

1 Introduction
Solar energy is the only renewable energy source that has sufficient capacity for the global energy need, and it is the only one that can address the issues of energy problems [1]. A huge amount of solar energy is harvested and stored via photosynthesis in plants, algae, and cyanobacteria for a period of more than 3 billion years. Photosynthesis converts the solar energy into chemical energy via photoinitiated multistep electron transfer between donors and acceptors in the reaction center.
Thus, pigment molecules such as chlorophylls and carotenoids absorb the visible light. These dyes are bound to light-harvesting protein complexes located in the photosynthetic membrane that surround the reaction centers and serve as an antenna. Photosynthesis is initiated by the absorption of a photon by an antenna molecule, which occurs in about a femtosecond and causes a transition from the electronic ground state to an excited state. Then, the excited state decays by vibrational relaxation to the first singlet excited state. The excited state energy is transferred to the reaction center, where the photon energy is converted into electronic energy. This process produces a charge-separated state across the membrane. This unique photosynthetic system has inspired many chemists to prepare artificial systems for solar fuel applications [2]. The preparation of photosynthetic mimics consists of two components. One of them acts as an energy donor and the other as an energy acceptor. The donor absorbs light and the energy is then transferred to the acceptor, which emits light with a longer wavelength. The efficiency of this transfer of energy depends on some factors such as the spectral overlap of the donor emission with the absorbance of the acceptor, the distance between the donor and the acceptor, and the relative orientation between the molecules [3].
In the first attempts to mimic photosynthesis, the reaction center components were studied but they did not serve as successful models for photosynthetic electron transfer, because it is difficult to control the electron transfer in unbound dyes [4]. To gain a better control of the electron-transfer process, different donor–acceptor linked molecules were studied. Thus, molecular dyads were used as the simplest type of such a device, where it is possible to generate a long-lived charge-separated state by tuning the electronic coupling between the two compounds and by changing the length and the type of their linkage [5]. Among various chromophores, porphyrins have been extensively used as light harvesting and electron-transfer units in artificial photosynthetic systems because of their resemblance to chlorophyll natural pigments [6]. Porphyrins can be structurally modified to tune their electronic properties and can form many complexes with metal ions. Moreover, they exhibit one strong Soret band and four moderately Q-bands at 400–800 nm region, whereas metalloporphyrins exhibit a Soret and one or two Q-bands at the same region. Also, they possess very rich redox chemistry that can be easily tuned. Although they exhibit many advantages as antenna chromophores, they do not tend to absorb strongly throughout the wavelengths useful for photosynthesis. The first covalently linked porphyrin-based donor–acceptor dimeric systems were described in 1972 [7]. Since then, a large variety of triads and multicomponent tetrapyrrole species have been prepared and studied [6b,8]. Moreover, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPYs) have attracted considerable attention in recent years as antenna molecules and as electron donors and electron acceptors [9]. BODIPY molecules are stable compounds, can be synthesized with a variety of structural modifications and handled easily because of their good solubility in many common organic solvents. Also, they exhibit many interesting properties such as sharp absorption and emission near 500 nm, high molar absorption coefficients, high fluorescence yields, low rates of intersystem crossing, relatively long excited state lifetimes, and excellent photostability [10]. It is because of these properties of porphyrins and BODIPYs that these compounds have complementary light absorbing properties. Therefore, conjugation of both molecules can absorb over a broad range of the visible light improving their light-harvesting efficiencies throughout the solar spectrum. In this review, we summarize porphyrin–BODIPY model compounds for artificial photosynthesis mainly prepared in our group; their synthesis, energy- and electron-transfer processes, and in some cases transient absorption and theoretical calculations are also presented.
2 Porphyrin–BODIPY assemblies
Various porphyrin–BODIPY conjugates have been synthesized and reported because of their use in various different applications, including mimics for the photosynthetic reaction center [11], as candidates for light sources [12] and photodynamic therapy [13], as well as in molecular electronics [6d]. Therefore, our first aim was to prepare artificial photosynthetic mimics using porphyrin and BODIPY as chromophores, linked via different bonds. These combined molecules absorb strongly in the blue-green region, possess complementary properties with porphyrins, and are highly fluorescent dyes. They can be covalently linked to the periphery of the porphyrin or noncovalently linked with axial attachment of BODIPY on the metalloporphyrin via coordination bonds. In this report, we will mainly focus on covalently or noncovalently linked porphyrin–BODIPY dimeric and trimeric compounds of porphyrin–BODIPY that have been synthesized in our laboratory.
2.1 Linkage via cyanuric chloride bridge
Cyanuric chloride is a very useful and inexpensive compound that has been extensively used in a variety of organic reactions [14] and as a template for the synthesis of macrocycles [15], dendrimers [16], and multiporphyrin arrays [17]. Its temperature-dependent reactivity allows the introduction of up to three different nucleophiles on an s-triazine unit through sequential substitution of its three chlorine atoms. As a result, it allows the use of one-pot protocol reactions, providing a modular synthetic route to the desired multiporphyrin arrays in good yields by avoiding extensive and expensive preparative and purification procedures. Thus, taking advantage of their properties, we prepared assemblies 4 and 5 (Scheme 1) in which a BODIPY chromophore is connected via a cyanuric chloride bridge with a free-base porphyrin or a zinc porphyrin, respectively [18]. The synthesis of dimers 4 and 5 is shown in Scheme 1. Reaction of porphyrin 1 with cyanuric chloride under low-temperature conditions afforded the monoadduct 2, which is stable enough to be purified by column chromatography. The BODIPY–porphyrin conjugate 4 was obtained by a reaction of 2 with 3 at 60 °C. At this temperature, the second chloride of the cyanuric chloride group can be substituted by an amine group. Reaction of the free-base porphyrin 4 with zinc acetate readily afforded the corresponding Zn derivative 5.
