

1 Introduction
Among metal-catalyzed cascade reactions, those initiated by palladium-based catalysts are undoubtedly the ones that have been the most intensively studied for more than the last 40 years [1]. Efficient processes have been developed to quickly synthesize valuable molecular scaffolds bearing various heteroatoms mostly including nitrogen [2] and oxygen [3]. In contrast to this intensive work, transformations involving organosulfur substrates have been far less studied, certainly because of the poisoning of the catalyst caused by the thiophilicity of palladium. However, in recent years, the number of palladium-catalyzed processes involving substrates bearing a sulfur functionality has significantly increased and elegant methodologies have emerged insufflating a real interest to the synthetic chemist community [4]. During the course of our studies on metal-mediated transformations of sulfur-containing substrates [5], we have recently reported a domino palladium-catalyzed access to original thiacycles, which are compounds of outstanding importance in particular for the pharmaceutical industry, starting from propargylic or alkynyl sulfides [6]. This sequence involves an initial cyclizing carbopalladation step followed by a cross-coupling reaction between the resulting vinyl-palladium species and a coupling partner (stannylated or borylated) (Scheme 1).
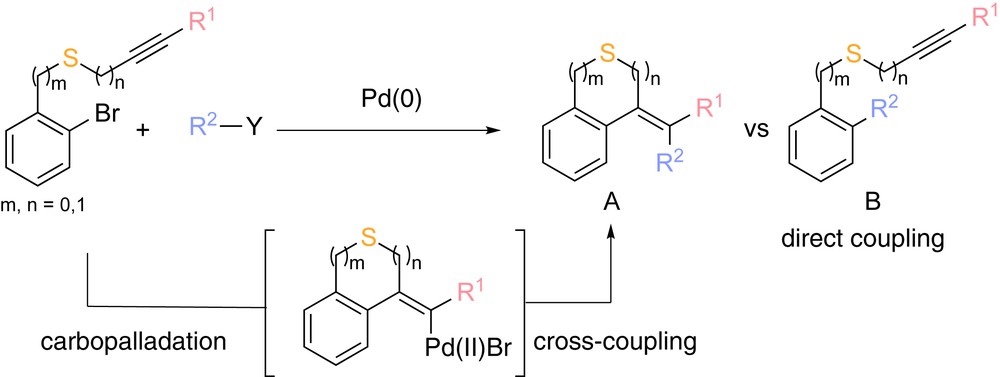
General strategy of a palladium-catalyzed domino reaction of alkynyl and propargyl sulfides.
However, in this preliminary report, only the most efficient cross-coupling reactions, namely the Stille and the Suzuki couplings, have been investigated and an additional effort had to be made to rationalize the behavior of such substrates under different palladium-catalyzed domino transformations. To do so, we are reporting here a complete study on the reactivity of three representative types of substrates (1a, 1b, and 1c) toward four distinct palladium-catalyzed domino reactions involving an initial cyclizing carbometalation step followed by the four most common cross-coupling reactions, respectively, the Stille reaction (organotin partner), the Suzuki–Miyaura reaction (organoboron partner), the Sonogashira reaction (alkyne partner), and the Mizoroki–Heck reaction (alkene partner) (Fig. 1).
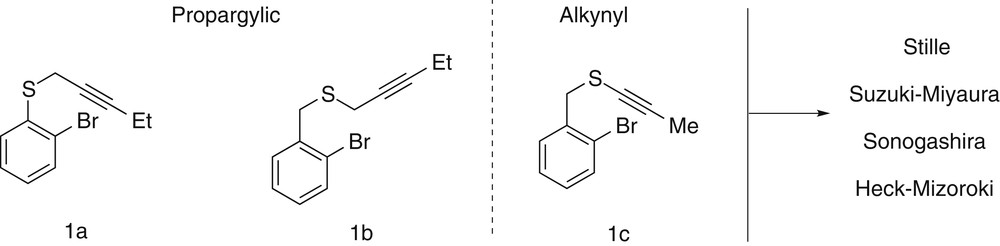
The 3 substrates and the 4 ending couplings used for the study.
2 Results and discussion
To rationalize the behavior of alkynyl and propargyl sulfides while submitted to these palladium-catalyzed domino transformations, we have first synthesized a set of three representative substrates namely propargyl aryl sulfide (1a), propargyl benzyl sulfide (1b), and alkynyl benzyl sulfide (1c). To access these three compounds, two different routes have been developed (Scheme 2).
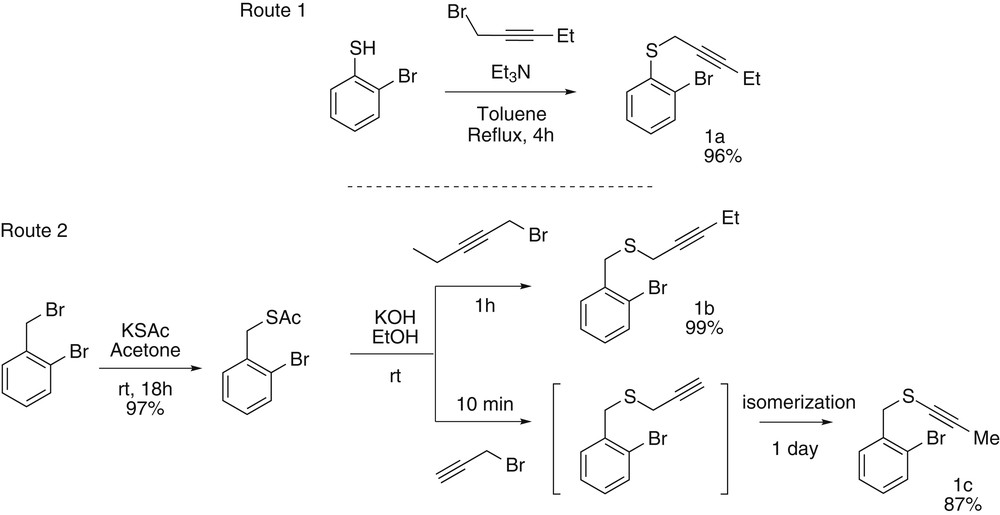
Substrate syntheses.
The first route starts from 2-bromothiophenol and is based on a classical alkylation reaction using triethylamine as base and 3-(ethyl)propargyl bromide as an alkylating agent. After 4 h under reflux the desired aryl propargyl thioether 1a was obtained almost quantitatively. The second route involves the in situ formation, by ethanolysis of a benzylic thioacetate, of a thiolate that can subsequently be alkylated. When 3-(ethyl)propargyl bromide is used as an alkylating agent, the thioether 1b is obtained quantitatively. However, when propargyl bromide is used, the alkylation occurs to form the intermediary propargyl thioether that can then undergo a zip-type isomerization to reach the targeted ynethioether 1c in a good 87% yield.
