

1 Introduction
The carbonsulfur (CS) bond formation holds a prominent position in the preparation of valuable and attractive synthetic intermediates, because many sulfenylated compounds are known to play a crucial role in primary metabolism of a wide range of living organisms [1] and are, at the same time, privileged structural motifs in many synthetic drugs [2], bioactive natural products [3], and functional materials [4]. Sulfenylated heterocycles have also attracted considerable attention from both industry and academia because of their presence in many biologically active small molecules and their therapeutic value against a variety of diseases [5]. Significant progress has been achieved throughout the years in forging CS bonds mainly exploiting metal-catalyzed and asymmetric organocatalytic strategies which, however, still make use of toxic and hazardous volatile organic solvents (e.g., benzene, toluene, THF, CH2Cl2, MeCN, Et2O, and so forth) with critical issues from an environmental point of view [6]. On the other hand, small organic molecules (e.g., secondary and tertiary amines, thiourea derivatives, phosphines, and so forth) have proven to be effective in promoting nonenantioselective organocatalytic CS bond formation such as sulfa-Michael additions [7], thiolysis of epoxides and aziridines [8], and thioesterification reactions [9] also in ionic liquids, in water, and under solvent-free conditions.
Recently, Coltart et al. [10] have reported an interesting asymmetric organocatalytic α-sulfenylation of ketones that proceeds in an umpolung fashion relative to conventional enolate/azaenolate methods [10]. In this transformation, simple thiols have been added in the presence of a catalyst to in situ–derived nitrosoalkenes 2, generated by treating the corresponding α-chlorinated oximes 1 with a base, to give α-sulfenylated oximes 3, which could be finally elaborated to the corresponding sulfenylated ketones or undergo other transformations (Scheme 1). The nature of the solvent was proven to be crucial for final product yield, and the best results were achieved using anhydrous CH2Cl2 and carrying out the reaction under a slight static pressure of argon.
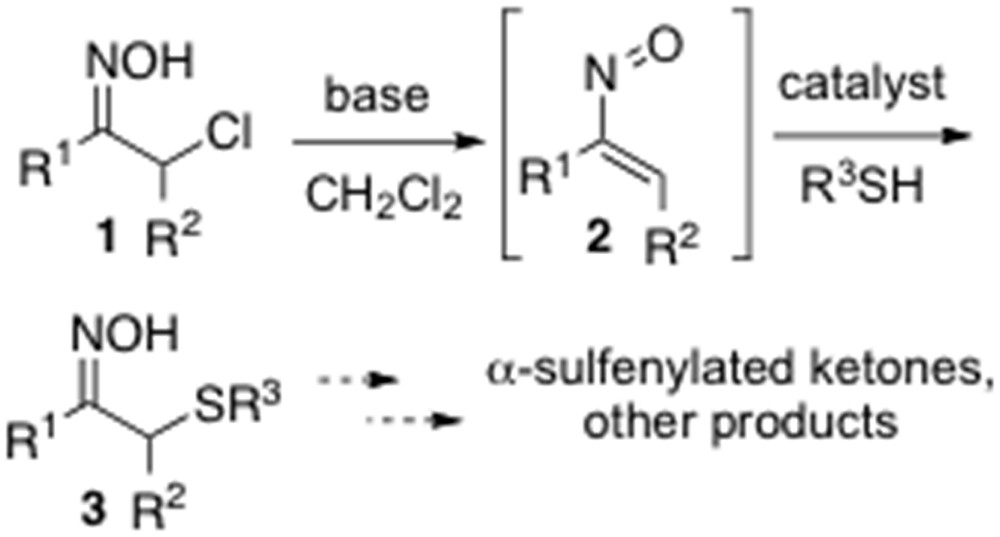
Catalytic umpolung sulfenylation of α-chloro oximes.
Natural deep eutectic solvents (DESs) represent an emerging generation of green and unconventional reaction media generally composed of two or three safe and inexpensive components, which are able to engage in hydrogen-bond interaction with each other to form a eutectic mixture with a melting point much lower than that of either of the individual components [11]. They are also known as “low transition temperature mixtures” (LTTMs) because many of them do not exhibit clear eutectic melting points, but glass transitions [11f]. Thanks to their extraordinarily biodegradability, biocompatibility, negligible vapor pressure, nonflammability, high thermal stability, tunable physicochemical properties, and the easiness of preparation by gentle thermal mixing, which requires no further purification, they are progressively replacing volatile organic solvents in many fields of science with interesting and somewhat unexpected results [11l–n], and thus they are capturing more and more the interest of academic and industrial community.