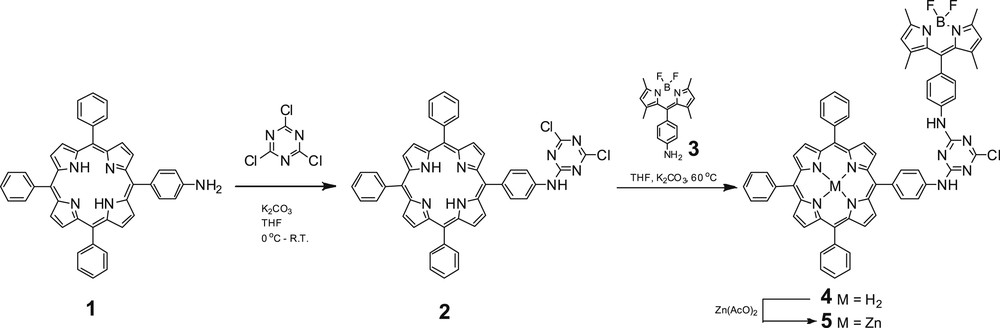
Synthetic procedure for the preparation of compounds 4 and 5.
The comparison of the absorption spectra and cyclic voltammograms of dyads 4 and 5 with those of their model compounds showed that the spectroscopic and electrochemical properties of the constituent chromophores were essentially retained, indicating negligible interaction between them in the ground state. Steady-state and time-resolved fluorescence studies clearly showed that initial excitation of the BODIPY unit at 490 nm was followed by rapid BODIPY to porphyrin energy transfer as shown by the greatly reduced BODIPY fluorescence quantum yields and lifetimes of 4 and 5 (Φ ≈ 0.01, τ ≈ 0.1 ns) with respect to the corresponding values of a free BODIPY control (Φ = 0.58, τ = 3.58 ns). Moreover, ultrafast transient absorption experiments revealed that excitation of 4 and 5 into their BODIPY-based π–π* excited states is followed by a rapid energy transfer to the lowest lying porphyrin-based singlet excited states with rate constants of 2.9 × 1010 s−1 and 2.2 × 1010 s−1, respectively. Thus, the BODIPY dye acts as an antenna and increases the light-harvesting potential of the porphyrin chromophore. The slightly faster energy transfer rate measured for 4 correlates well with the larger spectral overlap compared with that of 5. The observed BODIPY to porphyrin energy-transfer rates were considerably faster than those measured in other BODIPY–porphyrin dyads with nonconjugated bridges [19] and comparable with those found in dyads connected through an amide linkage [20] or a rigid conjugated ethynyl bridge [21]. Analysis of the experimental results with the assistance of density functional theory (DFT) calculations suggested a Förster-type dipole–dipole interaction as the most likely mechanism of BODIPY to porphyrin energy transfer. In another work, we used conjugates 4 and 5 as precursors and we substituted the third chlorine atom of the cyanuric chloride with a substituted fullerene that possess an ethylene glycol chain terminating in an amino group (Scheme 2) [22]. The new electron-donor/acceptor conjugates, namely 6 and 7 (Scheme 2), feature three different components linked through s-triazine. These compounds were also examined as artificial photosynthetic models in mimicking the energy- and electron-transfer processes of natural photosynthesis. The strategy for the synthesis of triads 6 and 7 represents a new and facile approach for the preparation of covalently linked multichromophoric systems with a star-type arrangement of three photo- and redox-active moieties around a central bridging unit. In addition to the facile synthesis, the star-type arrangement of the components around the central s-triazine unit permits direct interaction between the three components, in contrast to reported examples in which the three molecules are arranged in a linear fashion.
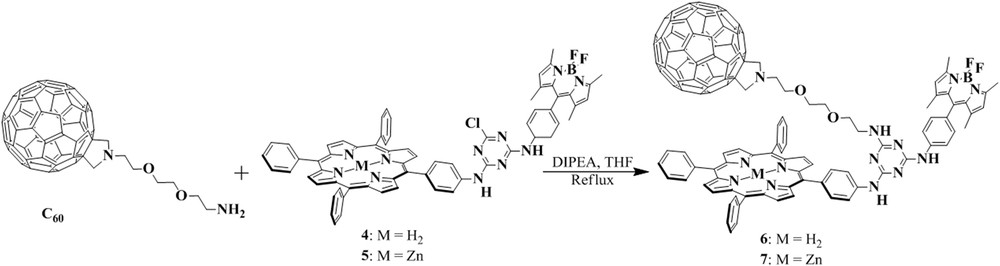
Synthesis of compounds 6 and 7.
Comparison of the absorption spectra and cyclic voltammograms of 6 and 7 with their reference molecules showed that the spectroscopic and electrochemical properties of the individual constituents are basically retained, although some appreciable shifts in terms of absorption indicate some interactions in the ground state.
Turning to the transient absorption studies of these triads, selective BODIPY excitation at 490 nm produces the first singlet excited state of BODIPY. A minor fraction decays through fluorescence to the ground state, whereas the major fraction (>99%) decays by energy transfer to the porphyrin singlet excited state with time constants of 32 and 26 ps, respectively.
In contrast to 4 and 5 where the porphyrin singlet excited state decays to the ground state by fluorescence and intersystem crossing to the triplet, in 6 and 7 it is followed by rapid intramolecular photoinduced electron transfer yielding the metastable radical ion pair states H2P+/C60− and ZnP+/C60−, which decay back to the singlet ground state with lifetimes of 3000 and 900 ps, respectively. It is worth noting that little evidence of direct electron transfer from BODIPY to C60 was observed in the transient spectra. Nevertheless, charge-separated state (BODIPY+/C60−) with a lifetime of 80 ps was confirmed as the dominant decay pathway of an analogous BODIPY-C60 dyad and it was studied as a control. The absence of electron transfer from BODIPY to C60 can be attributed to kinetic factors because BODIPY to H2P/ZnP (ca. 30–32 ps) energy transfer is faster than that of BODIPY to C60 electron transfer (80 ps). Therefore, the sequence of energy transfer from the BODIPY to the porphyrin and the subsequent electron transfer from the porphyrin to the fullerene acceptor in the present models mimics the events of natural photosynthesis.