With our substrates in hand, we have decided to first focus our interest on the sequences involving a cyclocarbopalladation/Stille coupling reactions. These reactions are usually highly efficient and their optimizations are classically easy and quick. Once optimized using the compound 1a as substrate [7] and 2-furyl tributylstannane as coupling partner, it appeared that the best results were obtained with 10% of palladium tetrakis(triphenylphosphine) with 1.5 equiv of stannane in benzene at 115 °C under microwave irradiation. After 3 h a complete conversion could be observed and the desired dihydro[b]benzothiophene derivative 2a resulting from the cascade reaction was obtained in 52% yield. Notably, if the benzyl ynethioether 1c led under the same conditions to the corresponding product dihydro[c]benzothiophene 2c in a similar 59% yield, the substrate 1b driving to a 6-membered heterocycle reacted in a better way and gave the targeted isothiochromane derivative 2b in a good 81% yield. From these results it appeared that the 6-exo-dig/Stille coupling sequence was more efficient than the cascades involving a 5-exo-dig cyclization. However, the reaction seemed insensible to the mode of linkage of the sulfur atom to the alkyne moiety as the ynethioether 1c and the propargyl thioether 1a gave similar results (Scheme 3).
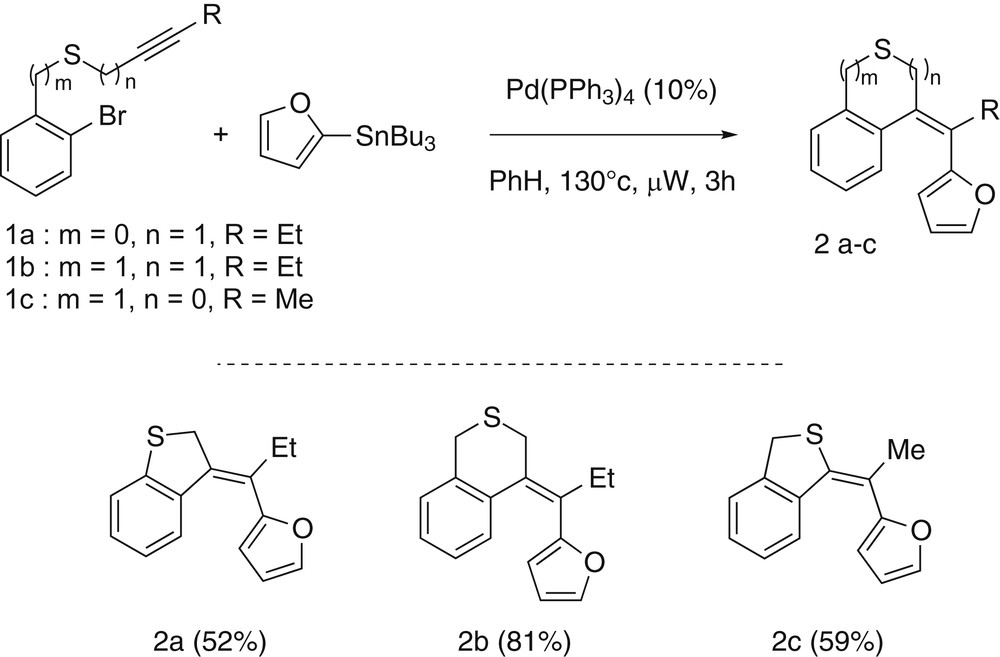
Results for the cyclocarbopalladation/Stille coupling domino reactions.
Then, we have been interested in comparing the reactivity of our three representative substrates in the case of a cascade ending with a Suzuki–Miyaura cross coupling after the initial cyclizing carbopalladation step. In that case a greater effort had to be made to optimize the reaction to obtain the cyclized molecules as sole products versus those coming from the direct coupling. When performing the reaction with 1a as a starting material and phenylboronic acid as a coupling partner, after having screened several reaction conditions, the best results were obtained with 10% of Pd(PPh3)4 and K3PO4 as a base in a mixture of 2-MeTHF and water (98/2) at 130 °C under microwave irradiation. Under these conditions, after 3 h, the substrate 1a reached the targeted compound 3a in a 66% yield. Remarkably, no trace of the corresponding product coming from a direct coupling has been detected. Then, the starting material 1c, having the sulfur atom directly linked to the alkyne has been investigated. Notably, in this case the reactivity appeared similar but the desired product 3c has been only obtained in a modest 30% yield mainly because of its instability. The benzyl propargyl thioether substrate 1b gave better results and led to the desired isothiochromane derivative 3b in a very good 91% yield. Once again the sequence involving a 6-exo-dig cyclizing step seemed to be more effective than the one based on a 5-exo-dig cyclization (Scheme 4).
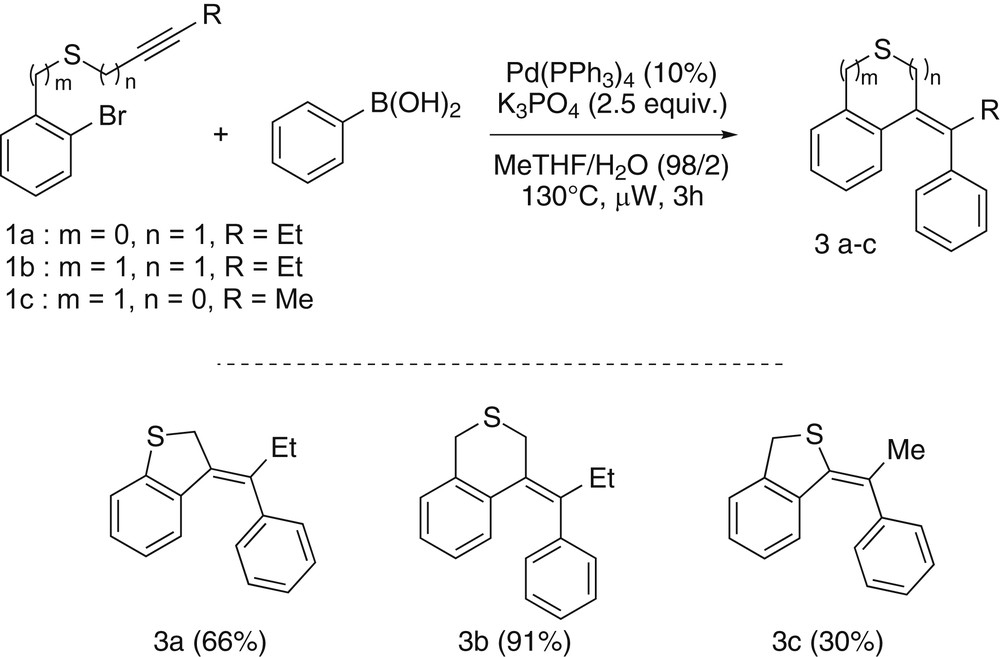
Results for the cyclocarbopalladation/Suzuki–Miyaura coupling domino reactions.