To the best of our knowledge, there are only a few examples of sulfenylation reactions run in DESs reported in the literature: (1) a CS bond formation via a one-pot reaction of alkyl halides, thiourea, and electron-deficient olefins using the choline chloride (ChCl)–urea DES both as the reaction medium and catalyst [12], (2) the ring opening of a variety of epoxides by thiols (and other nucleophiles) using the eutectic mixture ChCl–SnCl2 as a solvent and a catalyst [13], and a catalyst-free multicomponent synthesis of various sulfenylated 4H-chromenes in a ChCl-urea–based DES [14].
Inspired by the results of Coltart et al. [10], and building on our recent findings in using DESs as nonconventional reaction media for exploring novel paradigms in fields of organometallics, catalysis, and solar technologies [15], in this report we establish that sulfenylation of α-chlorinated oximes can be safely and conveniently carried out also using DESs as privileged and environmentally friendly solvents in reactions run at room temperature (RT) and under air.
2 Results and discussion
We initiated our study by investigating, as a model reaction, the one-pot sulfenylation of cyclic α-chloro oxime 1a with sodium thiophenolate 4a for the preparation of α-sulfenylated adduct 3a (Table 1). A preliminary reaction run in CH2Cl2 was done for comparison. After stirring a mixture of 1a (1.0 mmol), Et3N (1.1 mmol), and 4a (2.0 mmol) in CH2Cl2 (1.5 mL) at RT and under air, followed by quenching after 1.5 h with a saturated aqueous NH4Cl solution, the desired adduct 3a was formed in 75% yield. Under these conditions, however, a β-elimination reaction was also noted to compete, and α,β-unsaturated oxime 5 could be additionally isolated as a byproduct (20% yield) (Table 1, entry 1). Disappointedly, when the aforementioned mixture was vigorously stirred at RT and under air in a type III archetypal DES [11c], namely ChCl–glycerol (Gly) (1:2) (1.5 g per 1.0 mmol of 1a), the reaction resulted in only formation of 5 (50% yield; the remaining being starting material only), either after 1.5 h or after a prolonged reaction time of 24 h (Table 1, entries 2,3).
Survey of conditions for α-sulfenylation of α-chloro oxime 1aa.
| Entry | Solvent | T (°C) | t (h) | Base | Yield (%) 3ab |
| 1 | CH2Cl2 | 25 | 1.5 | Et3N | 75c |
| 2 | ChCl/Glyd | 25 | 1.5 | Et3N | NRe,f |
| 3 | ChCl/Glyd | 25 | 24 | Et3N | NRe,f |
| 4 | l-Tartaric acid/ChClg | 100 | 24 | Et3N | 10 |
| 5 | l-Lactic acid/d-glucoseh | 25 | 24 | Et3N | 30 |
| 6 | l-Lactic acid/l-alaninei | 25 | 24 | Et3N | 35 |
| 7 | l-Lactic acid/ChClj | 25 | 24 | Et3N | 49 |
| 8 | l-Lactic acid/ChClj | 50 | 24 | Et3N | 10 |
| 9 | l-Lactic acid/ChClj | 25 | 1.5 | NaHCO3 | 90 |
| 10 | l-Lactic acid/ChClj | 25 | 1.5 | NaHCO3 | 43k |
| 11 | l-Lactic acid/ChClj | 25 | 1.5 | –l | 80 |
| 12 | Urea/ChClj | 25 | 1.5 | –l | 80 |
| 13 | l-Lactic acid/ChClj | 25 | 1.5 | NaHCO3m | 44 |
| 14 | l-Lactic acid/ChClj | 25 | 1.5 | NaHCO3n | 54 |
| 15 | l-Lactic acid/ChClj | 25 | 1.0 | NaHCO3 | >98o |
a Reaction conditions: α-chloro oxime 1a (1.0 mmol), base (1.1 mmol), sodium thiophenolate 4a (2.0 mmol); DES mixture: 1.5 g per 1.0 mmol of 1a.