Cyanuric chloride was also used as a linker for the preparation of trimer 8 (Fig. 1) in which two BODIPY molecules were covalently attached via an s-triazine unit with a free-base meso-substituted porphyrin [23]. The synthesis of the triad was accomplished via stepwise amination reactions. Absorption spectroscopy and electrochemical studies of this triad revealed that there is a negligible electronic interaction between the porphyrin and BODIPY moieties in the ground state. However, steady-state fluorescence experiments suggested that there is an energy transfer from the BODIPY to the porphyrin molecule. Therefore, in this case the BODIPY units are acting as an antenna and the phenomenon is more pronounced as this triad possesses two BODIPY molecules, which can absorb more light. Triad 8 was used as a chromophore in dye sensitized solar cells [23] and in bulk heterojunction solution processed organic solar cells [24], exhibiting moderate power conversion efficiencies.
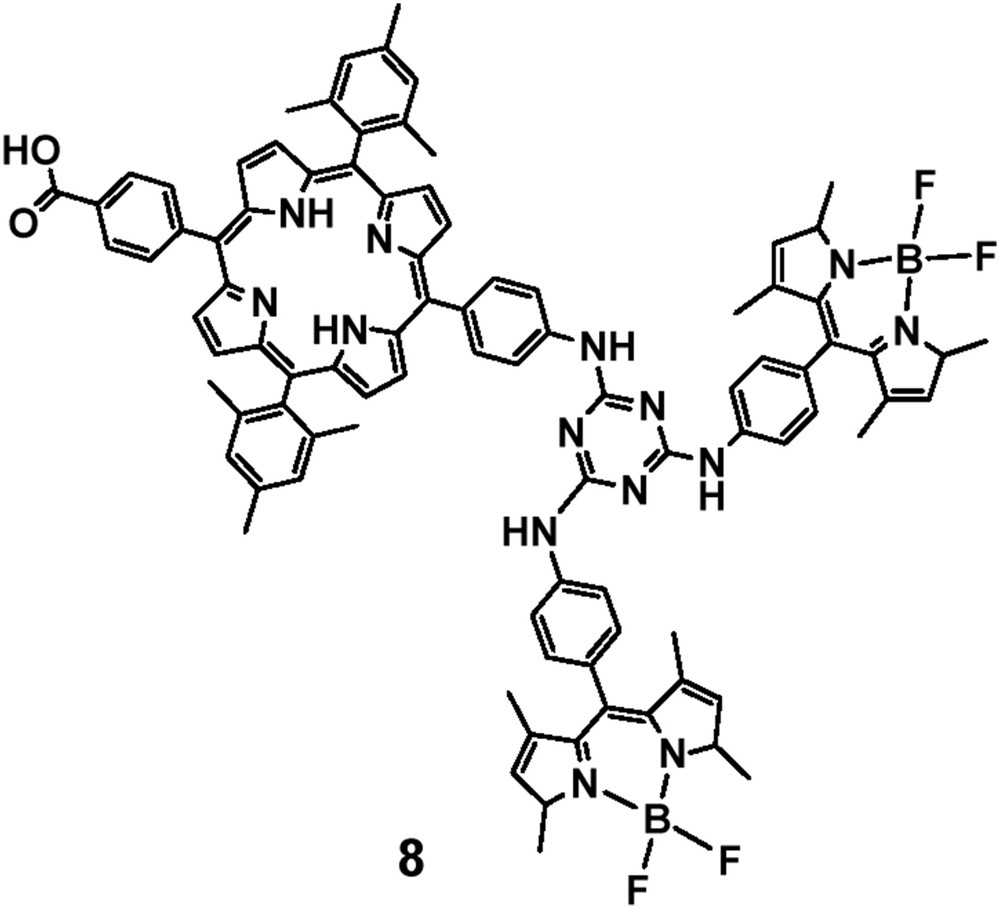
Structure of porphyrin–BODIPY triad 8.
2.2 Linkage via amino group
In this section, two BODIPY molecules were attached directly at the meso-position of the porphyrin ring via a palladium-catalyzed amination reaction [25]. This approach allowed the synthesis of a trimeric compound that absorbs light over most of the visible region by using a single halogenated porphyrin under mild conditions and high yields. The triad porphyrin 10 was successfully synthesized under palladium-catalyzed double amination conditions with coupling Zn-meso-dibromo porphyrin 9 and amino-BODIPY 3 in high yield (93%) (Scheme 3).
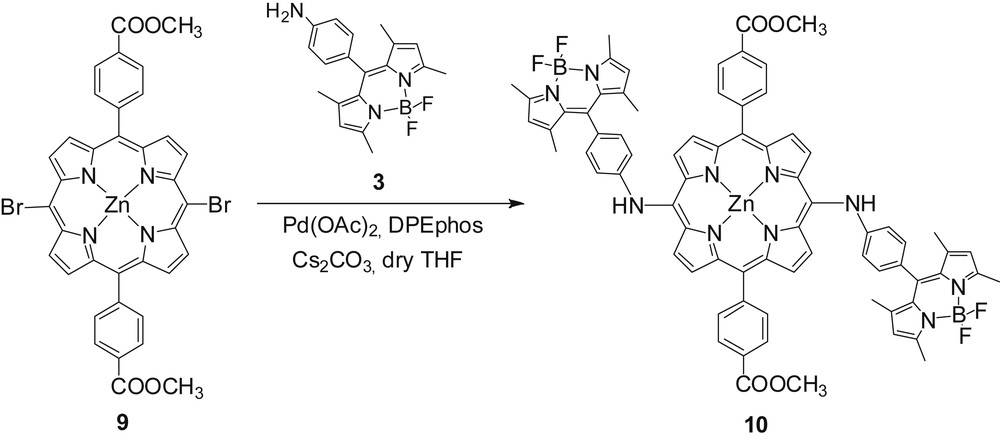
Synthesis of trimeric compound 10.
