

1 Introduction
UV and visible photons have an energy content comparable to that of chemical bonds and their absorption causes the promotion to an electronically excited state [1]. The deep change in the electronic structure and the high energy of these states open up different chemical paths and thus make possible the selective transformations of synthetic interest. Photochemical reactions occur under very mild conditions and avoid the use of heating and/or of aggressive chemicals, otherwise required to promote the desired reaction (as in thermal processes), and further reduce the attending waste produced. In turn, this simplifies work up and purification procedures [2].
The differences between photochemical and thermal reactions and the role of excited states have been recognized since the birth of photochemistry. As a pioneer in this area, Giacomo Ciamician remarked in his talks presented in front of the French Chemical Society in Paris in 1908 [3a] and at the VIII international meeting of Applied Chemistry in New York in 1912 [3b] that “The photochemical reactions follow the fundamental laws of affinity, but have a special character. They are especially notable for the small temperature coefficient and are, however, comparable - a fact which is not without technical importance - to the reactions which take place at very high temperatures. According to a brilliant idea of Plotnikow, luminous radiations produce a different ionization from that due to electrolytic dissociation; the separation of an ion requires a quantity of light which is determined by the theory of Planck and Einstein. The question is therefore related to the most recent and profound speculations of mathematical physics” [3b]. These peculiar characteristics were bound to give a fundamental contribution to synthesis. In “the field of organic chemistry, the reactions caused by light are so many that it should not be difficult to find some which are of practical value. The action of light is especially favorable to processes of reciprocal oxidation and reduction which give rise to or are associated with phenomena of condensation. Since the common condensation is that of the aldolic type there is much hope for the future, the aldolic condensation being the fundamental reaction of organic synthesis. […] The simplest case is that of the action of light on a mixture of acetone and methyl alcohol in which isobutylene glycol is produced. But this condensation which may be considered as a simultaneous process of oxidation and reduction, is accompanied by the reduction of the ketone to isopropyl alcohol and by the oxidation of the methyl alcohol to formaldehyde” [3]:
The above reaction is a condensation that does not require, contrary to its thermal counterpart, the addition of a base. This is because these reactions occur via excited states that may be considered electronic isomers of ground states and have their own structure and thermodynamics. The energy of such states is in the same order of chemical bonds and often the chemical bonds can be cleaved and intermediates are generated. In turn, the last species use their energy and form new bonds [1]. Since its birth as a discipline, photochemistry has consistently offered new paths to synthetic chemists, often complementary to thermal processes [4]. In the following, a short overview of two topics that have been developed in our lab in recent years is presented.
2 Photochemical generation of aryl cations and their application in arylation processes
Aromatic nucleophilic substitution is the key step in many synthetically useful processes via different intermediates (metal and (photo)redox assisted, both thermally and photochemically induced SRN1, nucleophilic substitution via benzyne intervention, etc.) [5]. Transition metal catalyzed reactions have been the most frequent approach to achieve this target, but still have to confront some limitations, such as the use of co-catalysts and auxiliaries, the (often) difficult work up and the recovery of the expensive catalyst. At any rate, it is important to have available simple alternative methods that avoid the use of metals for products to be used as food or drugs [6].
In most cases, substitution occurring via transition metal catalysis can be envisaged as involving the synthetic equivalent of an aryl cation, formed via oxidative insertion onto the metal center [7]. However, “real” aryl cations are not easily generated and a unimolecular SN1 mechanism is rarely operated in aromatic chemistry, in contrast to its important role in aliphatic chemistry.
2.1 Structure, generation and reactivity of aryl cations