Then, to compare the efficiency of the sequence involving a Stille or a Suzuki final step, 2-furylboronic acid was reacted with the best substrate in both transformations, namely the compound 1b. Surprisingly in that case, our optimized conditions led only to the product resulting from the direct coupling 2b′, whereas the Stille coupling gave only the targeted compound 2b. By substituting the Pd(PPh3)4 catalyst by PdCl2 as a metal source and 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPHOS) as a ligand, we obtained our best results consisting in an equimolar mixture of the desired cyclized product 2b and the direct coupling compound 2b′ (Scheme 5).
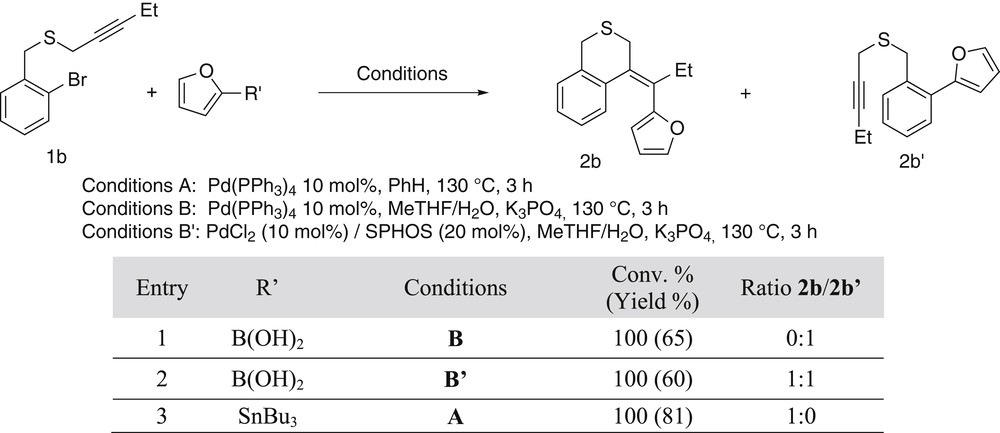
Comparison between the sequences ending with a Stille or a Suzuki–Miyaura coupling.
Then we attempted to introduce after the cyclocarbopalladation an alkynyl substituent via a Stille or a Sonogashira cross-coupling reaction with 1-trimethylsilyl alkynyl tributylstannane (method A) or with trimethylsilylacetylene (method C), respectively (Scheme 6) [8]. In the case of 1a (Scheme 6 table, entries 1 and 2), both of the methods gave disappointing results, with a low conversion (20%) and a 4a/4a′ ratio of 3/2 using Sonogashira coupling, and with a 4a/4a′ ratio of 5/1, but a low isolated yield because of the degradation of the products during purification, when Stille coupling was involved. When 1b was reacted in conditions C, the conversion was total and product 4b′ resulting from the direct cross-coupling Sonogashira reaction was obtained with almost total selectively (Scheme 6, entry 3). In contrast, under conditions A, sulfide 1b led to an inseparable mixture of products 4b and 4b′ in a ratio of 2/1 (Scheme 6, entry 4). Ynethioether 1c was then involved in the same two types of processes (Scheme 6 table, entries 5 and 6). The best selectivity in favor of the cyclic product 4c was obtained via the Stille reaction (ratio 4c/4c′, 3/1). Because of the inconvenient presence of stannane derivatives, it was not possible to isolate the products; however, we were able to characterize the compounds from their mixture resulting from the Sonogashira reaction (ratio 4c/4c′, 2/1; 41% yield).
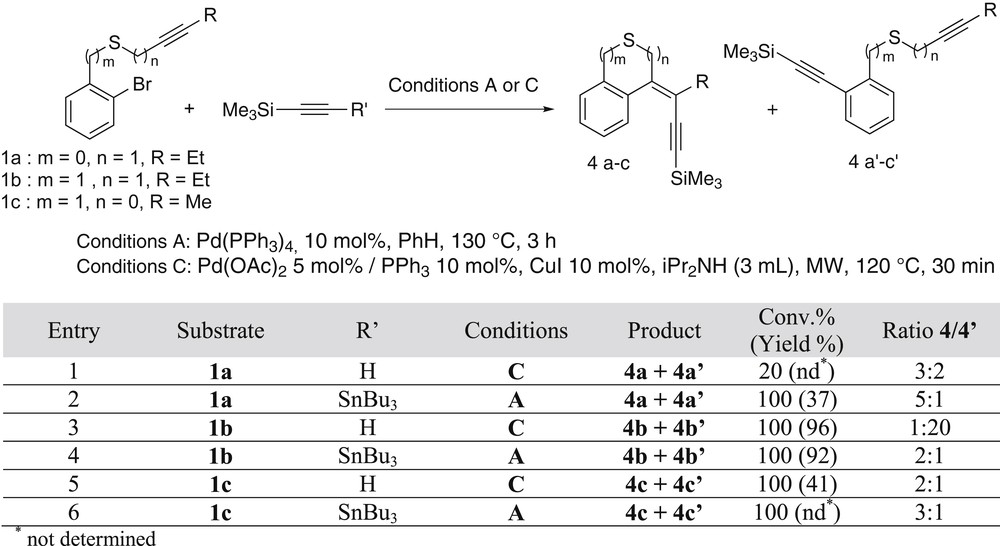
Comparison between the sequences ending with Sonogashira (C) or Stille (A) coupling.
To complete this comparative study, the Mizoroki–Heck coupling was investigated as a final step of the domino sequence. We started by exploring the reactivity of the aryl propargyl thioether substrate 1a. After optimization, we have determined that the best reaction conditions, when using methyl acrylate as a coupling partner, are 10% of Pd(PPh3)4 as a catalyst and potassium carbonate as a base in toluene for 18 h at 125 °C under classical heating (sealed tube). In that case the benzothiophene 5a″, from the isomerization of the targeted product 5a, was obtained in a 60% yield as an exclusive product. Pleasingly, no trace of the compound resulting from the direct coupling was detected. Nonetheless the ynethioether 1c was subjected to the same reaction conditions and gave the desired product 5c in a lowest 51% yield whereas the benzyl propargyl thioether led to the isothiochromane 5b in a 72% yield. It is interesting to note that in the case of this cyclocarbopalladation/Mizoroki–Heck domino sequence, the size of the newly formed cycle does not impact the efficiency of the overall process as substrates 1a and 1b gave similar results in terms of yields. However, it appeared clearly that a significant difference in reactivity exists between the substrate bearing an alkynyl or a propargyl thioether as the yield of the reaction decreases by 10% when the sulfur atom is directly linked to the alkyne moiety (Scheme 7).
