vibration features of CO2 directly observed on SM-Pd is shown in the rectangular dotted box, which demonstrates CO2 production. The inset shows the core-level C 1s spectra under the same conditions for SM-Pd.
4 Conclusion
This study, along with our previous reports [10,11,19,24], highlights that subsurface population of oxygen definitely plays an important role in the catalytic activity of Pd. The reaction temperature extends to ambient regimes upon SM. SM-Pd shows activity from 325 K onwards for NO + H2 + O2 reaction and for CO oxidation. However, we do not generalize this aspect that all of the reactions are catalyzed by Pd, and moreover investigations are pending to establish this aspect over ranges of Pd-based powder catalysts under practical catalysis conditions.
Acknowledgements
We dedicate this manuscript to Prof. Edmond Payen, UCCS, USTL, Lille. KR, RJ, and KR and RJ thanks to CSIR, New Delhi for research fellowship. MKG and KPR thanks to UGC, New Delhi for research fellowship. We thank SERB (SR/S1/PC-16/2012) and BRNS (2011/37C/36/BRNS) for funding the present research project.