Literature thermodynamic data show that the stability of aryl cation intermediates lays between that of a vinyl cation, C2H3+ and that of an ethyl cation, C2H5+ [8]. Another peculiarity is that these intermediates can exist in two different spin states, either the triplet or the singlet. DFT calculations on the parent phenyl cation provided evidence that in the singlet ground state (1, with π6σ0 orbital occupance) the positive charge is localized at the dicoordinated carbon, whereas in the triplet state the two unpaired electrons reside in orthogonal orbitals, viz. the σ orbital localized at the dicoordinated carbon and the π system (2, π5σ1 structure), with the positive charge delocalized over the entire ring. The geometry of the two states is also different and is affected by the nature and position of the substituent(s), if any. The singlets have a structure with a cumulene character in the C2–C1–C6 moiety, while the geometry of the ring can be planar or puckered. In contrast, a regular hexagon geometry has been computed for the ring of the corresponding triplet phenyl cations, independently from the position and the nature of the substituents present (Scheme 1a) [9].
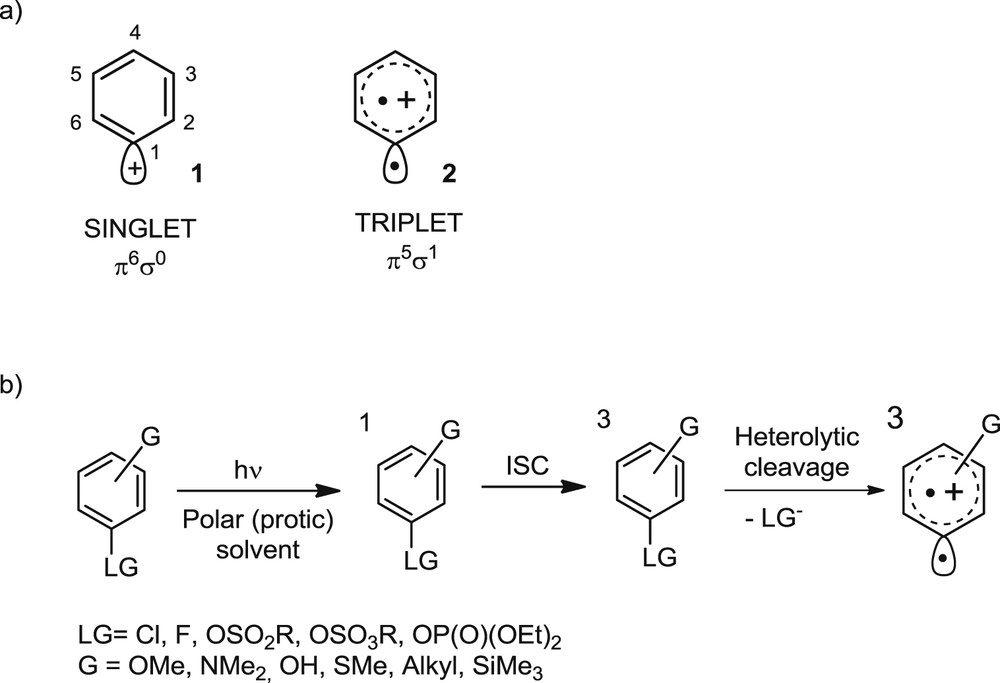
a) Electronic structure of singlet and triplet phenyl cations. b) Photochemical generation of triplet phenyl cations from electron-rich aryl halides and esters.
As one may guess, a largely differentiated reactivity has been likewise observed. Thus, singlet states behave as unselective electrophiles, often reacting with the (polar/protic) solvent in which they are generated, to result a formal solvolysis process. On the other hand, the diradical nature of triplet states causes a high selectivity towards π-bond nucleophiles (such as alkenes, alkynes and (hetero)aromatics) when present in the reaction mixture, thus leading to the formation of a new aryl–C bond, an appealing synthetic target [10].
As hinted above, the ArSN1 path is a rare occurrence in thermal chemistry, often requiring drastic conditions [11,12]. The presence of an excellent leaving group such as the nitrogen molecule makes aryldiazonium salts thermal precursors of aryl cations under mild conditions, but the generation of phenyl radicals via reduction of the starting salt efficiently competes with direct heterolysis [13]. However, even if it was possible to easily generate a phenyl cation through a thermal approach, this would lead to the singlet state, which is, as mentioned above, an unselective electrophile with limited synthetic appeal [11,12].
In contrast, the photochemical approach via an excited state starts from a high energy level and allows for the generation of intermediates with spin states different from those of the ground state (the singlet). In the last few years, we developed a method for the photochemical generation of triplet phenyl cations from aryl halides (mainly chlorides and, in selected cases, fluorides) and esters (sulfonates, sulfates and phosphates). Thus, irradiation of such compounds leads to their singlet excited state that then intersystem crosses efficiently to the triplet. When the reaction is carried out in polar and/or protic solvents, heterolysis of the aryl–halogen or aryl–oxygen bond is the favored pathway and the generated phenyl cation maintains the same spin state of its precursor (Scheme 1b) [9]. As for the solvent, polar aprotic acetonitrile is a convenient medium for substituted anilines, whereas protic methanol, 2,2,2-trifluoroethanol (TFE) and acetonitrile/water mixtures are required for the other precursors.
Heterolytic cleavage results in the Umpolung of the nucleophilic electron-rich aromatic, which is converted into an aggressive (but yet selective) electrophile. The nature and the position of the aromatic substituent(s) play a key role in the feasibility and efficiency of this process, which is maximized when an electron-donating moiety is present in the para-position with respect to the leaving group. A combined experimental/computational investigation on the feasibility of triplet aryl cation generation from differently para-substituted aryl halides (mainly chlorides) has been recently carried out by our group [14]. The data suggested that a few specific parameters in the excited triplet of the precursors allow us to predict whether the Ar–halogen bond will cleave heterolitically. These include, among the others:
- a) An out of plane deformation of the Carbon-Leaving Group bond;
- b) The development of a (partial) negative charge on the halogen atom in the triplet state of the aryl halide, when the Ar–halogen bond is elongated up to 4 Å.
The energy change associated with the Ar–X bond heterolytic cleavage nicely correlated with the substituent Hammett–Brown constant (σp+) supporting the development of a strong positive charge in the ring π system. Taking inspiration from the GSK solvent selection guide compiled by Henderson et al. [15], we represent the results obtained in our work in Fig. 1. The photogeneration of aryl cations is favored in the case of electron-rich aromatics (σp+ = −1.7÷−0.1; anilines, phenols and alkylbenzenes, the green part of the diagram in Fig. 1) but, under appropriate reaction conditions, can be extended also to substrates bearing neutral or moderate electron-withdrawing substituents (yellow) and the COOR group (σp+ = 0.49) can be taken as a reasonable edge. Thus, a large number of substrates can be used as precursors of phenyl cation intermediates [14].

