

1 Introduction
Converting solar light into a usable form of energy is a very worthy challenge, since sunlight is relatively equally distributed on the planet, very abundant, strictly nonpolluting, virtually inexhaustible and free. Taking inspiration from photosynthesis, scientists have early tried to develop artificial photosystems, capable of harvesting and converting light into chemical potential, storing the immaterial electromagnetic energy in the shape of chemical bonds.1–5 In other words, using light to drive highly endothermic reactions transforming abundant and low energy raw materials into high added value molecules is the ambitious purpose of a significant part of the scientific community.6–8 This is epitomized by the well-known and much desired water splitting reaction (equation (1)):
| (1) |
This would provide humanity with an inexhaustible source of H2, which is a clean, energy rich solar fuel.9–11
In natural photosynthesis, H2O is first oxidized to O2 and the electrons resulting from this redox process are used to generate the biological reductant NAD(P)H from NAD(P)+.12,13 Ultimately, NADH is implied in CO2 reduction into biomass. In a way, biomass, growth of the photosynthetic organisms, results from the reduction of CO2 by the electrons photo-extracted from water and O2 is nothing but a waste product. Therefore, photosynthesis promotes CO2 reduction into biomass by water, the energy necessary to drive this very endothermic reaction being provided by solar light. In a way, H2O can be seen as a sacrificial electron donor, fueling the natural photosystem with electrons.
Ideally, the same goal is envisioned for artificial photosynthesis but extracting electrons from water, low added value molecule par excellence, is particularly difficult and requires several processes (light harvesting,14 charge photoaccumulation,15,16 and catalysis17–19) which are at the very core of artificial photosynthesis research field. Crudely, the great ordeal to mimic a full photosystem lead scientists to study half photosystems separately, oxidative ones on one hand (models of PSII)7,20–23 and reductive ones on the other (models of PSI).3,24–40 Despite the obvious interest of oxidative artificial photosystems, we will in this review focus exclusively on the reductive ones. For the latter, simpler electron donors than H2O were used, to meticulously focus on the reductive processes. By “simple”, we mean that these electron donors can readily fuel a photosystem with electrons. The latter are the subject of this review, namely “waste” molecules, sacrificed in the course of the photochemical reactions, and used essentially for the practical study of the artificial reductive photosystems. These molecules are most often referred to as sacrificial donors (SDs) and are playing a pivotal role in peculiar artificial photosystems called “three-component system” (TCS).41
2 General operating mechanism of sacrificial reagents and thermodynamic considerations
Artificial photosystems other than TCSs are currently developed,22,42–47 but the latter is historically the first artificial photosystem.3,34,35 There are both oxidative21–23 and reductive TCSs,4,34,48 with SDs intervening in the latter one. Those three components are 1) the photosensitizer (PS) which must harvest solar light and convert it into chemical potential, i.e. into reductive or oxidative potential; 2) the sacrificial donor SD, providing electrons on photo-induced command by PS; 3) the reduction catalyst, accumulating electrons. A fully operational TCS is very often designed to photo-produce high added value molecules such as H2 from H+ or CO2 reduction compounds (CO, HCHO…)41 and this naturally requires appropriate catalysts (a molecular entity27,28,30,49–52 or an insoluble material).3,34,35,53 Redox relays shuttling electrons between PS and the catalyst are often required in a TCS.3,34,35 Thus, TCSs where there is no catalyst but only a redox shuttle have been extensively studied in order to comprehend the photo-induced redox mechanisms at stake, independently from catalysis requirements.54,55–57 In the rest of the text, TCSs with catalysts and TCSs without will not be differentiated because SDs are not primarily involved in corresponding steps. We will call SUB, like “SUBstrate”, the ensemble of molecules and materials which are the ultimate electron acceptors in a TCS, the photo-produced electrons' final destination, accumulating during photolysis.
The modus operandi of a TCS is the following: upon light absorption, PS is promoted to its excited state and acquires at the same time enhanced oxidative and/or reductive power(s). Thermodynamically speaking, E(PS*/PS−) is superior to E(PS/PS−) (E(PS*/PS−) = E(PS/PS−) + E00, where E00 is the lowest excited state energy) and PS* is therefore more prone to harvest an electron from the nearby donor SD (equation (2)). Similarly, E(PS+/PS*) is more negative than E(PS+/PS) and PS* is thus more prone to donate an electron to SUB (equation (3)).
| (2) |
| (3) |
In equation (2), PS ends up in a reduced state, which is why quenching of PS* by an electron donor is referred to as “reductive quenching” (RQ). On the other hand, PS* endures an “oxidative quenching” (OQ) in equation (3). Whether PS* will be implied in a reductive or an oxidative quenching depends on the nature of PS, SUB, and SDs and the external medium can play a major role too (pH, solvent …); all is governed by the thermodynamic and kinetic parameters at stake.
In a working TCS, both oxidative and reductive quenching pathways can be used: for OQ based TCSs, a photo-induced electron transfer from PS* to SUB entails the reduction of the latter, and the oxidation of the former. PS+ is then regenerated by the SD, while SUB accumulates upon repeating such cycles (Fig. 1, right). Conversely, PS* can abstract an electron from the SD first, and resulting PS− is regenerated by transferring an electron to SUB (Fig. 1, left). In the end, regardless of the mechanism, SUB was reduced by the SD using light as the sole source of energy to perform this otherwise endothermic reaction, and the reaction may theoretically proceed until all SDs have been oxidized.