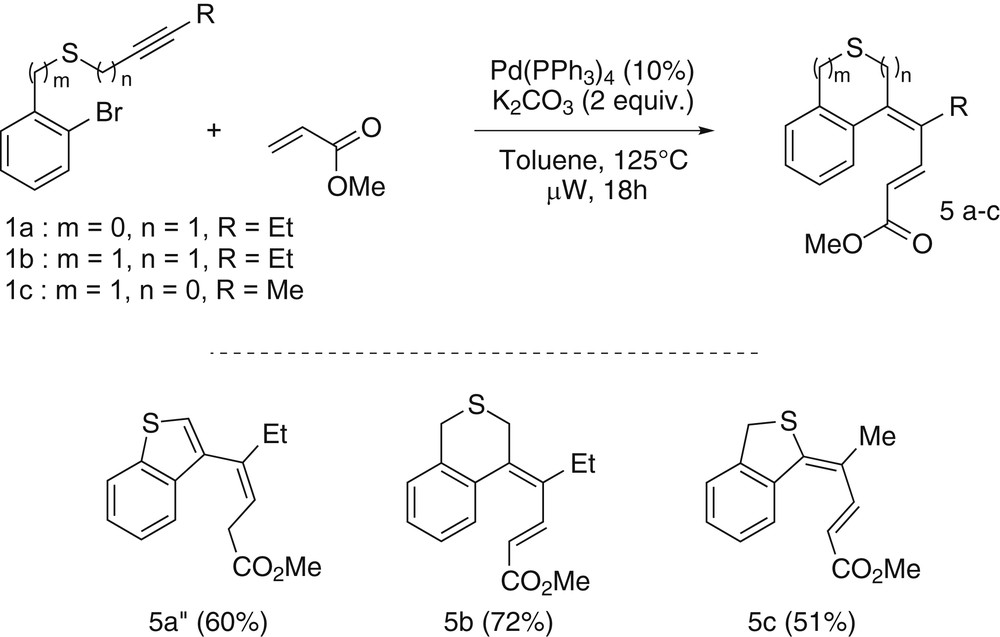
Results for the cyclocarbopalladation/Mizoroki–Heck coupling domino reactions.
3 Conclusions
This study demonstrates that palladium-catalyzed domino sequences are an efficient tool for the synthesis of valuable heterocycles containing a sulfur atom. We have been able to observe the behavior of three representative substrates when submitted to four distinct cyclocarbopalladation/cross-coupling domino reactions. During the course of this study, it clearly appeared that the domino sequences applied to the substrate driving to a 6-exo-dig initial cyclocarbopalladation reaction were constantly more efficient than the same transformations done on 5-exo-dig precursors. The sequences ending by Stille, Suzuki–Miyaura, or Mizoroki–Heck coupling have been shown to be efficient and highly selective to the cyclized products. Notably, when the introduction of an alkyne was targeted at the end of the cascade, it appeared that the Sonogashira coupling led every time to a mixture of the desired cyclic product with the product resulting from the direct coupling between the aryl moiety of the substrate and the alkyne used as partner. Finishing the domino sequence with a Stille coupling instead of a Sonogashira coupling allowed improving significantly the ratio of the mixture in favor of the desired cyclized compound.
4 Materials and methods
4.1 General consideration
All reagents, chemicals, and dry solvents were purchased from commercial sources and used without purification. Reactions were monitored by thin-layer silica gel chromatography using Merck silica gel 60 F254 on aluminum sheets. Thin-layer silica gel chromatography plates were visualized under UV light and revealed with acidic p-anisaldehyde stain or KMnO4 stain. Crude products were purified by flash column chromatography on Merck silica gel Si 60 (40–63 μm). All NMR spectra were recorded in CDCl3, C6D6, or CD2Cl2 on a Bruker Avance III 400 MHz BBFO+ probe spectrometer for 1H NMR and 100 MHz for 13C NMR, and a Bruker Avance 300 MHz dual probe spectrometer for 1H NMR. Proton chemical shifts are reported in ppm (δ), relatively to residual solvent. Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), broad signal (br s), and multiplet (m). Coupling constant values J are given in hertz. Carbon chemical shifts are reported in parts per million with the respective solvent resonance as the internal standard. 1H NMR and 13C NMR signals were assigned mostly on the basis of distortionless enhancement by polarization transfer (DEPT) and 2D-NMR (correlation spectroscopy (COSY), heteronuclear multiple-bond correlation spectroscopy (HMBC), and heteronuclear single-quantum correlation spectroscopy (HSQC)) experiments. High-resolution mass spectral analysis (HRMS) was performed using an Agilent 1200 rapid resolution liquid chromatography (RRLC) high performance liquid chromatography (HPLC) chain and an Agilent 6520 Accurate mass Quadrupole Time-of-Flight (QToF). Microwave irradiation was carried out with a microwave reactor from BIOTAGE using pressurized vials. Infrared (IR) spectra were recorded on a FT IR Thermo Nicollet ATR 380 Diamond Spectrometer. Microwave irradiations have been performed using a BIOTAGE Smith Creator apparatus.
4.2 Procedures and characterizations
4.2.1 (2-Bromophenyl)(pent-2-yn-1-yl)sulfide (1a)
To a solution of 2-bromobenzenethiol (1.5 g, 8 mmol, 1 equiv) in toluene (80 mL), triethylamine (1.2 mL, 8.3 mmol, 1.03 equiv) and then 3-(ethyl)propargyl bromide (1.84 g, 12.5 mmol, 1.5 equiv) were added. The reaction mixture was heated under reflux for 4 h. After filtration of the triethylammonium hydrobromide, the filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (eluent: pentane 100%, then pentane/ethyl acetate 95/5) to afford 1.96 g of sulfide 1a (7.7 mmol, 96%).