Feasibility of triplet phenyl cation photogeneration from 4-substituted aryl chlorides. Only functional groups for which both experimental and computational evidences were obtained have been reported in the diagram. See the text above for further details.
In contrast, phenyl cations cannot be accessed in the case of aryl chlorides bearing strong electron-withdrawing substituents, such as 4-nitrochlorobenzene. However, in some cases this limitation can be circumvented. This is the case of chloro-benzaldehydes and acetophenones (σp+ = 0.73), the photochemistry of which involves the carbonyl function exclusively. The conversion of these substrates into the corresponding 1,3-dioxolanes (σp+ = 0.132), however, reintroduced the heterolytic photofragmentation of the aryl–Cl bond [14].
2.2 Synthetic applications of aryl cations. Metal-free arylations starting from phenyl chlorides
Triplet phenyl cations are short lived species and, when generated in a neat solvent, mainly undergo hydrogen abstraction from the solvent to form the corresponding photoreduced product. Attack of the cation onto a molecule of the precursor (to form a substituted biaryl) [9] or intersystem crossing to the singlet state [16] followed by the nucleophilic addition of the solvent, are other suitable pathways. However, in the presence of an excess (from 4 up to 20 equiv with respect to the amount of the starting aromatic) of a π-bond nucleophile, trapping of the intermediate is the main or even the exclusive pathway observed. The use of poorly hydrogen-donating solvents (e.g., TFE or aqueous acetonitrile) was found to minimize photoreduction. This resulted in the formation of a new Ar–C bond and the process was exploited by our group in the development of a wide range of arylation reactions through a photo-ArSN1 mechanism. The added value of this procedure is the use of phenyl chlorides, otherwise of limited application in metal-free arylation protocols, as it is currently restricted to a few intramolecular processes and to the photoredox catalyzed reaction developed by Koenig et al. for electron-poor aromatics [17].
As for the reactivity of triplet phenyl cations with alkenes, both computational and spectroscopic evidences support the initial formation of a triplet adduct evolving into a phenonium ion. This σ-bridged intermediate can then follow different pathways, depending on the nature of the substrates involved (both the aromatic and the olefin), as well as on the solvent employed [18]. These include elimination of an electrofugal group (from either the benzylic or allylic position; Scheme 2, paths a and a′), nucleophilic addition of either the counter ion or the nucleophilic solvent (e.g., path b) and rearrangements involving either alkyl or hydride shift (e.g., path c).
- With simple alkenes, the chemistry of aryl cations often lacks selectivity [18]. However, the use of appropriate nucleophiles makes one of the pathways predominant over the others. Thus, a good electrofugal group makes its own elimination the main path. With allyltrimethylsilane, the leaving group Me3Si+ is lost and an allylarene is efficiently formed. This method has been applied to the synthesis of naturally occurring bioactive allylphenols and anisoles from the corresponding chlorides. An added bonus of this approach is that chlorophenols can be directly functionalized avoiding any preliminary protection of the OH moiety (Scheme 3) [19].
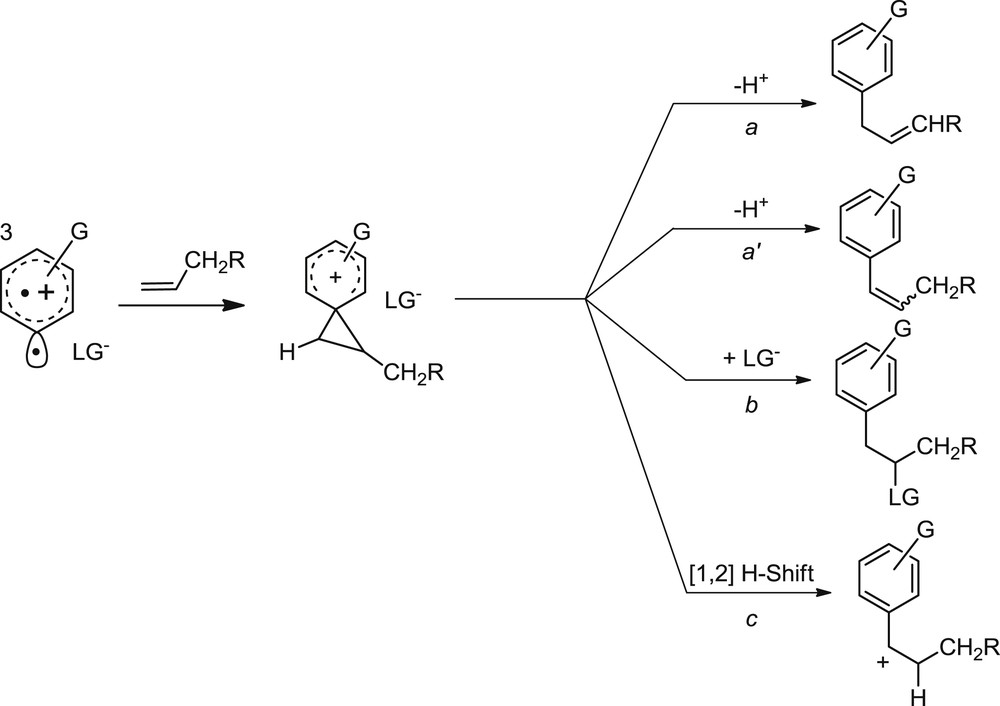
The formation of a phenonium ion via the addition of a phenyl cation onto an olefin opens several pathways.