The absorption spectrum of triad 10 (Fig. 2) shows features attributed to both of its components that had an intense absorption at 503 nm because of the two BODIPY dyes and a Soret at 445 nm and two Q-bands at 561 and 614 nm because of the metalated porphyrin. These results suggested that there is a weak electronic interaction between the two side BODIPY molecules and the central Zn–porphyrin in the ground state.
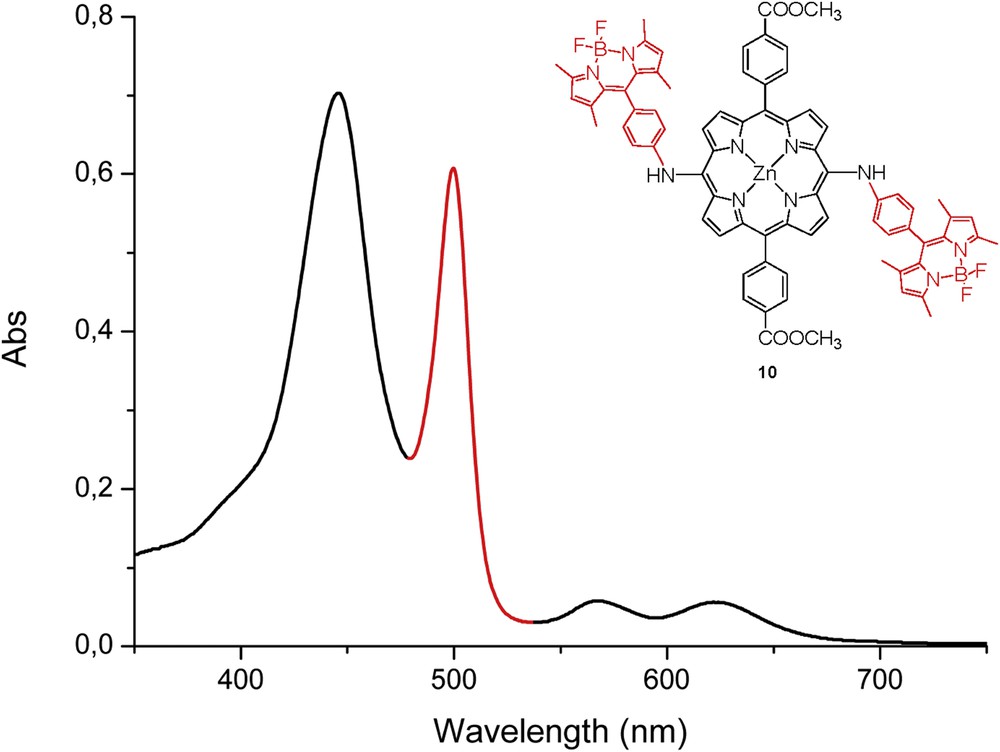
The absorption spectrum of trimer 10.
Emission spectroscopy studies of triad 10 showed that the fluorescence of BODIPY was strongly quenched compared with the free BODIPY molecule. Also, the excitation spectrum of 10 monitoring at the fluorescence of the porphyrin at 685 nm showed an intense BODIPY absorption at 499 nm. All of the aforementioned data support a rapid energy transfer from the BODIPY to the corresponding zinc–porphyrin singlet excited state.
2.3 Linkage via ethynyl bridge
A different type of linkage between the porphyrin and the BODIPY was examined via the ethynyl bridge. Therefore, in this study two BODIPY molecules were attached at the meso-position of the porphyrin through a triple bond to prepare trimeric compound 12 (Scheme 4) [26].
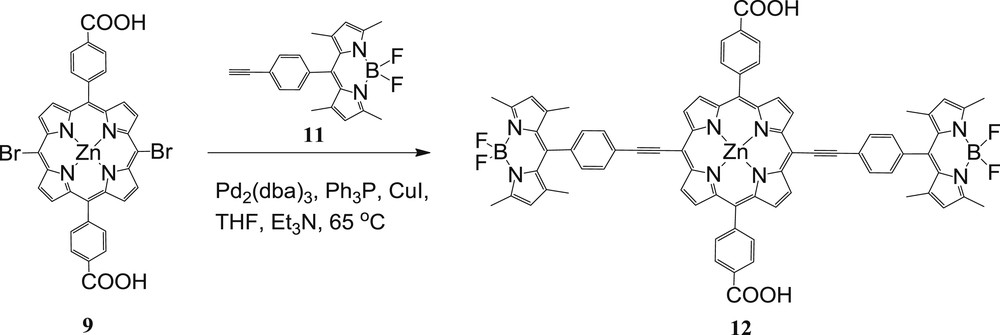
Synthesis of triad 12.
The absorption spectrum of triad 12 is the combination of absorption features from the two constituent chromophores. In addition, irradiation of compound 12 at 480 nm, corresponding to selective excitation of the two BODIPY units, resulted in emission from the Zn–porphyrin at 662 nm, as well as residual fluorescence from the two BODIPY groups at 517 nm, which was strongly quenched (Fig. 3). Also, the excitation spectrum of 12 monitoring at the fluorescence of the porphyrin moiety at 680 nm showed a pronounced BODIPY absorption feature at 503 nm (Fig. 4). Combination of steady-state and time-resolved emission spectra clearly supports photoinduced energy transfer from the 1π–π* excited state of the BODIPY chromophore to the lower lying singlet excited state of the porphyrin unit, leading to sensitized fluorescence from the latter. There are more examples in the literature where the BODIPY molecule is connected via a triple bond on the porphyrin ring, and in all cases there is sufficient energy transfer from the BODIPY unit to the porphyrin [6d].