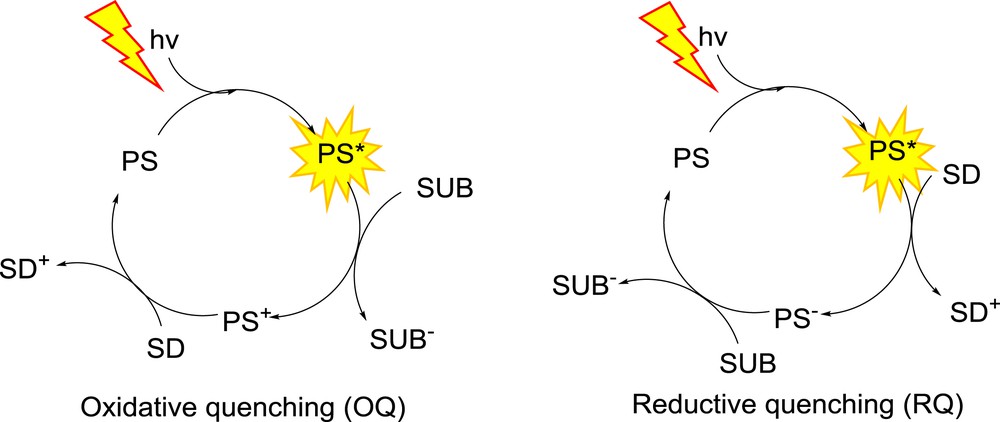
Mechanisms of RQ and OQ within a TCS.
From the mechanisms above, a few constraints can be deduced: in the case of OQ, photo-induced electron transfer from PS* to SUB must be thermodynamically allowed (E(PS+/PS*) < E(SUB/SUB−)) while the same goes for (dark) electron transfer from the SD to PS+ (E(PS+/PS) > E(SD+/SD)). In the case of RQ, the photo-induced electron transfer involves PS* and SD, and therefore E(SD+/SD) < E(PS*/PS−) while E(PS/PS−) < E(SUB/SUB−). RQ is considered to be a slightly preferred mechanism over OQ, because PS− is a more potent reducer than PS*, allowing the photo-reduction of a larger number of SUB, with more cathodic reduction potentials.
The above conditions on SDs are necessary but not sufficient. First and foremost, in order to allow SUB− accumulation, avoiding recombination reactions (4) and (5) is mandatory
| (4) |
| (5) |
SDs must therefore be irreversibly oxidized into inert molecules, unable to interfere with the proper running of the photochemical cycles. This deserves further comments: in a donor-acceptor system D/A, D and A must pre-assemble in an encounter complex [D---A] to allow photo-induced charge transfer to take place within the latter.25,58–60 As such, the photo-excitation of [D---A] yields the geminate pair [D+---A−]. Geminate charge recombination within this ion pair can be very fast, and competes with the dislocation of [D+---A−], which can be a difficult process due to possible attractive coulombic interactions between D+ and A−. This competition translates into the cage escape yield ηCE: the higher ηCE, the lesser the probability of charge recombination between D+ and A− and the larger the chance to accumulate A−. In the case of RQ, the encounter complex after photo-electron transfer is [SD+---PS−]; if the kinetics of SD+ irreversible transformations into inert entities are lesser than the kinetics of charge recombination, the TCS will fail because no accumulation of PS− and consequently of SUB− can take place.25,58
To summarize, a good SD must gather several important qualities: thermodynamic adequacy with PS (E(SD+/SD) < E(PS+/PS) for OQ and E(SD+/SD) > E(PS*/PS−) for RQ), irreversible transformation upon monoelectronic oxidation into inert molecules, and – if need be – high cage escape yield or faster degradation kinetics than charge recombination. This latter condition concerns reductive quenching mechanism; the same goes for oxidative quenching but will not be treated here since SDs are not involved in the corresponding encounter complex.
As will be shown in what follows, the reaction medium (solvent, pH), the concentration [SD], and the nature of PS can strongly control the overall efficiency of the solar fuel generation.34,35 To review all impacts of the variations of these conditions is not the purpose of the present article, nor is to give a comprehensive view of the innumerable TCS from late 1970s until today. We rather wish to present an overview of the different sacrificial donors which have been, and are still, most commonly used in reductive TCSs. We wish to pinpoint that redox organic photochemistry,61 despite its unquestionable interest, is irrelevant to artificial photosynthesis and will therefore be omitted in the present review.
Transition metal anions have been extensively used as sacrificial donors,62–64 some of them like PtCl42− can however be considered as substrates SUB rather than sacrificial (waste) donors and will therefore be omitted in this review.
3 Aliphatic amines as SDs
Tertiary aliphatic amines are probably the most used sacrificial donors to fuel photochemical reduction reactions. This class of molecules is epitomized by well-known triethylamine (TEA) and triethanolamine (TEOA). Both were employed in the pioneering works setting the bases for a TCS, with the general purpose of photoproducing hydrogen gas25,35 or photo-decomposing CO2.48,65,66 TEA and TEOA display very similar features: they exhibit irreversible oxidation potentials around 0.7 V vs. SCE24,67,35,65 making them thermodynamically able to be part of similar electron transfer processes. However, let us bear in mind that electrochemical irreversible processes are poorly described by a redox potential E(SD+/SD) and the anodic peak potential can only give a rough estimate of the thermodynamics lying behind the electron transfers within the SD+/SD couple.68 This is further demonstrated by the various reported oxidation potentials for the same species, differing sometimes by more than 100 mV.35,53 Besides, and equally important, TEA and TEOA are used in an impressive collection of various conditions (solvent, pH, concentration…) making a systematic study of the thermodynamics of OQ or RQ through the whole literature quite intricate. As such, redox potentials given in Table 1 are to be taken with great caution. Concerning the solvent, TEA or TEOA has been used in purely organic30,65,69 or aqueous media;35,70 very often though, mixtures of water with an organic solvent (DMF, MeCN, and THF) are reported,71–76 the reasons for this are given below.