b Isolated yield by column chromatography on silica gel.
c Compound 5 was also isolated in 20% yield.
d 1:2 mol/mol.
e No reaction.
f Compound 5 was isolated in 50% yield as the sole product.
g 1:2 mol/mol.
h 5:1 mol/mol.
i 9:1 mol/mol.
j 2:1 mol/mol.
k Thiophenol was used in place of 4a.
l Sodium thiophenolate 4a: 2 equiv.
m Sodium thiophenolate 4a: 1 equiv, NaHCO3: 1 equiv.
n Sodium thiophenolate 4a: 1 equiv, NaHCO3: 2 equiv.
o In this reaction, only 20 mol % DES was used under sonication.
As pointed out in Section 1, DESs are unconventional fluids characterized by strong intermolecular hydrogen-bonding interactions between their components. We anticipated that hydrogen bond catalysis provided by solvents with stronger H-bond donors might contribute to better increase the electrophilic character at the α-carbon of the in situ–formed nitrosoalkenes. Thus, a second set of experiments was performed in more acidic DES mixtures [11b]. After mixing the aforementioned reagents thoroughly in a l-tartaric acid–ChCl (1:2) DES mixture for 24 h at 100 °C (because of the high-viscosity medium), sulfenylated oxime 3a was formed in 10% yield but as the sole product (Table 1, entry 4). Such a percentage could be progressively increased up to 49% using l-lactic acid–based LTTMs in combination with d-glucose, l-alanine, and ChCl, and working at RT (Table 1, entries 5–7). On the other hand, a higher temperature (50 °C) proved to be strongly detrimental for the chemical yield of 3a (10%) (Table 1, entry 8). Remarkably, upon switching from Et3N to NaHCO3 as the base, the desired adduct 3a could be isolated in 90% yield from a l-lactic acid–ChCl (2:1) LTTM [11g] working at RT and after a reaction time of only 1.5 h (Table 1, entry 9); no other byproducts could be detected. It is worth noting that by replacing 4a with simple thiophenol, a significant drop in the formation of 3a was noticed: 43% (Table 1, entry 10). By reacting 1a with 4a (2 equiv) without any base, the expected adduct 3a was again formed, albeit in a slightly reduced yield (80%) (Table 1, entry 11). A similar result in terms of yield was obtained upon switching to a more basic eutectic mixture, such as ChCl–urea (2:1) (Table 1, entry 12). By running the reaction with an equimolar amount of both NaHCO3 (1 equiv) and 4a (1 equiv), adduct 3a was formed in 44% yield only. The latter yield could be increased to 54% by increasing the amount of base up to 2 equiv (Table 1, entries 13,14). Thus, both the electrophilic character of the substrate and the nucleophilicity/amount of the thiolate, jointly with the correct choice/amount of the base to generate the putative nitrosoalkene, are all important factors to allow the sulfenylation reaction to take place in high yields in an acidic l-lactic acid–based LTTM at RT and under air.
With the optimized conditions in hand, we next turned our attention to investigate the scope of this atom-economy reaction with various combinations of thiolates and α-chloro oxime 1a, and results are reported in Scheme 2. We were pleased to find that assorted aliphatic and heteroaromatic thiolates 4b–e all proved to be competent partners straightforwardly providing the expected α-sulfenylated adducts 3b–e in very good yields (70–90%) at RT and under air (Scheme 2).