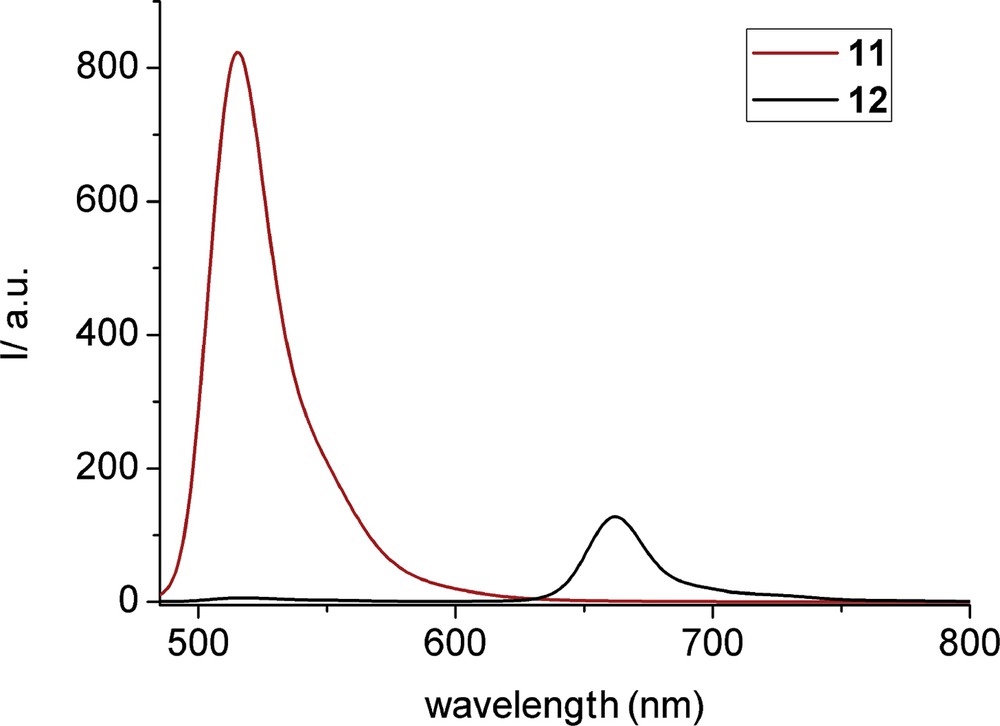
Room-temperature fluorescence spectra of isoabsorbing (A = 0.1, at 480 nm) tetrahydrofuran (THF) solutions of 11 and 12 exciting at 480 nm.
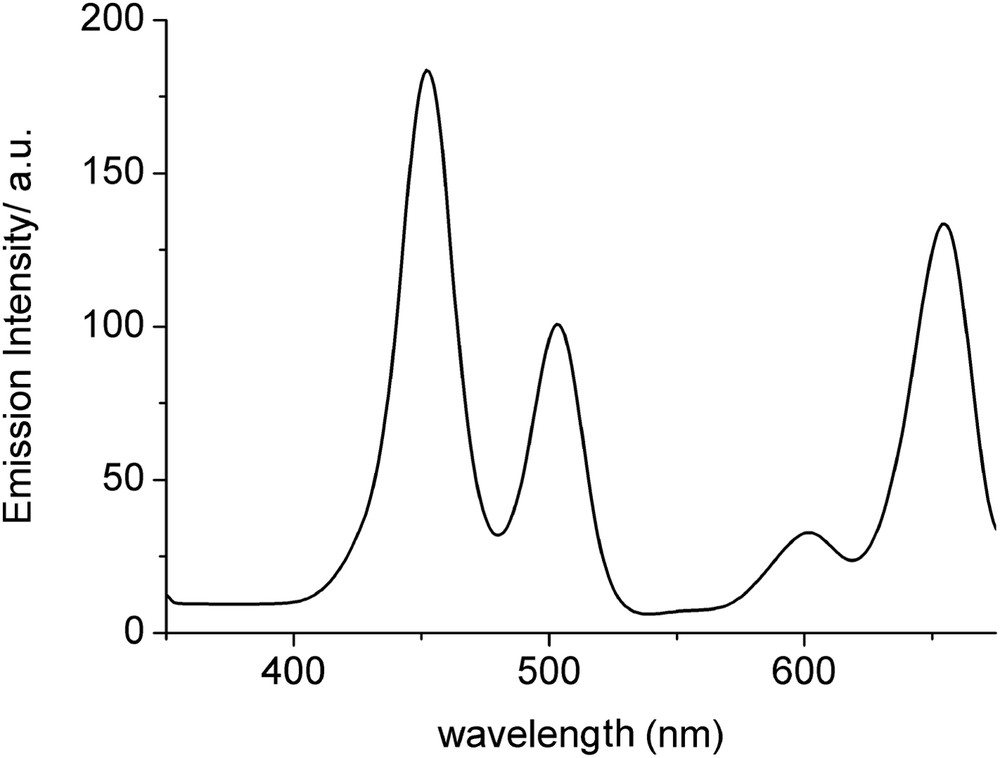
The excitation spectrum of 12 in tetrahydrofuran (THF) at 298 ± 3 K monitoring at 680 nm.
2.4 Linkage via metal
Although there are numerous studies focused on BODIPY–porphyrin dyads, only a few examples are dealing with axial coordination of the BODIPY unit to the metal center of the porphyrin. This synthetic approach offers the benefit of avoiding the elaborate and multistep synthetic procedures usually involved in the preparation of unsymmetrically substituted porphyrins. The organic periphery of the porphyrin is thus left free for further chemical functionalization. Shiragami et al. [27] reported the first BODIPY–porphyrin dyad synthesized through axial substitution. They prepared a series of dyads (13–15) using hydroxyphenyl-BODIPY with different lengths linked with an antimony tetraphenylporphyrin (Sb(TPP)) and a triad (16) consisting of two BODIPY units coordinated to the Sb(TPP) (Fig. 5).