Structures, names and properties of usual sacrificial donors. Redox potentials given in V vs. SCE.
| Structure | Name of general use | Eox | Medium |
| TEA | 0.6967 | Aqueous | |
| TEOA | a0.5735–0.8253 | Aqueous | |
| EDTA | b0.5782–0.9253 | Aqueous | |
| Amine 1102 | n.r. | – | |
| DMA | 0.81103 | MeCN | |
| DMT | 0.71103 | MeCN | |
| BNAH | 0.57115 | MeCN | |
| BIH | 0.33153 | MeCN | |
| Ascorbic acid (H2A) | a0.46154 (HA−/HA•) | Aqueous | |
| Oxalate | – | – | |
| Benz-S− | −0.1149 | MeCN | |
| Dtc− | 0.0554 | MeCN | |
| Xan− | 0.2154 | MeCN | |
| Dtb− | 0.1954 | MeCN | |
| Dtp− | 0.7154 | MeCN | |
| PPh3 | 0.98151 | MeCN |
a Values published vs. NHE, obtained vs. SCE by removing 0.25 V.
b Values for EDTA are deduced from ref.82
The main appeal concerning TEA and TEOA is their degradation pathway,24,77–79 which is described in Fig. 2 for TEOA, but is very similar for TEA. Upon monoelectronic oxidation (1), a positively charged aminyl radical is formed. The latter is a good oxidant which could in principle react with any reduced species (PS− in the case of RQ, or SUB− in the case of OQ), i.e. lead to counter-productive back electron transfer. However, deprotonation of the aminyl radical by TEOA itself leads to a rearrangement into a carbon centered radical displaying a significant reductive power (E(b/a) = 1V, Fig. 2)77. The latter is beneficially used to reduce PS or SUB or a redox mediator shuttling between them, and therefore contributes to increase the overall yield of SUB− photoproduction.35,80 The iminium species which results from this “dark” electron transfer process is either the final product in the absence of water, or further degrades into (hydroxy)ethanal and secondary amine by hydrolysis of the iminium in aqueous media. This last step is often reported to be very important48,77,81 to insure the buildup of reduced SUB− but accumulation of reduced species upon photolysis in a purely organic medium (acetonitrile, dimethylformamide) has been reported too.80,81 We believe this strongly depends on the kinetics at stake in the cage escape process. All in all, aliphatic tertiary amines are endowed with good cage escape yields,82 but this is highly dependent on the photosensitizer and the occurrence of a reductive quenching mechanism.
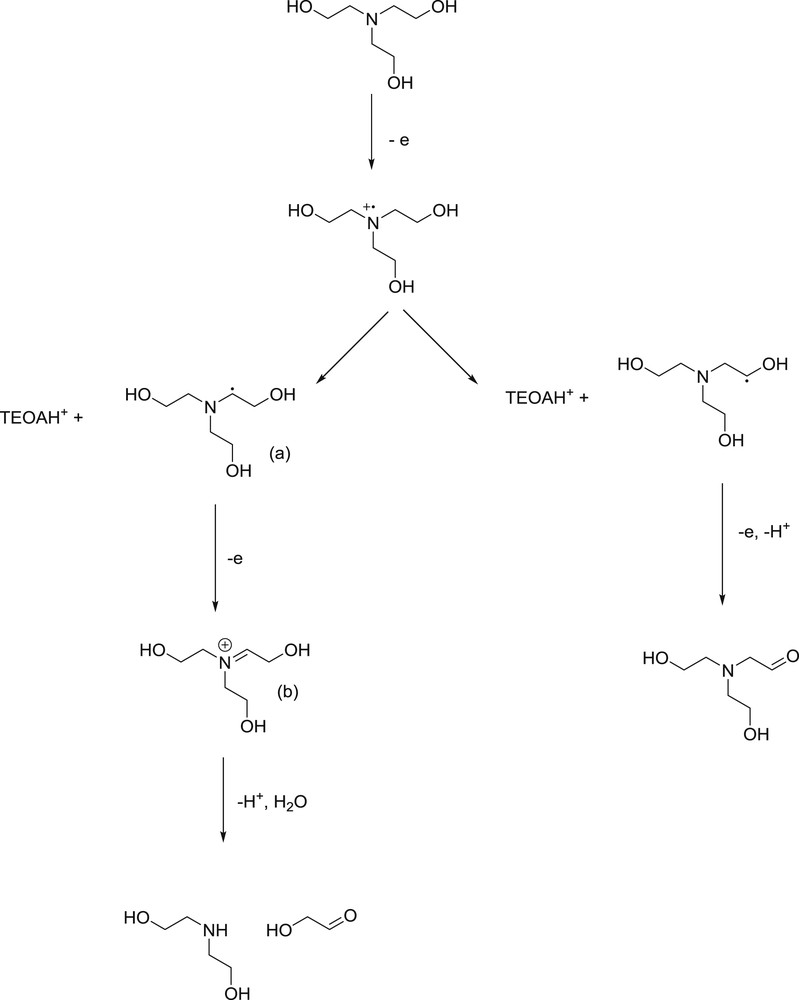
Degradation pathway of TEOA upon monoelectronic oxidation.