1H NMR (400 MHz, CDCl3) δ 1.08 (t, J = 7.5 Hz, 3H, CH3), 2.13–2.20 (m, 2H, CH2), 3.65 (t, J = 2.3 Hz, 2H, SCH2), 7.06 (td, J = 7.7, 1.6 Hz, 1H), 7.30 (td, J = 7.7, 1.5 Hz, 1H), 7.42 (dd, J = 7.9, 1.6 Hz, 1H), 7.55 (dd, J = 7.9, 1.5 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 12.5 (CH3), 13.7 (CH2), 22.1 (SCH2), 73.9 (CH2CC), 85.8 (CH2CC), 123.5 (Cq), 127.0 (CH), 128.6 (CH), 130.7 (CH), 133.2 (CH), 137.2 (Cq). HRMS (ESI, 120 eV) calculated for C11H11BrS [M]+ 253.9763, found 253.9764.
4.2.2 S-(2-Bromobenzyl)ethanethioate (precursor of 1b and 1c)
To a solution of 2-bromobenzyl bromide (3.5 g, 15 mmol, 1 equiv) in acetone (100 mL), potassium thioacetate (2.05 g, 18 mmol, 1.2 equiv) was added. The reaction mixture was stirred at room temperature overnight. After filtration of the potassium bromide, the filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel to afford 3.5 g of product (97% yield).
1H NMR (400 MHz, CDCl3) δ 2.34 (s, 3H, CH3), 4.24 (s, 2H, SCH2), 7.11 (td, J = 7.7, 1.6 Hz, 1H), 7.25 (td, J = 7.7, 1.4 Hz, 1H), 7.45 (dd, J = 7.5, 1.5 Hz, 1H), 7.54 (dd, J = 8.0, 1.4 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 30.5 (CH3), 34.2 (CH2), 124.7 (Cq), 127.8 (CH), 129.2(CH), 131.4 (CH), 133.0 (CH), 137.3 (Cq), 195.1 (CO).
4.2.3 (2-Bromobenzyl)(pent-2-yn-1-yl)sulfide (1b)
To a solution of KOH (336 mg, 6 mmol, 1.5 equiv) in ethanol (60 mL), S-(2-bromobenzyl)ethanethioate (980 mg, 4 mmol, 1 equiv) and then 3-(ethyl)propargyl bromide (882 mg, 6 mmol, 1.5 equiv) were added. The reaction mixture was stirred at room temperature for 1 h. After evaporation of the solvent, hydrolysis by water, and extraction with ether, the organic phases were separated, dried (MgSO4), and the solution was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (pentane/Et2O 9/1) to afford 1.06 g of sulfide 1b (99% yield).
1H NMR (400 MHz, CDCl3) δ 1.17 (t, J = 7.5 Hz, 3H, CH3), 2.21–2.28 (m, 2H, CH2), 3.15 (t, J = 2.3 Hz, 2H, SCH2), 3.98 (s, 2H, SCH2), 7.12 (td, J = 7.7, 1.8 Hz, 1H), 7.26 (td, J = 7.5, 1.4 Hz, 1H), 7.38 (dd, J = 7.6, 1.8 Hz, 1H), 7.57 (dd, J = 7.9, 1.4 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 12.7 (CH3), 14.2 (CH2), 19.7 (SCH2), 35.9 (SCH2), 75.0 (C), 85.7 (C), 124.8 (Cq), 127.5 (CH), 128.8 (CH), 130.9 (CH), 133.4 (CH), 137.4 (Cq). HRMS (ESI, 120 eV) calculated for C12H13BrS [M]+ 267.9938, found 267.9921.
4.2.4 (2-Bromobenzyl)(prop-1-yn-1-yl)sulfide (1c)
To a solution of S-(2-bromobenzyl)ethanethioate (980 mg, 4 mmol, 1 equiv) in ethanol (60 mL), KOH (448 mg, 8 mmol, 2 equiv) and then propargyl bromide (1.2 g of 80% solution in toluene, 12.5 mmol, 1.5 equiv) were added. The reaction mixture was stirred at room temperature for 24 h. After filtration of the potassium bromide, the filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (pentane 100%) to afford 835 mg of sulfide 1c (87% yield).
1H NMR (400 MHz, CDCl3) δ 1.92 (s, 3H, CH3), 4.00 (s, 2H, SCH2), 7.15 (dt, J = 7.4, 1.0 Hz, 1H, H4), 7.29 (dt, J = 7.7, 1.8 Hz, 1H, H5), 7.37 (dd, J = 7.5, 1.8 Hz, 1H, H6), 7.58 (dd, J = 8.0, 1.1 Hz, 1H, H3). 13C NMR (100.6 MHz, CDCl3) δ 5.0 (CH3), 40.2 (SCH2), 66.8 (SC), 91.8 (CMe), 124.5 (CBr), 127.3 (C5), 129.2 (C6), 131.1 (C4), 133.1 (C3), 136.3 (C1). HRMS (ESI, 120 eV) calculated for C10H9BrS [M]+ 239.9595, found 239.9608.
4.2.5 General procedure A (cyclocarbopalladation/Stille domino reaction)
In a 2–5 mL microwave vial were added a solution of sulfide 1 (0.4 mmol, 1 equiv) and Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv) in benzene (3 mL). The vial was sealed with a Teflon cap and the 2-furyl tributylstannane (0.6 mmol, 1.5 equiv) was added, then the mixture was irradiated in the microwave for 3 h at 115 °C. The reaction mixture was then filtered through Celite to eliminate the metal traces and then concentrated under reduced pressure. The product was purified by flash column chromatography on silica gel (eluent: heptane 100%).
4.2.6 General procedure B (cyclocarbopalladation/Suzuki–Miyaura domino reaction)
In a 10–20 mL microwave vial were added a solution of sulfide 1 (0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), K3PO4 (212 mg, 1 mmol, 2.5 equiv), and boronic acid (0.6 mmol, 1.5 equiv) in a mixture of 2-methyltetrahydrofurane (5 mL) and water (0.1 mL). The vial was sealed with a Teflon cap and the mixture was irradiated in the microwave for 3 h at 130 °C. The reaction mixture was then evaporated and heptane was added to dissolve the product. The liquid phase was filtered through silica gel (previously treated by triethylamine) to eliminate the metal traces and then concentrated under reduced pressure. The ratio A/B was measured in the crude mixture by 1H NMR. The product was purified by flash column chromatography on silica gel (eluent: heptane 100%, then heptane/diethyl ether 99/1).