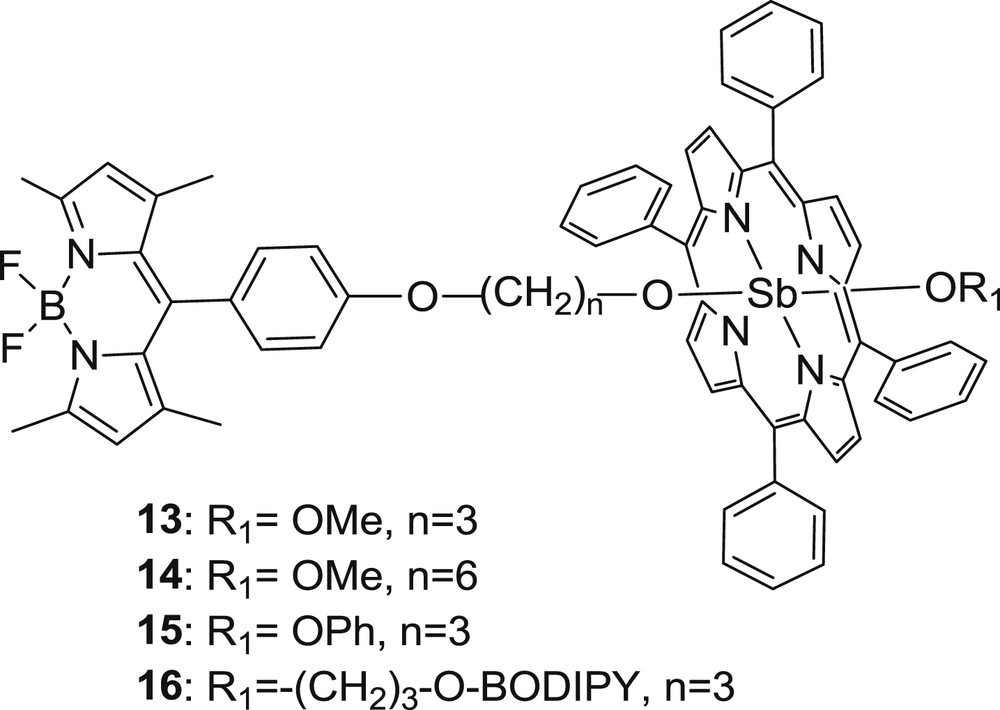
Structure of compounds 13–16.
Energy transfer from the BODIPY unit to the Sb(TPP) chromophore occurred and the porphyrin-based singlet excited state formed was quenched by the phenoxy ligand via nonradiative processes involving electron transfer. In another approach, Khan and Ravikanth [28] synthesized noncovalent BODIPY–porphyrin trimers 17–20 using metal to ligand interaction throughout the pyridine group (Fig. 6). The formation of triads 17–20 was confirmed by significant upfield shifts of pyridyl protons because of the ring current effect of metalloporphyrin. The nuclear magnetic resonance studies also revealed that BODIPY–RuTPP(CO) triads (17–18) are more stable than BODIPY–ZnTPP compounds (19–20).
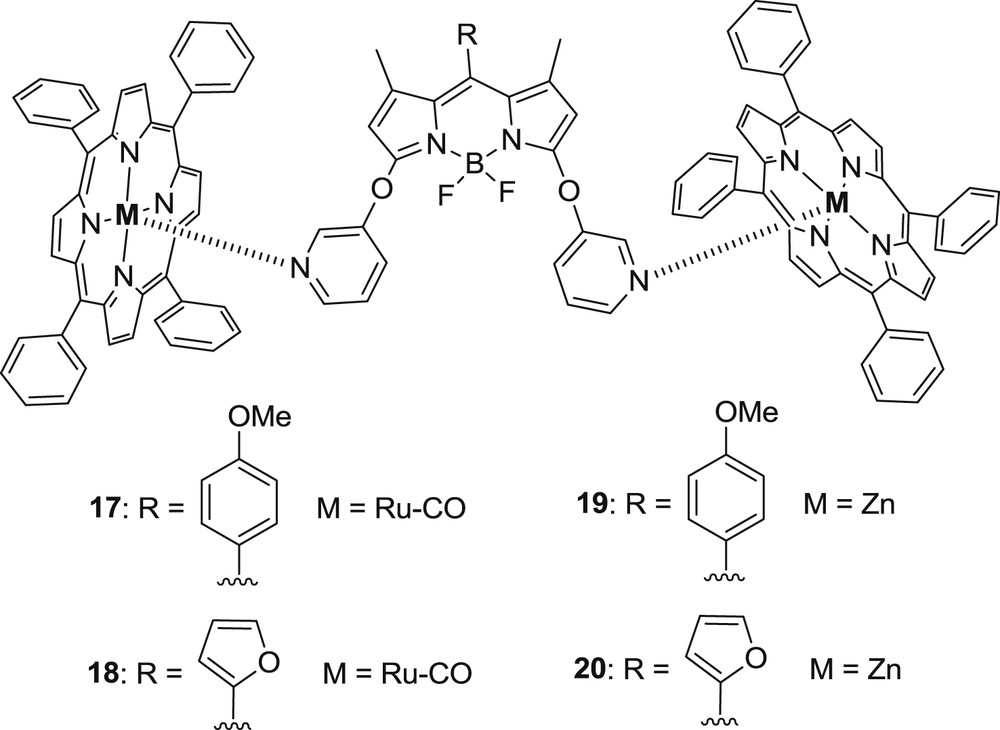
Structure of trimers 17–20.
In this work, there is an indication that excited-state interaction exists because the BODIPY-based emission was significantly quenched after coordination with the RuTPP(CO) unit. The BODIPY-ZnTPP-based triads were not stable enough at low concentrations, and for this reason it was not possible to study their absorption and emission properties.