When one considers the chain of reactions depicted in Fig. 2, deprotonation of the aminyl radical (or hydrogen abstraction) by a regular amine molecule is mandatory in order to form the reductive carbon centered radical, and prevent charge recombination.35,83 Knowing that the pKa of TEOA and TEA is 7.9 and 10.7, respectively, this deprotonation step is less and less likely as pH decreases.35,70 Consequently, most reported works implying TEOA based TCSs are performed at pH values above 8. This is particularly important to pinpoint since the optimal working conditions of catalysts in a TCS may depend on pH. Additionally, since pH is an ambiguous parameter in a purely organic medium, let us nevertheless mention that TEOA or TEA are mostly used in excess compared to SUB to insure the irreversible aminyl transformation,25 and to improve the overall yield of the TCS.84
The concentration of amine in the medium varies a lot (from less than 10 mM to more than 1 M) from a TCS to another. The higher the concentration of SDs, the higher the probability to observe RQ vs OQ.75,77,85 Although RQ was reported with common [Ru(bpy)3]2+,81 OQ is more often observed with this milder photo-oxidant.30,86–88 Alterations of the structures of bpy (bipyrazine bpz, bipyrimidine bpm,58 or carboxyester-tethered bpy78) allowed to increase the photo-oxidizing power of ruthenium polypyridine complexes and RQ was thus monitored (Fig. 3).
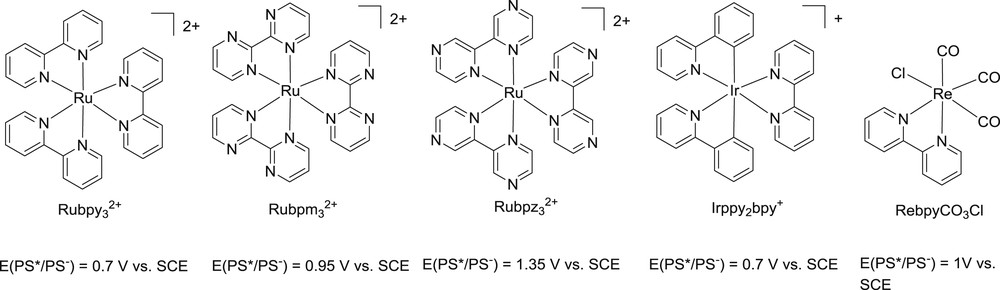
Some metal based photosensitizers with various photo-oxidizing powers (Re(bpy)CO3Cl,65,93 Irppy2bpy+ and [Ru(bpy)3]2+, 87 [Ru(bpm)3]2+ and [Ru(bpz)3]2+)82
RQ with TEA or TEOA is often reported with iridium complexes as PS, even though its thermodynamics are not often very different from those of ruthenium systems,24,69,70,85,89–92 underlying the importance of kinetics and bringing further evidence that even a careful study of redox potentials for SDs and PS* is not enough to discard an SD over another. Admittedly though, strongly photo-oxidizing PS (like rhenium complexes, Fig. 3) tends to allow RQ.50,80
Let us lay stress on the fact that the effect of changing the ratio MeCN/H2O within the frame of H2 photogeneration with TEA74 and TEOA84 as SDs was investigated and it was proven that the TCS worked better when the ratio MeCN/H2O was increased. The improved solubility of TEA in the solvent mixture, modifications of the redox potentials and changes of dielectric constant of the medium were invoked to explain these observations. The action of SDs on the catalyst is not excluded but is irrelevant to the objectives of this review. All in all, those experimental observations are very difficult to support the given complexity of a TCS.
TEA and TEOA are clearly able to provide two reductive equivalents for each photon absorbed by PS.34,80 Importantly, quenching efficiencies are therefore theoretically equal to 2, but are more or less comprised between 0.1 and 0.5 at best. Loss in the overall efficiency is thought to be grounded in back electron transfer occurring within the geminate pair ([PS---SD] for RQ, [PS---SUB] for OQ).35 This highlights again the importance of having TCSs with significant cage escape yields. Importantly, in the case of H2 photogeneration from H+, protons stemming from TEOA degradation are reported to be implied in H2 evolution.80
The use of TEA vs. TEOA is difficult to rationalize. For instance, Bernhard et al. described the superiority of TEA over TEOA for their photocatalytic system for stability reasons.71,94 Chang et al. observed an improved efficiency with TEA rather than TEOA,69 whereas Zou et al. reported the improved reductive quenching rates with TEOA vs. TEA.70 The conditions are nevertheless rather different from one experiment to another, depending on the requisites of each property (solubility, activity of the catalysts…) in the considered TCS. There is therefore no clear-cut consensus, TEA and TEOA having very similar properties. Let us mention nevertheless that alcohol groups in TEOA necessarily improve miscibility in water, and that a lower pKa for TEOA than TEA allows working at lower pH.
Additionally, the amines are Lewis bases too, likely to play a role in some other steps of the intricate TCS's operation, and may for instance take part in the catalysis itself (coordination of the metal centers in a molecular catalyst, or participation in the overall mechanism69,95) or even poison nanoparticulate catalysts.94
Other aliphatic tertiary amines have been used: increasing the carbon atom number of the alkyl chain from ethyl to pentyl, passing from symmetrical NR3 to unsymmetrical NR2R′, and altering the steric bulk was studied.48,66 Like TEA and TEOA, irreversible oxidation of the amines leads to dealkylation (secondary amine) and aldehydes if water is present.79 It is not the purpose of the present article to review the various results obtained with those SDs. Nevertheless, in the particular case of CO2 photoreduction in CO and H2 by [Ru(bpy)3]2+ and CoII ions, NPr3 (where Pr means n-propyl) was reported to be a better SD than TEA66 and yet the great majority of nowadays TCSs are based on TEA rather than NPr3.