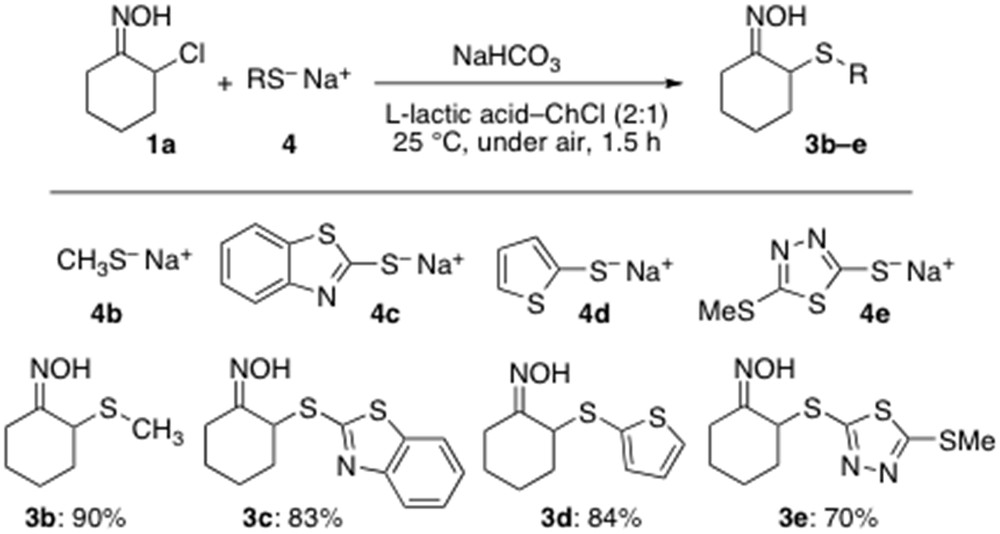
Scope of the α-sulfenylation reaction with various thiolates.
Recently, there has been a growing interest in finding novel applications of DESs both as sustainable media and catalysts for organic transformations [11j,k]. Indeed, hydrogen bond catalysis promoted by “noninnocent” DESs has been proven to be responsible for the increase in reaction rate observed in several multicomponent and heterocyclodehydration processes as well as in conjugate addition reactions [16]. Thus, we next investigated the possibility of conducting a one-pot sulfenylation by considerably reducing the amount of the aforementioned DES. On the basis of this, the formation of 3a was reinvestigated by using only 20 mol % of l-lactic acid–ChCl (2:1) and without any additional cosolvent. Because of the high viscosity of the reaction medium at RT, the reaction was run with the aid of a sonicating bath [17]. The complete disappearance of the starting oxime took place after 1 h of stirring, as revealed by GC–MS analysis. Pleasingly, the desired adduct 3a could be isolated in almost quantitative yield (>98%) by simple dilution of the mixture with equal volume mixture of water and Et2O, followed by separation of the organic layer and removal of the solvent under reduced pressure (Table 1, entry 15). It is worth noting that the same reaction run without the aforementioned LTTM, under solvent-free conditions, gave only starting materials after 1 h. This observation is consistent with the catalytic effect provided by LTTM in this reaction. A plausible mechanism of formation of compounds 3 from α-chloro oximes 1 and thiolates 4, highlighting the active role played by LTTM components in increasing the electrophilic character of the in situ-formed nitrosoalkenes 2, is depicted in Scheme 3.
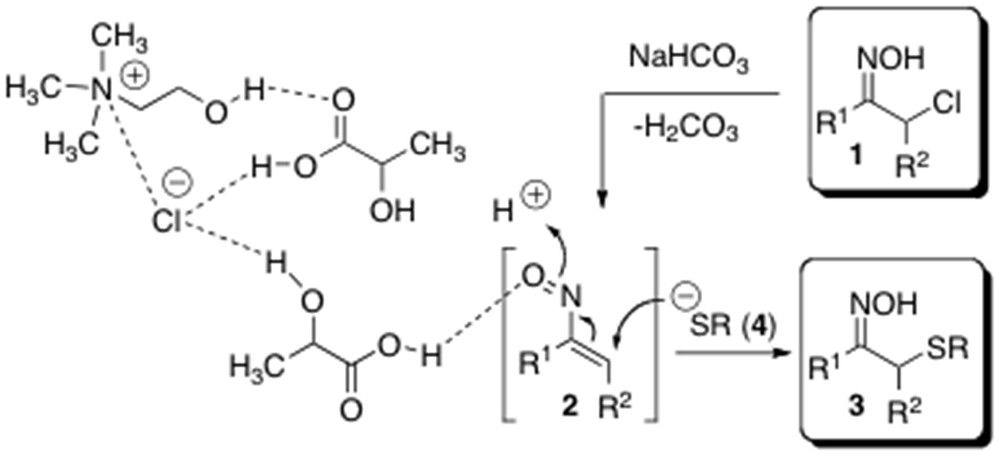
Plausible mechanism for the formation of sulfenylated products 3 under LTTM catalysis.