To this end, our group investigated the effect of axial substitution on tin and indium porphyrins by BODIPY units during electron- and energy-transfer processes [29]. In both cases, the linkage between the porphyrin and the BODIPY chromophore was through a phenolate or a benzoate bridge. The BODIPY–tin–porphyrin triads 23 and 25 were prepared by a reaction of tin porphyrin 21 with hydroxyl or carboxy BODIPY (22 or 24), respectively (Scheme 5) [29a].
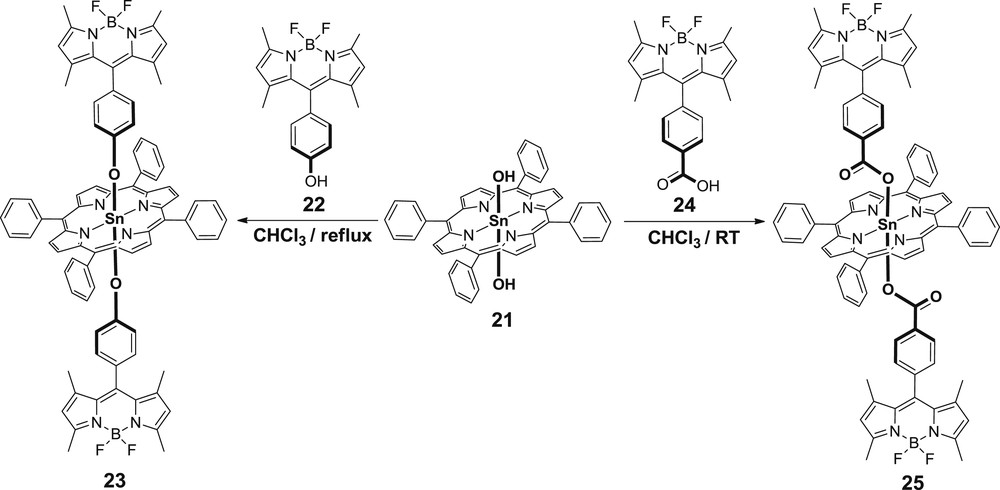
Synthetic approach for the compounds 23 and 25.
The electrochemical data coincide with the DFT calculations revealing that the highest occupied molecular orbital is mainly located on the BODIPY unit and the lowest unoccupied molecular orbital possesses predominantly porphyrin character. The absorption studies showed weak ground state interactions; however, emission measurements suggested that an efficient excited state interaction is possible to take place. Indeed, the transient absorption experiments proved the aforementioned hypothesis confirming that energy- and electron-transfer processes prevail. In both cases (23 and 25) selective excitation of the BODIPY (490 nm) resulted in rapid BODIPY to porphyrin unit energy transfer with rates = 5.0 × 1011 s−1 and = 2.9 × 1011 s−1, respectively, although in the case of 23 the phenolate ligand is responsible for the conversion of the porphyrin-based singlet state to a charge-separated state because of electron transfer ( = 2.5 × 1011 s−1) (Fig. 7, left). The rapid deactivation of the porphyrin singlet excited state because of this electron transfer is the leading factor and as such, no porphyrin-based fluorescence is detected at room temperature. On the contrary, the benzoate linker in 25 stabilizes the singlet excited state of the porphyrin unit and the deactivation process arise either by fluorescence (radiative decay) or by intersystem crossing to the formation of the triplet excited state of the porphyrin ( = 7.1 × 108 s−1) (Fig. 7, right).
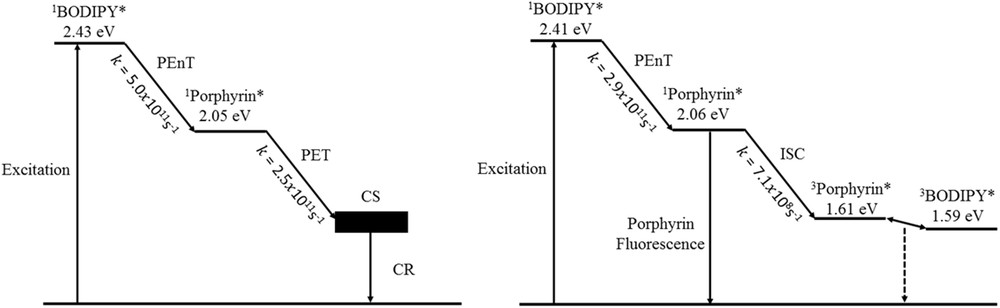
Jablonski diagrams of 23 (left part) and 25 (right part) illustrating the processes that take place after photoexcitation of the BODIPY at room temperature. * refers to the excited state.
The most recent example in the literature, regarding the axial substitution, is the synthesis of five-coordinate indium porphyrins with BODIPY units as ligands [29b]. A series of BODIPY–porphyrin dyads (27 and 28) is presented in this work consisting of an indium porphyrin 26 with hydroxy or carboxy BODIPY (22 or 24) (Scheme 6).
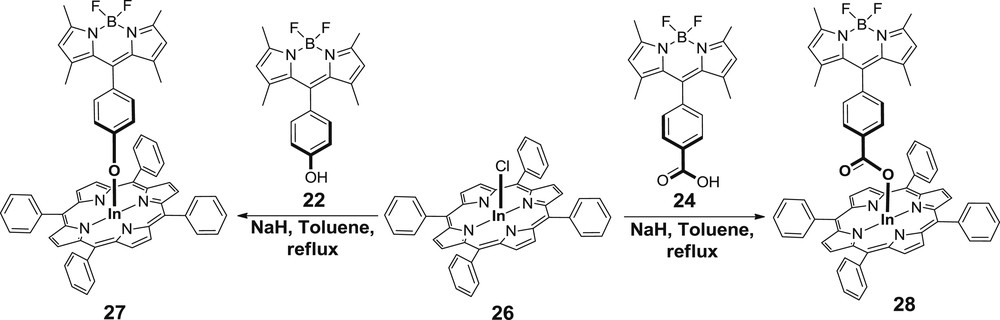
Synthetic route for the dyads 27 and 28.