At low pH, protonated amines cannot behave anymore as sacrificial donors. Some of them however with lower pKa, can be used in a relatively acidic medium, for instance N-ethyl morpholine (pKa = 6.8) or famous ethylenediaminetetraacetic acid (EDTA).57,96,97 EDTA is a tertiary diamine with four carboxylic functions, and logically displays a series of 6 pKa (0, 1.5, 2.0, 2.7, 6.1, 10.2).55 Usually employed between pH 5 and pH 7, the so called “EDTA” is as a matter of fact utilized as its disodium salt. It is a two-electron donating sacrificial donor like TEA or TEOA98,99 mostly utilized in an aqueous medium, and degradation of EDTA upon monoelectronic oxidation is well described.99
Like the previously reported amines, EDTA can be involved in reductive and oxidative quenching, depending on the oxidative power of PS*. We could consider that the reaction of SDs with traditional [Ru(bpy)3]2+* may serve as a diagnostic for the efficiency of reductive quenching in a TCS (this makes sense since [Ru(bpy)3]2+ is by far the most used chromophore in artificial photosynthesis). In that case, EDTA displays very high oxidation potentials for [Ru(bpy)3]2+* to photo-oxidize it. Incidentally, chemical engineering of the ligands coordinating ruthenium (from bpy to bipyrazine or bipyrimidine, Fig. 3) allowed to increase the photo-oxidizing power of corresponding ruthenium complexes and reductive quenching was monitored. Like in the case of TEA or TEOA, transformation of monoreduced EDTA into a carbon centered reductive radical is very rapid and competes efficiently with charge recombination.100
Regarding the influence of pH, EDTA may operate at lower pH than TEA or TEOA as mentioned above. Efficient reductive quenching at pH as low as 5 can be found in the literature34 or lower.101 However, slower kinetics for reductive quenching are reported at lower pH (see for instance, PS = [Ru(bpz)3]2+, pH = 4.7).98,99 Importantly, EDTA has excellent coordinating ability and has exhibited deleterious interactions with methylviologen, which are pH-dependent.99
Defying the usual, fair comments about the polluting and “useless” nature of most SDs, amine 1 (see Table 1) was reported as a renewable SD: after photo-induced oxidation and further degradation (implying photoproduced reduced carbon dioxide), the amine can be regenerated in its initial state by mere catalyzed hydrogenation, and can therefore be used again.102
Conclusively, picking an amine SD over another depends on many parameters. With the exception of pH considerations (which favor EDTA over TEA and TEOA for lower pH vide supra), the choice of a suitable SD among all amines presented in Table 1 is mostly empirical, depending on each property of the TCS and the overall reaction medium.
4 Aromatic amines as SDs
The latter point is clearly exemplified by the case of N,N-dimethylaniline (DMA, E ≈ 0.8 V vs. SCE). Although well-known as a potent SD in the early literature,62,103,104 DMA performed poorly in a H2 photoproduction TCS by Bernhard et al. Fast charge recombination, energy transfer and unfavorable interactions with the catalyst were invoked to rationalize this result.24 On the other hand, Brewer et al. demonstrated the efficient photoproduction of H2 from a heterotrinuclear triad ruthenium–rhenium photocatalyst with DMA as a sacrificial donor.32,105,106 Different photocatalytic systems and different conditions (concentrations and composition of the solvent mixture) can easily account for those discrepancies, and underline again the importance of serendipitous experimental work in those intricate, multi-component artificial photosystems. Noteworthily, Brewer's system used highly concentrated DMA (between 1 and 3 M) in a water/DMF or water/MeCN mixture. High [SD] as mentioned above tends to favor RQ.
A number of other aromatic amines have been studied as potential sacrificial donors.103,107–109 It was concluded that tertiary aromatic amines were superior to secondary and primary amines to achieve reductive quenching of the excited state of [Ru(bpy)3]2+ in MeCN. However, in the event of an RQ mechanism, the charge recombination within the geminate pair [PS−---SD+] was often observed.103,107,108,110 Hence dimethyl paratoluidine (DMT) displays very similar properties to DMA, with a 100 mV less anodic oxidation potential and was successfully used as the SD.103,111 Advantageously, the irreversible transformation of DMT upon oxidation is well documented112 (the DMT•+ radical loses a proton to DMT resulting in a carbon centered radical which dimerizes) and leaves little room for counter-productive interactions (equations (4) and (5)).