With the aim to extend the scope of this protocol even more, thiolates 4a–e were also reacted with the acyclic α-chloro oxime 1b exposing the mixture to the aforementioned catalytic sulfenylation conditions, under sonication at RT and under air for 1 h. All reactions proved to be extraordinarily clean affording the anticipated adducts 3f–j in very good yields (60–90%) and with no other byproducts (Scheme 4).
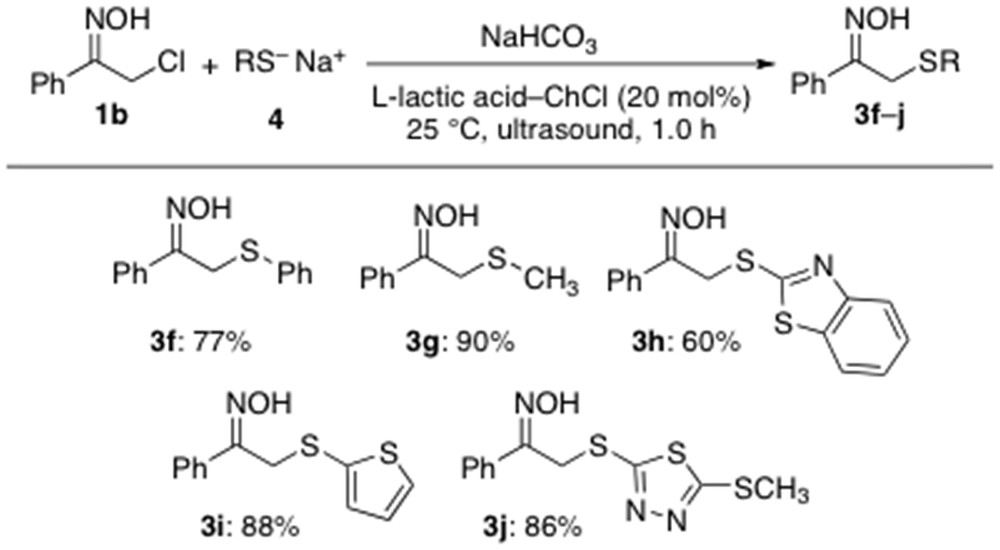
Scope of the α-sulfenylation reaction under catalytic conditions.
Finally, we have also briefly explored the feasibility of using sulfinate ions, which are ambident nucleophiles but with a reactivity much smaller than that of thiolates, as partners in CS bond-forming reactions [18]. They are known to react with various electrophiles either at the sulfur atom (soft nucleophilic center) to give sulfonyl derivatives or at the oxygen end (hard nucleophilic center) to lead to sulfinate esters. Thus, sulfinate ion 6 was reacted at RT and under air with α-chloro oximes 1a,b synergistically combining ultrasound and the LTTM l-lactic acid–ChCl (2:1) (20 mol %) in the presence of NaHCO3. Both reactions occurred at the sulfinate sulfur, thereby providing sulfone derivatives 7a,b in high yields (72% to >98%) (Scheme 5). As far as we are aware, these represent the first examples of direct sulfonylation of organic substrates performed in low melting mixtures under catalytic conditions.
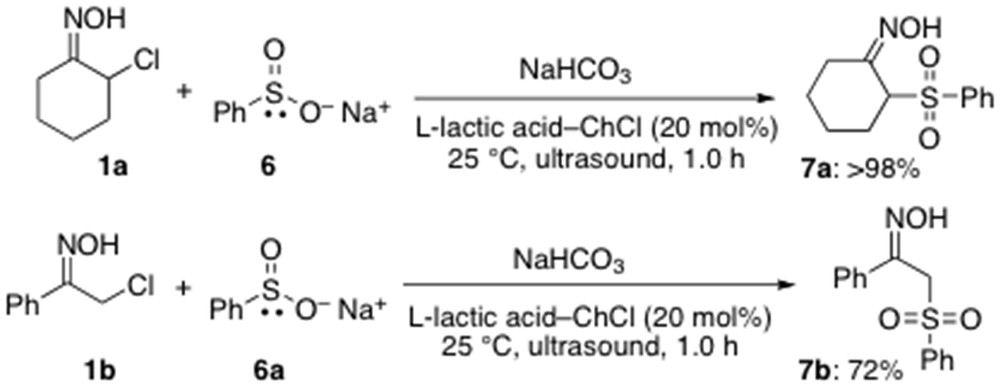
Direct sulfonylation of α-chloro oximes under catalytic conditions.