Synthesis of allylphenols and allylanisoles via phenyl cations [19].
When using a polar, non-nucleophilic medium, the phenonium ion reacts with the counter ion which is thus incorporated into the end product, forming β-chloroalkylarenes (Scheme 2, path b, LG = Cl) [9,18]. Likewise, the addition of an aryl cation onto ethyl vinyl ether (or related compounds) results in the formation of a stabilized carbocation, where the positive charge is delocalized onto the alkoxy group present in the α-position. The addition of the (nucleophilic) alcoholic solvent resulted in a three-component process, which afforded the acetals of α-arylaldehydes or ketones in good yields (Scheme 4) [20].

Three-component synthesis of acetals via phenyl cations.
Treating the photolyzed solution with an equimolar amount of hydrochloric acid resulted in the deprotection of the carbonyl group leading to the one-pot synthesis of α-arylaldehydes and ketones in good yields (an example is shown in Scheme 5a) [21]. The same targets are also accessible through the reaction of phenyl cations with silyl enol ethers [22]. As an example, the irradiation of 4-chloro-N,N-dimethylaniline in the presence of (cyclohexenyloxy)triethylsilane afforded the corresponding 2-arylcyclohexanone in 80% yield (Scheme 5b) [22].

One-pot synthesis of α-arylketones via the reaction of triplet phenyl cations with: a) 2-methoxypropene and b) (cyclohexenyloxy)triethylsilane.
Furthermore, the reaction of aryl cations with the ketene silyl acetal prepared from methyl propanoate afforded α-aryl propionates in discrete to good yields (Scheme 6). The latter products are of interest, since α-arylpropionic acids are a well-known class of nonsteroidal antiinflammatory agents [22,23]. In the case of 4-chloro-N,N-dimethylaniline, that absorbs significantly within the solar emission spectrum, the reaction has been carried out on a gram scale under exposition to sunlight and the corresponding 4-N,N-dimethylaminophenyl propionate was isolated in 58% yield after only 6 h sunlight exposure.