4.2.7 General procedure C (cyclocarbopalladation/Sonogashira domino reaction)
In a 2–5 mL microwave vial were added sulfide 1 (1 equiv, 0.166 mmol), Pd(OAc)2 (0.05 equiv), copper iodide (0.1 equiv), and PPh3 (0.1 equiv). The vial was sealed with a Teflon cap and the reaction mixture was then dissolved in distilled diisopropylamine (3 mL). The reaction mixture was placed under argon, freezed in liquid nitrogen, and put under vacuum. The O2 liberation proceeds when the temperature rises back to ambient. The operation was repeated two times. Then, the trimethylsilylacetylene (1.5 equiv) was added to the reaction mixture. The vial was irradiated in the microwave for 30 min at 120 °C. The reaction mixture is then filtered through Celite to eliminate the metal traces and then concentrated under reduced pressure. The product was purified by flash column chromatography (eluent: heptane 100%).
4.2.8 General procedure D (cyclocarbopalladation/Mizoroki–Heck domino reaction)
In a sealed tube (or a microwave vial) were added a solution of sulfide 1 (0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), potassium carbonate (110 mg, 0.8 mmol, 2 equiv), and then methyl acrylate (69 mg, 0.8 mmol, 2 equiv) in toluene (3 mL). The vial was sealed with a Teflon cap and the mixture was stirred at 130 °C for 7–18 h. The reaction mixture was then evaporated and heptane was added to dissolve the product. Afterward, the reaction mixture was allowed to cool to room temperature and filtered through a pad of Celite. The solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography on silica gel (eluent: heptane 100%, then heptane/diethyl ether 95/5).
4.2.9 (E)-2-(1-(Benzo[b]thiophen-3(2H)-ylidene)propyl)furan (2a)
The general procedure A was followed using sulfide 1a (102 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), and 2-furyl tributylstannane (0.6 mmol, 1.5 equiv). Product 2a was isolated as a yellow oil (50 mg, 52% yield).
1H NMR (400 MHz, CDCl3) δ 1.06 (t, J = 7.5 Hz, 3H, CH3), 2.44 (q, J = 7.5 Hz, 2H, CH2), 4.16 (s, 2H, SCH2), 6.28 (dd, J = 3.3, 0.7 Hz, 1H), 6.43 (m, 1H), 6.46 (m, 1H), 6.80 (m, 1H), 7.05 (dt, J = 7.5 Hz, 1.2 Hz, 1H), 7.18 (m, 1H), 7.43 (dd, J = 1.9, 0.8 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 12.2 (CH3), 29.2 (CH2), 36.4 (SCH2), 108.3 (CH), 111.3 (CH), 122.8 (CH), 123.8 (CH), 125.7 (CH), 127.2 (Cq), 128.5 (CH), 136.3 (Cq), 139.0 (Cq), 141.5 (CH), 145.6 (Cq), 153.2 (Cq). HRMS (ESI, 120 eV) calculated for C15H14OS [M]+ 242.0765, found 242.0768.
4.2.10 (E)-2-(1-(Isothiochroman-4-ylidene)propyl)furan (2b)
Stille coupling: procedure A was followed using sulfide 1b (107 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), and 2-furyl tributylstannane (0.6 mmol, 1.5 equiv). Product 2b was isolated as a yellow oil (82 mg, 81% yield).
Suzuki coupling: procedure B was modified as following: sulfide 1b (107 mg, 0.4 mmol, 1 equiv), PdCl2 (7 mg, 0.04 mmol, 0.1 equiv), SPHOS (32 mg, 0.08 mmol, 0.2 equiv), potassium phosphate (212 mg, 1 mmol, 2.5 equiv), and 2-furylboronic acid (67 mg, 0.6 mmol, 1.5 equiv). The yield was 60%. Compounds 2b and 2b′ were obtained as an inseparable equimolar mixture.
1H NMR (400 MHz, CDCl3) δ 1.16 (t, J = 7.5 Hz, 3H, CH3), 2.64 (q, J = 7.5 Hz, 2H, CH2), 3.59 (s, 2H, SCH2), 3.67 (s, 2H, SCH2), 5.78 (d, J = 3.3 Hz, 1H), 6.19 (dd, J = 3.6, 1.8 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 7.10 (m, 1H), 7.20 (m, 3H). 13C NMR (100.6 MHz, CDCl3) δ 14.1, 24.9, 29.1, 29.4, 109.5, 110.9, 126.5, 126.7, 127.3, 128.6, 129.6, 130.7, 137.4, 140.5, 141.2, 154.2. HRMS (ESI, 120 eV) calculated for C16H16OS [M]+ 256.0917, found 256.0921.
4.2.11 (E)-2-(1-(Benzo[c]thiophen-1(3H)-ylidene)ethyl)furan (2c)
The general procedure A was followed using sulfide 1c (96 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), and 2-furyl tributylstannane (0.6 mmol, 1.5 equiv). Product 2c was isolated as a yellow oil (53 mg, 59% yield).
1H NMR (400 MHz, CDCl3) δ 1.93 (s, 3H, CH3), 4.10 (s, 2H, SCH2), 6.03 (dd, J = 3.3, 0.8 Hz, 1H), 6.21 (m, 1H), 6.54 (d, J = 8.1 Hz, 1H), 6.80 (m, 1H), 6.91 (dt, J = 7.4, 1.1 Hz, 1H), 7.16 (dd, J = 1.9, 0.7 Hz, 1H), 7.21 (m, 1H). 13C NMR (100.6 MHz, CDCl3) δ 24.4 (CH3), 36.5 (SCH2), 107.7 (CH), 111.4 (CH), 124.6 (CH), 125.4 (CH), 126.8 (CH), 127.6 (CH), 140.9 (CH), 128.6 (Cq), 137.8 (Cq), 142.2 (Cq), 143.4 (Cq), 154.2 (Cq). IR (neat) ν (cm−1): 2919, 1619, 1485, 1469, 1153, 1004, 905, 759, 736, 595. HRMS (ESI, 120 eV) calculated for C14H12OS [M]+ 228.0628, found 228.068.
4.2.12 (E)-3-(1-Phenylpropylidene)-2,3-dihydrobenzo[b]thiophene (3a)
The general procedure B was followed using sulfide 1a (102 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), potassium phosphate (212 mg, 1 mmol, 2.5 equiv), and phenylboronic acid (73 mg, 0.6 mmol, 1.5 equiv). The product was isolated as a yellow oil (66 mg, 66% yield).