5 1-Benzyl-1,4-dihydronicotinamide BNAH as a SD
The biological reductant NADH is formed –for instance- during the photosynthetic process, en route towards CO2 reduction into biomass. Reports of NADH employed as a SD in artificial photosystems can be found in the literature.113,114 However, the manipulation of biological materials can be difficult. Inspired by the original NAD+/NADH couple, an efficient model has been developed: BNA+/BNAH (Table 1 and Fig. 4). Similar to NADH, BNAH stores 2 electrons which are released in the presence of a (photo)oxidant (E(BNA+/BNAH) is often reported to be equal to = 0.57 V/SCE in acetonitrile,115 but the irreversibility of the process makes it difficult to give such a precise value and it seems fairer to assign a 0.6–0.8 V vs. SCE range).68 Importantly, a proton is released upon oxidation of BNAH into BNA+, a feature that earned BNAH the reputation of a hydride donor.115–117
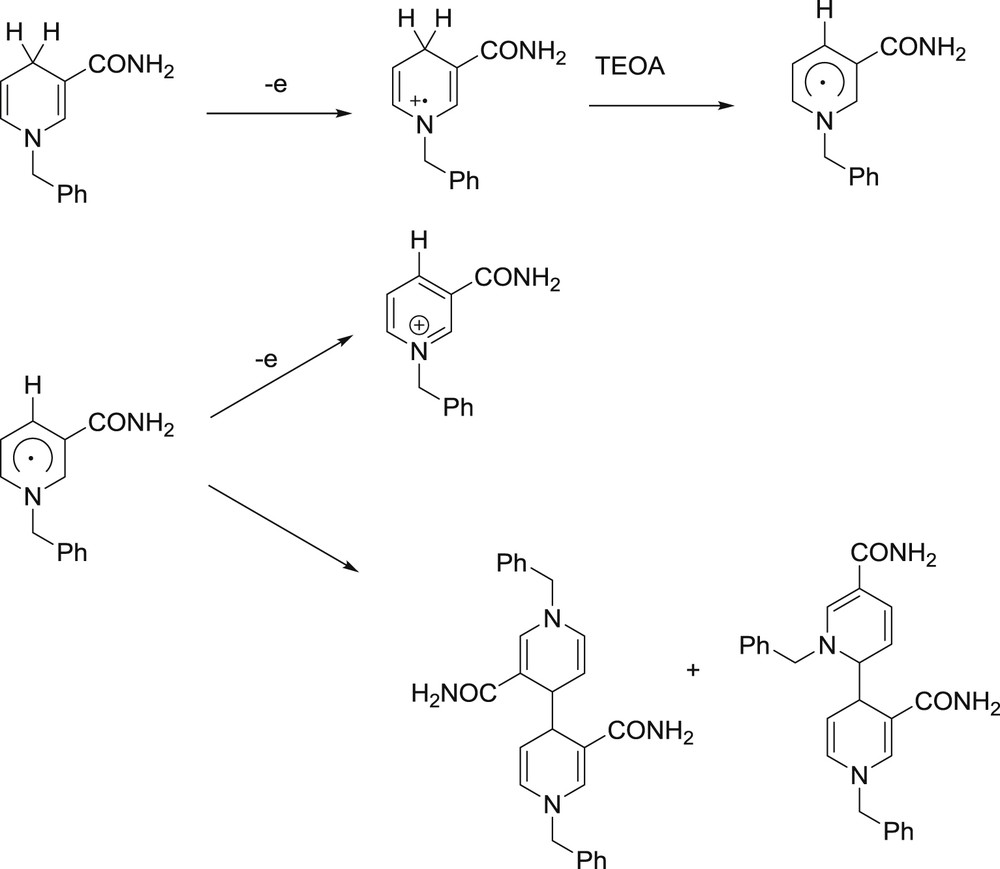
Mechanism of BNAH degradation upon monoelectronic oxidation.
BNAH is an efficient SD which is particularly famous for achieving the reductive quenching of [Ru(bpy)3]2+*,118 although the driving force for this process is rather weak, underlining the paramount importance of kinetics. This is a very significant appeal for BNAH as a SD because reduced [Ru(bpy)3]2+ is more reductive than excited [Ru(bpy)3]2+* by more than 500 mV.108 This strong reductive power and hydride donating character were used to reduce substrates such as olefins118,119 and aromatic ketones.120 Activation of CO2 reduction catalysts (which are known to display quite negative onset reduction potentials)121-123 is reported as well.124–128 In this case, a typical TCS designed for CO2 reduction with BNAH as the SD is set in a mixture of organic solvents such as DMF or MeCN with a base, typically TEOA. The mechanism is represented in Fig. 4. As mentioned above, BNAH can be a one- or two-electron donor.56,120 The first electron transfer is photo-induced in nature, and yields BNAH+• and PS− within the frame of a reductive quenching process. BNAH+• being very acidic (pKa below 1)56 can donate a proton, hence the importance of the base in the mixture. The absence of the latter (or any proton acceptor) entails a patent weakening of the quantum yield of the photochemical reaction.127 A neutral radical BNA• is then capable of providing another electron to the photosystem (in a dark process) resulting in the formation of a stable pyridinium species; or, it merely dimerizes into BNA2,127 which happens to be a potentially good SD too.129 However, when the kinetics of the formation of BNA2 are more favorable than the electron transfer from BNA• to PS or SUB, the photo-induced cycle reaches the end and BNAH behaves as a one-electron donor.126,129,130
Mild variations of the molecular structure of BNAH were performed in order to shift the oxidation potential to less positive values115,129 (electron donating groups on the benzyl moiety, such as methoxy) and consequently increase its reactivity with PS*, yielding improved photochemical yields.