Synthesis of methyl α-arylpropionate esters.
When the alkene contained a tethered chain with a neutral nucleophilic moiety, as in the case of alkenoic acids or alkenols, intermolecular trapping of the aryl cation was followed by intramolecular cyclization to give a benzyl γ-lactone (an example is shown in Scheme 7, path a) [24] or a benzyltetrahydrofuran, respectively (Scheme 7, path b) [25].

Reaction of 4-chlorophenol with: a) an alkenoic acid or b) an alkenol.
The reaction of triplet phenyl cations with alkynes was found to follow a path similar to that observed with alkenes. Thus, trapping of the intermediate by a terminal C–C triple bond led to a ring-closed vinylenephenonium ion that in turn gave the corresponding arylalkyne after proton loss (Scheme 8a). The reaction can be considered as a metal-free analogue of the Sonogashira reaction [26]. When the starting aromatic bore a hydroxyl substituent in the ortho-position with respect to the leaving group, trapping of the cation by the alkyne was exclusively followed by intramolecular cyclization to give the corresponding benzofuran, an heterocyclic moiety largely diffuse among natural compounds and pharmaceutically active molecules (see Scheme 8b) [27].

Reactivity of phenyl cations with alkynes.
Turning to aromatics, the generation of aryl cations in the presence of benzene or symmetric methylbenzenes (e.g., p-xylene, mesitylene and 1,2,4,5-tetramethylbenzene) led to (crowded) biphenyls (Scheme 9) [28]. Five-membered heterocycles, such as pyrrole and thiophene, likewise underwent selective arylation in position 2, while the corresponding 2,5-dimethyl derivatives were smoothly arylated in position 3 [29].

Synthesis of biphenyls via phenyl cations.
Finally, triplet aryl cations are useful intermediates in the synthesis of benzonitriles, an important class of compounds that finds application among agrochemicals and pharmaceuticals. As an example, irradiation of electron-donating substituted phenyl halides in a water/acetonitrile 1:3 (or 1:1) solution of KCN allowed us to synthesize the desired nitriles in good yields [30].
2.3 Metal-free arylation of aryl esters
Besides from aryl chlorides, phenyl cations have been conveniently generated also from aryl sulfonates (mesylates, trifluoromethanesulfonates [31] and nonafluorobutanesulfonates [32]), sulfates [33] and phosphate [31] esters, allowing for the conversion of an Ar–O bond into an Ar–C bond. However, the use of these substrates introduced a new competing path, since the singlet excited state of the precursors may undergo homolysis of the S–O or P–O bond faster than ISC to the triplet, thus leading to a phenoxy radical in place of the desired cation. However, in the presence of a triplet sensitizer, such as acetone, the triplet state is directly populated, avoiding to pass through the singlet and thus preventing phenoxy radical formation.
Recently, we developed a protocol for the one-pot arylation of phenols, via the in-situ formation of the corresponding aryl nonaflates. Thus, a MeCN solution of the chosen phenol was treated under stirring with Cs2CO3 and C4F9SO2F for 15 min, then TFE, acetone and the desired π-bond nucleophile were added to the mixture and the resulting solution was nitrogen-purged and irradiated for 24 h. This afforded the expected arylated products in satisfactory yields (as an example, see the synthesis of methoxy-substituted biaryls in Scheme 10) [32].

One-pot conversion of phenols to biaryls.
2,2,2-Trifluoroethoxy aryl sulfates (a class of compounds rarely used in cross-coupling processes until now) were recently employed as precursors of phenyl cations. These substrates were prepared in-situ by dissolving the corresponding imidazolium sulfonate salts in basic 2,2,2-trifluoroethanol. The resulting solution was then irradiated in the presence of a π-bond nucleophile, smoothly affording the expected arylated product (Scheme 11) [33].