1H NMR (400 MHz, CDCl3) δ 1.02 (t, J = 7.5 Hz, 3H, CH3), 2.46 (q, J = 7.5 Hz, 2H, CH2), 4.21 (s, 2H, SCH2), 6.15 (d, J = 8.0 Hz, 1H), 6.58 (t, J = 7.8 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 7.13–7.18 (m, 3H), 7.32–7.42 (m, 3H). 13C NMR (100.6 MHz, CDCl3) δ 11.6 (CH3), 31.1 (CH2), 35.8 (SCH2), 122.7 (CH), 123.5 (CH), 126.0 (CH), 127.3 (CH), 127.8 (CH), 128.6 (2CH), 129.2 (2CH), 134.1 (Cq), 136.8 (Cq), 139.2 (Cq), 142.1 (Cq), 145.1 (Cq). IR (cm−1): 2963, 2927, 1577, 1486, 1370, 1455, 1438, 1274, 1161, 1134, 1065, 750, 727, 702, 687. HRMS (ESI, 120 eV) calculated for C17H16S [M]+ 252.0973, found 252.0984.
4.2.13 (E)-4-(1-Phenylpropylidene)isothiochroman (3b)
The general procedure B was followed using sulfide 1b (107 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), potassium phosphate (212 mg, 1 mmol, 2.5 equiv), and phenylboronic acid (73 mg, 0.6 mmol, 1.5 equiv). The product was isolated as a yellow oil (98 mg, 91% yield).
1H NMR (400 MHz, CD2Cl2) δ 0.93 (t, J = 7.5 Hz, 3H, CH3), 2.52 (q, J = 7.50 Hz, 2H, CH2), 3.50 (s, 2H, SCH2), 3.59 (s, 2H, PhSCH2), 6.53 (d, J = 8.0 Hz, 1H), 6.72 (t, J = 7.5 Hz, 1H), 6.86 (dd, J = 7.1 Hz, 1.7 Hz, 2H), 6.93 (t, J = 7.5 Hz, 1H), 6.98–7.01 (m, 4H). 13C NMR (100.6 MHz, CD2Cl2) δ 13.0 (CH3), 27.8 (CH2), 28.6 (SCH2), 28.9 (SCH2), 125.9 (CH), 126.0 (CH), 126.3 (CH), 126.4 (CH), 127.67 (2CH), 129.3 (2CH, Cq), 129.7 (CH), 138.1 (Cq), 139.9 (Cq), 140.9 (Cq), 142.8 (Cq). IR (cm−1): 2963, 2928, 1478, 1451, 1441, 1372, 1192, 1054, 907, 754, 697. HRMS (ESI, 120 eV) calculated for C18H18S [M]+ 266.1129, found 230.1124.
4.2.14 (E)-1-(1-Phenylethylidene)-1,3-dihydrobenzo[c]thiophene (3c)
The general procedure B was followed using sulfide 1c (96 mg, 0.4 mmol, 1 equiv), Pd(PPh3)4 (46 mg, 0.04 mmol, 0.1 equiv), potassium phosphate (212 mg, 1 mmol, 2.5 equiv), and phenylboronic acid (73 mg, 0.6 mmol, 1.5 equiv). The product was isolated as a yellow oil (29 mg, 30% yield).
1H NMR (400 MHz, C6D6) δ 2.27 (s, 3H, CH3), 3.93 (s, 2H, SCH2), 6.66 (dd, J = 6.6, 7.84 Hz, 1H), 6.76–6.82 (m, 3H), 7.06–7.15 (m, 5H). 13C NMR (100.6 MHz, CDCl3) δ 27.3 (CH3), 36.0 (SCH2), 125.4 (CH), 125.9 (CH), 126.5 (Cq), 126.8 (CH), 127.3 (CH), 127.5 (CH), 129.0 (2CH), 129.6 (2CH), 137.7 (Cq), 138.9 (Cq), 143.7 (Cq), 144.5 (Cq). IR (cm−1): 2922, 2850, 1688, 1593, 1489, 1471, 1454, 1440, 1264, 1026, 903, 757, 734, 698. HRMS (ESI, 120 eV) calculated for C16H14S [M]+ 238.0816, found 238.0824.
4.2.15 (E)-(3-(Benzo[b]thiophen-3(2H)-ylidene)pent-1-yn-1-yl)trimethylsilane (4a)
A small amount of the pure titled compound was isolated from the crude mixture obtained by method A (eluent: 100% heptane) and characterized by NMR spectroscopy.
1H NMR (400 MHz, CDCl3) δ 0.33 (s, 9H, SiMe3), 1.25 (t, J = 7.3 Hz, 3H, CH3), 2.30–2.35 (m, 3H, CH2), 4.16 (s, 2H, SCH2), 7.08–7.31 (m, 3H, H), 8.71 (d, J = 8.0, 1H). 13C NMR (100.6 MHz, CDCl3) δ 0.02 (SiMe3), 12.2 (CH3), 28.6 (CH2), 35.8 (SCH2), 102.8 (CCSi), 105.2 (CCSi), 117.5 (Cq), 122.4 (CH), 123.6 (CH), 125.8 (CH), 129.2 (CH), 136.3 (Cq), 144.1 (Cq), 146.0 (Cq).
Trimethyl((2-(pent-2-yn-1-ylthio)phenyl)ethynyl)silane (4a′). The titled compound was not isolated, as it was obtained by method C (via Sonogashira) or A (via Stille) only in a lesser amount, in mixture with compound 4a. One signal at δ = 3.69 (SCH2) was assigned to this compound in 1H NMR (CDCl3).
4.2.16 (E)-(3-(Isothiochroman-4-ylidene)pent-1-yn-1-yl)trimethylsilane (4b)
We were unable to isolate a pure sample of the titled compound; however, it was characterized from its mixture with compound 4b′ (obtained by method A).
1H NMR (400 MHz, CDCl3) δ 0.10 (s, 9H, SiMe3), 1.17 (t, J = 7.5 Hz, 3H, CH3), 2.34–2.40 (m, 3H, CH2), 3.57 (s, 2H, SCH2), 3.64 (s, 2H, SCH2), 7.13–7.26 (m, 3H, H), 7.86 (d, J = 7.2, 1H, H). 13C NMR (100.6 MHz, CDCl3) δ 0.05 (SiMe3), 13.7 (CH3), 26.3 (CH2), 28.4 (SCH2), 29.5 (SCH2), 97.9 (CCSi), 106.2 (CCSi), 121.2 (Cq), 125.9 (CH), 126.4 (CH), 127.7 (CH), 129.8 (CH), 136.9 (Cq), 137.2 (Cq), 138.6 (Cq).
4.2.17 Trimethyl((2-((pent-2-yn-1-ylthio)methyl)phenyl)ethynyl)silane (4b′)
The pure titled compound was isolated from the crude mixture obtained by method C (eluent: heptane/ethyl ether 99:1) and characterized by NMR spectroscopy.