3 Conclusions
In this work, we report a simple and effective green protocol to perform a direct addition of thiolates, at RT and under air, to in situ–derived nitrosoalkenes from α-chloro oximes using, for the first time, an LTTM formed by lactic acid and ChCl as an environmentally friendly unconventional solvent. This umpolung sulfenylation reaction, which can also be carried out under catalytic conditions (20 mol % LTTM) with the aid of ultrasound, allows the preparation in very good yields (up to >98%) of a variety of α-sulfenylated oximes (aliphatic, aromatic, and heteroaromatic) starting from cyclic and acyclic scaffolds. Furthermore, this procedure also offers a new scope for umpolung sulfonylation at the α-carbon position of carbonyl compounds. Studies of the development of an asymmetric carbonsulfur bond formation using eutectic mixtures as eco-friendly and sustainable solvent/catalyst systems in organic transformations are underway and results will be reported in due course.
4 Experimental section
4.1 Synthesis of α-sulfenyl oximes in DESs or LTTMs: general procedure
To a mixture of α-chlorinated oxime (0.67 mmol) and NaHCO3 (0.74 mmol) in 1 g of DES or LTTM, sodium thiolate (1.35 mmol) was added at RT, under air, and vigorous stirring. After 1.5 h, saturated aqueous NH4Cl (3 mL) was added and the reaction mixture was extracted with Et2O (3 × 5 mL), and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by chromatography over silica gel using 20:80 EtOAc/hexane as the solvent to give the desired α-sulfenylated oximes 3a–j.
4.2 Ultrasound-mediated synthesis of α-sulfenyl oximes under solvent-free conditions: general procedure
α-Chlorinated oxime (0.7 mmol), NaHCO3 (0.77 mmol), sodium thiolate (1.4 mmol), and 20 mol % of DES or LTTM were mixed in a 10 mL round-bottomed flask. The reaction mixture was irradiated in a water bath of an ultrasonic cleaner (frequency, 40 kHz; nominal power, 50 W) for 1 h at RT, and saturated aqueous NH4Cl (3 mL) was added. The reaction mixture was extracted with Et2O (3 × 5 mL), and the combined organic extracts were washed with a saturated aqueous solution of NaCl, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by chromatography over silica gel using 20:80 EtOAc/hexane as the solvent to give the desired α-sulfenylated oximes 3a,3f–j.
4.3 Synthesis of α-sulfonylated oxime 7a: typical procedure
α-Chlorinated oxime 1a (148 mg, 1 mmol), NaHCO3 (92 mg, 1.1 mmol), sodium benzenesulfinate (328 mg, 2 mmol), and l-lactic acid–ChCl (1:2 mol/mol) (64 mg, 0.2 mmol) were mixed in a 10 mL round-bottomed flask. The reaction mixture was irradiated in a water bath of an ultrasonic cleaner (frequency, 40 kHz; nominal power, 50 W) for 1 h at RT, and then saturated aqueous NH4Cl (3 mL) was added. The reaction mixture was extracted with Et2O (3 × 5 mL), and the combined organic extracts were washed with a saturated aqueous solution of NaCl, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by chromatography over silica gel using 20:80 EtOAc/hexane as the solvent to give the desired α-sulfonylated oxime 7a in a quantitative (>98%) yield (271 mg, 0.985 mmol).
4.4 Spectroscopic data
α-Chlorinated oximes 1a,b [19] and α-sulfenylated oximes 3a [19a], 3b [20], and 3d [19a] had analytical and spectroscopic data identical to those previously reported. Configuration of α-sulfenylated and α-sulfonylated oximes was determined by FT-IR experiments run in CHCl3 at a variable concentration of oxime. In the case of Z-isomers, the intramolecular hydrogen bonding stretching band with the sulfur atom did not change its position significantly even at high dilution, and thus was not altered by a change in concentration.