The use of sulfate esters as phenyl cation precursors.
3 C–C bond formation under decatungstate photocatalyzed conditions
At the onset of photochemistry, irradiations were carried out by exposing the samples to solar light and, although convenient UV sources have been later developed (in particular long lived low-pressure mercury arcs, emitting at 254 nm or at a longer wavelength through a phosphor), this surely remains a still convenient “green” choice. However, simple aliphatic compounds (such as hydrocarbons) are transparent to excitation at wavelengths > 200 nm and direct irradiation is impractical. Again, this point has been highlighted by early photochemists, such as Ciamician, who observed that many compounds underwent photochemical reactions of industrial importance, but did not react when exposed to solar light: “Several [processes] are known, which are caused by ultraviolet radiations and which might eventually take place under the influence of ordinary radiations, provided suitable sensitizers were discovered” [3]. In order to overcome this limitation, indirect strategies, such as photosensitization (where a (dark) excited state of the reagent is populated through an energy transfer step from an excited sensitizer) and photocatalysis [34–36], have been developed. In the latter case, the photocatalyst (PC) absorbs light and activates the actual substrate of the reaction through a chemical step. An example of such activation involves the transfer of an electron to/from the substrate to give, respectively, the corresponding radical-anion/cation through a Photoinduced Electron Transfer (PET) step (Scheme 12, path a). These intermediates can be directly exploited in synthesis or may undergo a further step, viz. the loss of a charged group, thus giving an indirect access to a radical derivative (path a′). A second alternative involves the transfer of an atom, most commonly a hydrogen atom (resulting in a Hydrogen Atom Transfer (HAT) step) [35], to give a radical species (Scheme 12, path b). A further requirement of photocatalytic reactions is that the photocatalyst is regenerated at the end of the process by one of the involved intermediates, so that it can be used in a sub-stoichiometric amount.

The modes of action of a photocatalyst (PC).
The activation of substrates through an electron transfer step, by far the most common approach described in the literature, has recently known an impressive development in synthesis and is generally known under the heading “photoredox catalysis” [36]. By contrast, only a handful of reactions have been reported for the HAT counterpart, since photocatalysts that are able to effectively perform this kind of reaction, while not being consumed through side reactions, are not easily found.
Our research group has been involved in the optimization of synthetic strategies based on the photocatalytic activation of organic substrates. In recent years, we focused our attention on a robust, inorganic photocatalyst, namely TetraButylAmmonium DecaTungstate, TBADT, a colorless salt with the molecular formula (nBu4N)4[W10O32]. The anion pertains to the family of polyoxometalates (POMs), viz. assemblies of inorganic oxyanions linked together by shared oxygen atoms. This photocatalyst is capable of absorbing light in the near-UV region and shows a strong absorption band centered at 323 nm (ε323 = 13,500 M−1 cm−1) [37].
Time-resolved spectroscopic measurements demonstrated that, upon excitation, the decatungstate anion interacts with organic derivatives through different mechanisms depending on the actual properties of the chosen substrate. Thus, in the presence of oxidizable compounds, PET from the substrate to TBADT occurs and gives a radical-cation that splits off a cation to give a radical intermediate. Alternatively, a HAT step is also feasible, giving directly a radical [38]. In turn, these intermediates have been exploited in radical conjugate addition reactions onto electron-poor olefins or related substrates (e.g., azodicarboxylates). In the following, a brief overview of the diverse synthetic applications of decatungstate photocatalyzed processes is offered.
3.1 The decatungstate anion as a photoredox catalyst
The generation of stabilized benzyl radicals under PET conditions relies on the ease of removal of an electron from the aromatic ring. The so formed radical-cation then splits off a positively-charged group from the benzylic position to give the desired intermediate. The ideal approach would be starting from alkylaromatic derivatives (e.g., toluene), where the loss of a proton should occur at the radical-cation stage. However, this process can be favored by having recourse to suitable electro-auxiliary groups, capable of lowering the oxidation potential of the substrate and acting as suitable electrofugal groups. The use of the trimethylsilyl (–SiMe3) group is quite common, as in the case of benzylsilanes. A further issue to be confronted with is trapping of the resulting benzyl radicals, since dimerization of these relatively stable species usually competes.
Our group recently optimized a reaction protocol for the benzylation of electron-poor olefins under photocatalytic conditions mediated by the decatungstate anion. It was found that the reaction is favored by the use of a nucleophilic co-solvent, such as water, and by the presence of an electrolyte (a 0.5 M LiClO4 5–1 acetonitrile/water mixture was actually used), for the role of stabilizing the charged intermediates involved in the process and of enhancing the efficiency of the fragmentation step. The electron-poor olefin used here had the double role of a radical trap and an electron acceptor, as shown in Scheme 13. Accordingly, the olefin was responsible for the regeneration of the starting photocatalyst by accepting an electron from [W10O32]5−, while generating the corresponding radical-anion. The latter was in turn a more efficient trap than the original olefin and coupled with the benzyl radical, giving the final product upon protonation and hindering the homodimerization of the benzyl radical. Interestingly, similar results were obtained when using easily oxidizable benzylsilanes [39], as well as alkylaromatics [40], as radical precursors. In the latter case, a detailed spectroscopic investigation supported that the activation of the benzylic C–H bond occurred via transfer of a hydrogen atom within an intermediate polar exciplex between decatungstate and the alkylaromatic derivative (Scheme 13) [40].