6 1,3-Dimethyl-2-phenylbenzimidazoline BIH
BIH (Fig. 5) shares common features with BNAH. Originally used to reduce organic compounds,131 it was recently applied in a TCS for CO2 photoreduction.130 Like BNAH, BIH is a two electron, one proton source (hydride donor)132 which can react as well with the excited state of [Ru(bpy)3]2+ as PS in a reductive quenching process, yielding PS− and oxidized BIH•+. BIH•+ is very acidic and loses a proton to a base in the medium. This step is important to limit back electron transfer between reduced PS− and BIH•+.130 It is relevant to note that deprotonation of BIH•+ is 100 times faster than deprotonation of BNAH•+ (data in DMF). Thus, besides knowing that E(BIH•+/BIH) is more than 200 mV less anodic than E(BNAH•+/BNAH), this suggests that BIH is a more potent SD than BNAH, at least in DMF. BI• is besides a strong reductant (E(BI+/BI•) = −1.5 V/SCE)130,132 and can transfer a second electron (dark process) either to PS or SUB. BIH is thus a two-electron, one-proton releasing system, which requires BNAH in the presence of a base (such as TEOA).

Mechanism of BIH degradation upon monoelectronic oxidation.
7 Ascorbic acid
l-Ascorbic acid and related ascorbate ions are famous anti-oxidant natural preservative agents. As such, they are likely candidates for SDs and were successfully used in the 80s.49,51,133 One particular appeal of ascorbic acid and related anions is its water solubility, and the possibility to work in a neutral and acidic medium, compared to traditional TEA and TEOA which require basic conditions. By ascorbate, one mostly means monodeprotonated ascorbic acid. If H2A designates plain ascorbic acid, HA− is indeed the predominant form within the usual pH window for TCSs (between 5 and 9, pKas 4.0 and 11.3)134. HA− is reported to be implied in both reductive and oxidative quenching processes.
Mechanistically speaking, HA− is first oxidized to HA• (E ≈ 0.5 V/SCE),134 which is a much stronger acid than HA−51,135 and then quickly dissociates into A•− and H+.136 A•− disproportionates into dehydroascorbic acid A and ascorbate A2− (Fig. 6).27,50–52,137 The couple A/A•− exhibits a potential of roughly −0.3 V/SCE134 and it is therefore very unlikely that A•− may further reduce any species in the medium (PS or SUB). Besides, E(A/HA-) lies around 0.28 V, in other words A is a mild oxidant thermodynamically able to oxidize PS− or reduced SUB (charge recombination, equations (4) and (5)), thus counter-productively interfering within the photochemical cycle.50,134,138 Moreover, HA• was reported to merely recombine with PS− where PS stands for the classic [Ru(bpy)3]2+ chromophore and therefore was considered as non-sacrificial, albeit being an efficient quencher of PS*.82 As a matter of fact, the chemical reversibility of the couple A/H2A was mentioned by Creutz,134 and that justifies the use of ascorbate HA− rather than ascorbic acid H2A for a TCS.
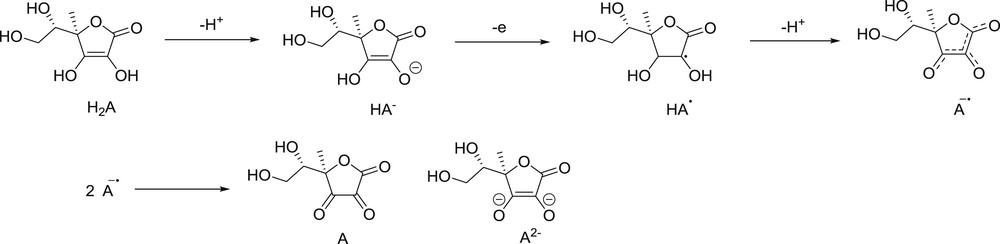
Mechanism of ascorbic acid degradation upon monoelectronic oxidation.
8 Carboxylic acids
Lactic acid is intensively used as a sacrificial donor in TCSs involving particularly quantum dots as PS and H2 photoproduction or organic substrates reduction, in the presence of water.139–141
The redox properties of oxalate naturally lead to attempts to use it as a sacrificial donor. The one-electron reduction of oxalate entails the formation of gaseous CO2 leaving the medium and very reductive radical CO2•−. The double advantage of oxalate oxidation is the evolution of CO2 setting at naught the risks of charge recombination,82 and the highly reductive power of CO2•− (E(CO2/CO2•−) < −2 V) which is capable of further reducing substrates (PS, SUB…) in a dark process.142
Oxalates (pKa 1.2 and 4.2)82 can be employed in water, over a wide range of pH from acidic pH (3) to basic (11).58,82,101 Nevertheless, its rather high oxidation potential imposes conditions on the reductive power of PS* (high E(PS*/PS−) for reductive quenching). No quenching of the excited state of PS* was monitored for instance in the case of classic [Ru(bpy)3]2+;143 more photo-oxidizing [Ru(bpz)3]2+ (Fig. 3) was necessary.58 Additionally, the anionic nature of oxalates came in very handy to form ionic pairs with cationic acceptors such as methylviologen, and photolysis of the latter system in UV efficiently leading to charge separation.142
9 Thiols
Thiols and thiolates are very relevant monoelectronic sacrificial donors because of their low oxidation potential and the irreversibility of the oxidation process yielding inert disulfide bridged dimers.144,145 Reports of chromophore emission quenching by cysteine (and other amino acids) date back to late 1970s.146
Cysteine was for instance utilized in a number of TCSs,35,82,147,148 formation of the radical Cys• (and concomitant deprotonation) occurred first, followed by dimerization. In another contribution,82 cysteine and various alkylthiols were involved in reductive quenching of [Ru(bpy)3]2+ excited state in water. Those SDs singled out among other donors, thanks to their reducing ability which was maintained even at lower pH. At higher pH (12), RS− is predominant over RSH (e.g., pKa (benz-SH/benz-S− = 9.4) and it was reported that oxidized thiolates displayed very low cage escape yields yielding to cumbersome charge recombination within the geminate pair PS−/SD+. This fact was justified by electrostatic attractive interactions between [Ru(bpy)3]2+ and RSSR•−, formed by the reaction of RS− and photoproduced RS•. RSSR•− is furthermore a rather good reductant (E = 0.65 V vs. ENH)145 which can easily react with PS−. Importantly, R–S− can play the role of ligands, or nucleophiles which could interfere with the smooth operation of a TCS.