1H NMR (400 MHz, CDCl3) δ 0.26 (s, 9H, SiMe3), 1.15 (t, J = 7.5 Hz, 3H, CH3), 2.20–2.26 (m, 2H, CH2), 3.15 (s, 2H, SCH2), 4.00 (s, 2H, SCH2Ar), 7.18 (t, J = 7.7 Hz, 1H, H), 7.25–7.34 (m, 2H, H), 7.47 (d, J = 7.7, 1H, H). 13C NMR (100.6 MHz, CDCl3) δ 0.08 (SiMe3), 12.7 (CH3), 14.2 (CH2), 19.6 (SCH2), 34.1 (SCH2Ar), 75.1 (CCEt), 85.2 (CCEt), 100.1 (CCSi), 102.8 (CCSi), 122.9 (Cq), 126.9 (CH), 128.5 (CH), 129.0 (CH), 132.8 (CH), 140.6 (Cq).
4.2.18 (E)-(3-(Benzo[c]thiophen-1(3H)-ylidene)but-1-yn-1-yl)trimethylsilane (4c)
We were unable to isolate a pure sample of the titled compound; however, it was characterized from its mixture with compound 4c′ (obtained by method C).
1H NMR (400 MHz, CDCl3) δ 0.26 (s, 9H, SiMe3), 2.05 (s, 3H, CH3), 4.31 (s, 2H, SCH2), 7.25–7.29 (m, 2H, H), 7.29 (dd, J = 4.6, 1.8 Hz, 1H, H), 8.78 (dd, J = 4.4, 4.8 Hz, 1H, H). 13C NMR (100.6 MHz, CDCl3) δ 0.2 (SiMe3), 24.2 (CH3), 36.5 (SCH2), 99.9 (CSiMe3), 103.7 (CCMe), 106.9 (CCSiMe3), 124.8 (CH), 125.7 (CH), 126.8 (CH), 128.2 (CH), 138.2 (Cq), 143.2 (Cq), 149.5 (SCC).
4.2.19 Trimethyl((2-((prop-1-yn-1-ylthio)methyl)phenyl)ethynyl)silane (4c′)
A small amount of the titled compound was isolated from the crude mixture obtained by method C (eluent: 100% heptane) and characterized by NMR spectroscopy.
1H NMR (400 MHz, CDCl3) δ 0.24 (s, 9H, SiMe3), 1.89 (s, 3H, CH3), 4.02 (s, 2H, SCH2), 7.22 (dt, J = 6.6, 1.6 Hz, 1H, H), 7.27 (dt, J = 7.3, 1.8 Hz, 1H, H), 7.33 (dd, J = 6.8, 1.8 Hz, 1H, H), 7.47 (dd, J = 7.3, 1.6 Hz, 1H, H). 13C NMR (100.6 MHz, CDCl3) δ 0.1 (SiMe3), 5.3 (CH3), 39.0 (SCH2), 67.5 (SC), 91.7 (CMe), 100.3 (CSiMe3), 102.8 (CCSiMe3), 123.3 (Cq), 127.5 (CH), 128.6 (CH), 129.4 (CH), 132.7 (CH), 139.6 (Cq).
4.2.20 Methyl (Z)-4-(benzo[b]thiophen-3-yl)hex-3-enoate (5a″)
The general procedure D was followed starting from sulfide 1a (102 mg, 0.4 mmol, 1 equiv). The entitled product was isolated as a yellow oil (63 mg, 60% yield).
1H NMR (400 MHz, CDCl3) δ 1.15 (t, J = 8 Hz, 3H, CH3), 2.51 (q, J = 8 Hz, 2H, CH2), 3.37 (d, J = 8 Hz, 2H, CH2), 3.78 (s, 3H, OCH3), 5.86 (t, J = 8 Hz, 1H), 7.36–7.43 (m, 3H), 7.88–7.92 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ 13.3, 25.2, 33.7, 52.1, 121.1122.4, 122.9, 123.3, 124.2, 124.4, 138.6, 139.1, 140.0, 140.5, 172.4. IR (cm−1): 2965, 1735, 1455, 1169, 761, 735. HRMS (ESI, 120 eV) calculated for C15H16O2S [M]+ 260.0871, found 260.0883.
4.2.21 Methyl (E)-4-((E)-isothiochroman-4-ylidene)hex-2-enoate (5b)
The general procedure D was followed starting from sulfide 1b (107 mg, 0.4 mmol, 1 equiv). The entitled product was isolated as a yellow oil (69 mg, 72% yield).
1H NMR (400 MHz, CDCl3) δ 1.15 (t, J = 8 Hz, 3H, CH3), 2.51 (q, J = 8 Hz, 2H, CH2), 3.60 (s, 2H, SCH2), 3.62 (s, 2H, SCH2), 3.70 (s, 3H, OCH3), 6.01 (d, J = 16 Hz, 1H), 7.12 (m, 1H), 7.21 (m, 1H), 7.28–7.32 (m, 2H), 7.54 (d, J = 16 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 13.8, 21.9, 29.1, 29.4, 51.7, 117.8126.94, 126.98, 128.7, 130.4, 135.1, 137.8, 137.9, 140.6, 144.5, 168.1. IR (cm−1): 2962, 1708, 1603, 1464, 1434, 1249, 1163, 1048, 815. HRMS (ESI, 120 eV) calculated for C16H18O2S [M]+ 274.1028, found 274.1038.
4.2.22 Methyl (2E)-4-(benzo[c]thiophen-1(3H)-ylidene)pent-2-enoate (5c)
The general procedure D was followed starting from sulfide 1c (91 mg, 0.4 mmol, 1 equiv). The entitled product was isolated as a yellow oil (40 mg, 51% yield).
1H NMR (400 MHz, CDCl3) δ 2.10 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 4.36 (s, 2H, SCH2), 5.90 (d, J = 16 Hz, 1H), 7.31–7.37 (m, 3H), 7.87 (d, J = 8 Hz, 1H), 8.35 (d, J = 16 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ 20.0, 36.9, 51.7, 115.2, 125.9, 126.5, 127.7, 128.4, 137.9, 142.0, 144.4, 146.5, 150.5, 168.7. IR (neat) ν (cm−1) 2359, 2341, 1710, 1600, 1455, 1293, 1260, 1166, 759. HRMS (ESI, 120 eV) calculated for C14H14O2S [M]+ 246.0715, found 246.0700.
Acknowledgments
This project was supported by the “Université de Strasbourg” (IDEX grant for T.C.) and the “Centre national de la recherche scientifique (CNRS)”.
Vous devez vous connecter pour continuer.
S'authentifier