Benzylation of electron-poor olefins via decatungstate photocatalysis.
3.2 Hydrogen atom transfer reactions promoted by decatungstate
3.2.1 Radicals from unactivated C–H bonds
The direct C–H to C–C conversion of unactivated C–H bonds in alkanes still remains a highly challenging task, often requiring harsh conditions. By contrast, the alkylation of a range of Michael acceptors by cyclic alkanes has been obtained by using 2 mol% of TBADT as the photocatalyst under mild conditions. As an example, cyclohexane (used in a 5-fold excess with respect to the olefin) added to methyl vinyl ketone and 3-methylene-2-norbornanone to give the corresponding adducts in 55 and 49% isolated yield, respectively, after 16 h of irradiation with artificial UV light (λ = 310 nm) in a process with 100% atom economy (see Scheme 14a) [41]. Further, notice that the absorption spectrum of TBADT covers a large fraction of the UV-A region and extends up to the VIS region limit, which makes this photocatalyst effective also under sunlight irradiation. Accordingly, the alkylation reported above could be conveniently carried out by simply exposing to solar light the desired reaction mixture in a Pyrex vessel on a window ledge. This approach further enhanced the sustainability of the process since no artificial energy was required during the synthesis and the products were isolated by mere evaporation of the solvent followed by bulb-to-bulb distillation. As an example (see Scheme 14b), a TBADT solution (100 mL) in acetonitrile containing cyclohexane (5 equiv.) and dimethyl maleate (0.1 M) was exposed to sunlight for 5 days (8 h per day irradiation) thus allowing to obtain dimethyl 2-cyclohexylsuccinate in a satisfactory yield (68% isolated yield) in a gram scale process (1.55 g) [42]. In a related case, the same approach has been exploited for the C–H to C–N conversion by using diisopropyl azodicarboxylate as the reaction partner [43].

Decatungstate-photocatalyzed alkylation of electron-poor olefins by alkanes.
When the same reaction was carried out under a CO atmosphere (80 atm), a selective three-component process resulted, allowing for a C–H to C–C(O)–C overall conversion. In the process, the initially formed alkyl radical added to CO to give an acyl radical that further added to the electron-poor olefin giving the final product (see Scheme 15a) [44].

a) Three-component synthesis of unsymmetrical ketones. b) β-functionalization of cyclopentanones.
The excellent properties of the decatungstate anion in terms of hydrogen atom abstraction have also shown interesting selectivities. As a matter of fact, excited decatungstate shares electronegative oxygen-centers with a partial radical character. Thus, when the decatungstate anion abstracts a hydrogen atom from a C–H bond, the transition state should be polar in order to balance the positively charged carbon atom. Therefore, when the C–H moiety is conjugated with an electron-withdrawing group (e.g., it is in the α-position with respect to a carbonyl group), an unfavorable situation occurs. In other words, TBADT shows a marked preference for nucleophilic, as opposed to electrophilic, sites. This particular behavior has been recently exploited for the regioselective β-alkylation of cyclopentanones with electron-deficient alkenes (Scheme 15b). An added bonus of this protocol is that it can be conveniently carried out under solar light irradiation [45]. Furthermore, a similar behavior has also been observed in the activation of the C–H bonds in aliphatic nitriles [46].
3.2.2 Radicals from aldehydes
Acyl radicals are valuable intermediates in synthetic chemistry and the most straightforward approach for their generation is the direct activation of the formyl C–H bond in aldehydes. Indeed, in recent years TBADT has been successfully used in the photogeneration (under artificial, as well as solar light) of acyl radicals from both aliphatic [42,47] and aromatic [42,48] aldehydes. The thus formed radicals were in turn trapped by suitable electron-poor olefins, paving the way to an alternative preparation of unsymmetrical ketones (Scheme 16a). The use of sunlight as the light source allows for a significant increase in the concentration of the reactants (up to five times the concentrations employed when performing the same reaction in a traditional multilamp reactor equipped with low-pressure Hg lamps), thus resulting in a dramatic drop of the amount of waste generated during the process (from 45 down to 7.8 kg of waste for a kg of desired product) and in a minimization of the energetic cost of the process (see Scheme 16b) [42]. When secondary or tertiary aldehydes were used as the substrates, the resulting acyl radicals showed a marked tendency to decarbonylation in competition with the desired trapping. However, the use of low temperatures (down to – 20 °C) allowed to suppress this undesired process and limited the competitive alkylation of the olefin (see Scheme 16c).