Using thiols and thiolates as SD in organic medium is possible too. Reductive quenching of the excited state of [Ru(bpy)3]2+ and subsequent accumulation of [Ru(bpy)3]2+ was observed in acetonitrile in presence of benzylthiolate (Table 1, benz-S−).149 The anodic peak potential for benzylthiolate E(benz-S−/bens-S•) is −0.1 V vs. SCE, therefore even low photo-oxidizing systems (i.e. E(PS*/PS−) weakly positive) are susceptible to react with benz-S−, making the latter a candidate of choice for reductive quenching. Once again, premium was put on the inertness of dimer (benz-S)2, which reduction potential was below −2 V vs SCE.
Within the same frame, diethyldithiocarbamate (Table 1, dtc−, Eox = 0.05 V vs. SCE), ethylxanthate (xan−, Eox = 0.21 V vs. SCE) and p-methoxydithiobenzoate (dtb−, Eox = 0.19 V vs. SCE) were studied for reductive quenching with [Ru(bpy)3]2+*.54 Those deprotonated carbodithioic acids tend to dimerize like thiolates when oxidized. It was proven that the reaction of photoproduced [Ru(bpy)]+ with a reducible substrate (such as anthraquinone) was quicker than the charge recombination involving dtc2 dimers.
A number of quenching experiments have been performed with other sacrificial donors, such as triphenylphosphine in an aqueous organic medium.150 The monoelectronic oxidation of PPh3 (Eox = 0.98 V vs. SCE)151 generates a radical which decomposes in the well-known inert triphenylphosphine oxide. The mechanism is represented in Fig. 7.
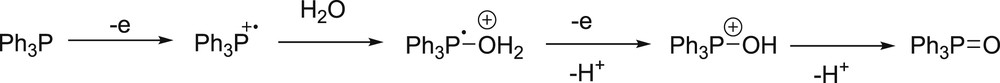
Mechanism of degradation of PPh3 upon monoelectronic oxidation.
One notices that PPh3, like tertiary amines above, is a two-electron donor triggered by monoelectronic photo-induced transfer. One PPh3 molecule can therefore theoretically participate in two reduction processes sparked off by one photon.152
10 Miscellaneous
Other sacrificial donors can be found in the literature. For example amino acids,155 and phenols156 which are hazardous pollutants whose photo-induced degradation is considered a positive boon. Besides, this is somehow reminiscent of the famous action of TyrZ in photosystem II,13 although the latter is a redox relay and not a sacrificial reagent. The use of sugars (such as glucose or fructose) in basic or acid aqueous media144,157,158 is reported too. An impressive list of organic molecules as potential SDs is available in the articles of Krasna et al.,144,157 where hydrogen production from water using solar light is attempted in a TCS. Only few SDs could assist H2 photogeneration.
11 Conclusion
Apart from the redox potential considerations presented in part 1, knowing though the difficulties related to accurately defining a potential for an irreversible step, the choice of an SD over another is not easy to rationalize. Accordingly, aliphatic and aromatic amines are well ahead of all other sacrificial donors in the field of artificial photosynthesis, from the sheer experimental point of view. As mentioned above, the properties of SD candidates may depend on the pH and nature of the medium, and in three-component systems the mixture can quickly become painfully complex (because in particular changing one parameter such as the solvent will have drastic effects on PS, SUB and SD altogether). To develop a working TCS and to optimize are therefore largely an empirical work. Nevertheless, trends can be observed. In particular, hydrophobic amines seem to necessitate the presence of an organic solvent, without nonetheless excluding the presence of water which is necessary to achieve irreversible degradation of the photo-produced radicals. The choice of an SD is likewise conditioned by pH and environment. Oxalates or ascorbates are more adapted to an aqueous, possibly acidic medium. The ambivalence of TEA and TEOA may precisely be the reason why they are so popular sacrificial donors.
However, the era of SD will reach an end, since a typical TCS is admittedly not a viable approach for large scale and economical production of solar fuels. Such TCSs are accordingly only proofs of concept and can be seen as experimental laboratory-scale tools, not futuristic solar fuel generators. Using water as the SD, just like photosynthetic organisms, would lead to the proverbial water splitting: generating hydrogen and oxygen gas with nothing else but water and light would put an end to the energy crisis and pollution. But the efficient oxidation of water (into molecular oxygen) and the control of all the side reactions is a very difficult task. An interesting alternative is the elaboration of photoelectrochemical devices (PEC), where sacrificial donors or acceptors are replaced by biased electrodes.7,37,159,160,22,36,38,161 Even more relevant are the tandem devices where the bias is no more necessary.162–164 As mentioned before though, TCSs with artificial SDs will be necessary to design the efficient PS and catalysts which will be implemented in a PEC, justifying why SDs still have a comfortable few decades ahead.
Acknowledgments
The authors wish to thank Dr. Alain Deronzier for very fruitful discussions.