The targeted compounds (27 and 28) were prepared by reacting porphyrin 26 with the corresponding BODIPY using NaH as the base. To explore the electronic properties and the molecular structures of the synthesized dyads, DFT calculations were performed. Similarly to the Sn(TPP) analogues, the detailed investigation of the absorption and electrochemical properties of the dyads (27 and 28) revealed weak interaction in the ground state. However, after the selective excitation of the BODIPY unit energy transfer from the BODIPY to the porphyrin chromophore occurs. The simplified energy level diagram, in which the energies were estimated from the emission spectra at 77 K, denotes that upon photoexcitation of the BODIPY (487 nm) the BODIPY-based singlet excited state is formed and then followed by partly deactivation directly back to the ground state (S0) and partly by energy transfer to the singlet excited state of the porphyrin unit. The porphyrin-based singlet excited state then decays by fluorescence and intersystem crossing to the triplet excited state (Fig. 8).
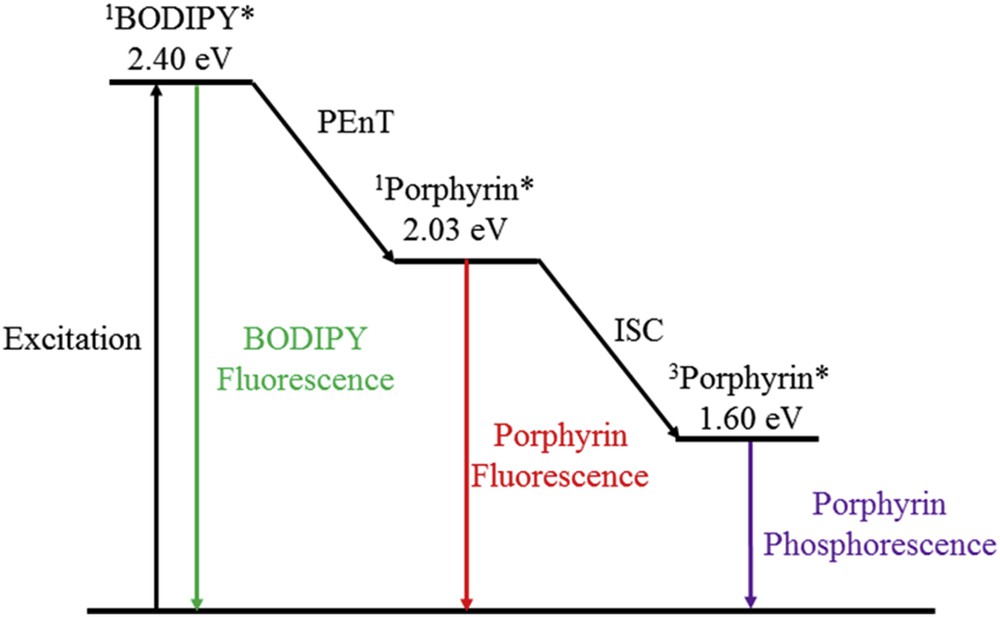
Simplified energy level diagram showing the possible events, which follow after excitation of the dyads (27 and 28) to the BODIPY unit. The energy levels are estimated from the emission spectra in the rigid matrix at 77 K. * refers to the excited state.
In this example, the BODIPY fluorescent, although not strongly quenched, the photoinduced energy transfer in 27 and 28 is less efficient than in the tin analogues 23 and 25. Nonetheless, in both systems the BODIPY unit can effectively sensitize the highly redox active singlet excited state of the porphyrin, therefore can potentially be used as a supramolecular antenna system.
3 Summary and outlook
As demonstrated in this review, covalently or noncovalently linked dimeric and trimeric porphyrin–BODIPY moieties were examined, regarding the impact of different linkages in the energy transfer efficiency. In all reported cases herein, BODIPY acts as an energy donor, transferring energy to porphyrin, because the singlet state energy level of the BODIPY is higher compared to the porphyrin. It was verified that the linkages, namely (1) cyanuric chloride bridge, (2) amino group, (3) ethynyl bridge, and (4) metal coordination, strongly affect the efficiency in the energy-transfer process. In fact, the most efficient energy transfer process (∼99% quenching) was detected for compounds linked via a cyanuric chloride bridge (4, 5, 6, 7, and 8) and via an ethynyl bridge for triad 12, where an ethynyl bridge was used as a linkage between the two chromophores. On the other hand, although the linkage with amino group did not provide similar results because the observed quenched process was calculated ∼92%. Concerning the last category, that is, the linkage via metal, the results were completely dependent on the bond that was formed between the metal and the BODIPY molecule. In the case of tin porphyrin triads (23 and 25), we observed that there was an efficient BODIPY to porphyrin energy transfer (99% quenching) in the excited state. Although for indium dyads (27 and 28) the quenching process either slightly or dramatically decreased, 90% and 60% quenching for dyads 28 and 27, respectively. Overall, the linkage through a cyanuric chloride bridge appears to be the most appropriate choice in constructing porphyrin–BODIPY assemblies for light harvesting applications. Moreover, these kinds of compounds can be prepared in high yields with the use of a facile temperature dependent synthetic procedure. Consequently, this review can possibly provide a strategy for the enhancement of light harvesting ability of porphyrins–BODIPY moieties making them attractive for various applications.
Acknowledgments
Α.G.C. acknowledges FP7-REGPOT-2008-1, project BIOSOLENUTI No. 229927, from European Commission, Heraklitos grant from Ministry of Education of Greece, the Special Research Account of the University of Crete and GSRT and IKY fellowships of excellence for postgraduate studies in Greece–Research programs of excellence IKY–Siemens.
The authors would like to express their special thanks to Kallia Katsampoxaki-Hodgetts, English teacher of the Chemistry Department, for her valuable comments for this manuscript.