Decatungstate photocatalyzed generation of acyl radicals and ensuing processes.
3.2.3 Radicals from amides
Amides are often considered as solvents in synthetic planning due to their excellent solvating properties and very low reactivity, while their use as building blocks is quite rare. However, the activation of these substrates is an interesting topic, since they have several bonds prone to be activated, including the C–H bond α- to the nitrogen atom (in secondary and tertiary amides), the formyl C–H bond in formamides, the C–H bond α- to the carbonyl in higher homologues and the N–H bond (in primary and secondary amides). Furthermore, the first two kinds of bonds have very similar Bond Dissociation Energies and, accordingly, the activation step has to be highly chemoselective if the reaction has to be driven toward a specific goal [49].
Interestingly, TBADT was demonstrated to offer a straightforward access to C-centered radicals via photocatalyzed hydrogen abstraction from unusual H-donors such as amides. The activation step was highly chemoselective, often leading to the activation of a single site in the starting molecule. The course of the reaction, however, was found to depend on the structure of the employed substrate. As an example, the activation of DMF (a tertiary amide) involved exclusively the N–C–H moiety, while in N-methyl formamide a completely different reaction course was observed, leading to the selective activation of the formyl C–H group (Scheme 17). The use of carbamates (actually protected amines) offered an interesting approach for functionalizing amines that cannot be directly activated under TBADT-photocatalyzed conditions [49].

Chemoselective radical generation from amides promoted by excited decatungstate.
3.2.4 Radicals from oxygenated derivatives
Hydrogen abstraction from an oxygenated derivative often follows an easily predictable course, since the lone pairs of the oxygen atom(s) greatly stabilize a radical site in the α-position, directing the generation of the desired intermediate. Recently, TBADT was successfully employed for the photoactivation of C–H bonds in a variety of O-containing derivatives, including ethers and acetals. Thus, both oxetane [50] and tetrahydrofuran [51] could be added to isopropylidenemalononitrile to give the corresponding adducts in 64 and 75% isolated yields, respectively (Scheme 18a).

Decatungstate-photocatalyzed C–H to C–C conversion in oxygenated derivatives.
The reaction could be likewise applied to acetals. In particular, the direct functionalization of benzodioxoles is of great significance since this moiety is present in many compounds endowed with biological activity. Thus, the mild activation of the methylene hydrogens has been obtained by using 2 mol% TBADT for the role of the photocatalyst, leading to the preparation of 2-substituted-1,3-benzodioxoles (an example is shown in Scheme 18b) with no interference from the presence of benzene ring substituents [52].
The versatility of TBADT as a photocatalyst has also been recently exploited for the facile PEGylation of carbon nanotubes by the photomediated addition of various PEG derivatives (PEG 200 to 600) onto the surface of the nanostructure, thus improving its dispersibility in water [53].
4 Conclusions
Synthetic chemists are becoming increasingly aware of the contribution that the photochemical approach gives, thanks to the unparalleled mild conditions employed and the possibility to start from non- (or minimally) functionalized precursors, often allowing one to adopt more atom- and step-economical pathways [4,35]. In our opinion, the two topics summarized in this review are also significant examples of the greenness of the photochemical approach [2]. As for photochemical arylations via phenyl cations, several synthetic targets can be obtained at room temperature under metal-free conditions, starting from easily available/synthesized aryl chlorides and esters and non-activated π-bond nucleophiles (alkenes, alkynes and aromatics). On the other hand, decatungstate photocatalysis allows for the direct activation of C–H bonds in a wide range of substrates including hardly reactive alkanes, avoiding any previous activation/functionalization of the substrate. The thus generated carbon-based radicals have been employed in quantitative atom-economical conjugate additions and an added bonus is that the reaction can be carried out under sunlight irradiation, minimizing the energetic cost of the process.
Acknowledgments
We are grateful to Carlotta Raviola, Stefano Crespi and Edoardo Torti for their precious work in the fields of photochemical and photocatalyzed synthesis.