4.4.1 (Z)-2-(Benzothiazol-2-ylthio)cyclohexanone oxime (3c)
Yellow solid, mp 168–170 °C, 83% yield. 1H NMR (600 MHz, CDCl3): δ 1.66–1.69 (m, 1H), 1.83–1.88 (m, 2H), 2.09–2.13 (m, 1H), 2.30–2.34 (m, 1H), 2.47–2.48 (m, 1H), 2.68–2.80 (m, 2H), 4.85 (t, J = 5.4 Hz, 1H), 7.29 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 8.91 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 22.8, 23.5, 25.4, 34.2, 49.5, 120.9, 121.9, 124.4, 125.9, 135.5, 153.1, 158,2, 165.0. FT-IR (film, cm−1): 3274, 2932, 2859, 2361, 1654, 1455, 1427, 1261, 980. HRMS (ESI): calcd for C13H14N2OS2 [M+Na]+, 301.0440; found, 301.0482.
4.4.2 (Z)-2-[5-(Methylthio)-1,3,4-thiadiazole-2-ylthio]cyclohexanone oxime (3e)
White solid, mp 154–156 °C, 70% yield. 1H NMR (600 MHz, CDCl3) δ 1.66–1.73 (m, 3H), 1.81–1.86 (m, 1H), 1.96–2.01 (m, 1H), 2.32–2.36 (m, 1H), 2.53–2.57 (m, 1H), 2.77 (s, 3H), 2.79–2.84 (m, 1H), 4.50 (dd, J = 6.6, 4.8 Hz, 1H), 7.85 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 16.4, 22.9, 23.6, 25.3, 34.0, 50.6, 158.1, 163.0, 167.5. FT-IR (film, cm−1): 3323 (OH), 2942, 2864, 1648, 1602, 1387, 1043, 974, 728. HRMS (ESI): calcd for C9H13N3OS3 [M+H]+, 276.0299; found, 276.0315.
4.4.3 (Z)-2-(Phenylthio)acetophenone oxime (3f)
White solid, mp 77–78 °C, 77% yield. 1H NMR (600 MHz, CDCl3): δ 4.26 (s, 2H), 7.22–7.24 (m, 1H), 7.26–7.29 (m, 2H), 7.37–7.41 (m, 3H), 7.42–7.45 (m, 2H), 7.59–7.61 (m, 2H), 8.98 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 28.5, 126.5, 127.0, 128.6, 128.9, 129.6, 131.0, 134.4, 135.4, 155.3. FT-IR (film, cm−1): 3244, 3062, 2922, 1735, 1583, 1573, 1439, 1407, 950. HRMS (ESI): calcd for C14H13NOS [M+H]+, 244.0800; found, 244.0788.
4.4.4 2-(Methylthio)-1-phenylethanone oxime (3g)
Colorless oil, inseparable mixture of diastereoisomers, dr E:Z = 1:1. 1H NMR (600 MHz, CDCl3): δ 2.11 (s, 3H), 2.16 (s, 3H), 3.56 (s, 2H), 3.88 (s, 2H), 7.39–7.47 (m, 6H), 7.53–7.55 (m, 2H), 7.68–7.70 (m, 2H), 8.69 (br s, 1H, exchanges with D2O), 9.07 (br s, 1 H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 15.1, 15.9, 26.2, 38.0, 126.4, 128.1, 128.3, 128.6, 129.4, 129.6, 132.1, 134.5154.8, 155.6. FT-IR (film, cm−1): 3303, 2919, 1631, 1435, 1304, 992, 750, 695. GC–MS (70 eV): m/z (%) diastereoisomer with minor tR 181 (M+, 19), 135 (100), 103 (71), 91 (15), 77 (28); GC–MS (70 eV): m/z (%) diastereoisomer with major tR 181 (M+, 35), 135 (100), 103 (95), 91 (17), 77 (36). HRMS (ESI): calcd for C9H11NOS [M+H]+, 182.0634; found, 182.0635.
4.4.5 (Z)-2-(Benzo[d]thiazol-2-ylthio)-1-phenylethanone oxime (3h)
White solid, mp 176–178 °C, 60% yield. 1H NMR (600 MHz, CDCl3): δ 4.83 (s, 2H), 7.10–7.12 (m, 2H), 7.26–7.28 (m, 2H), 7.44–7.48 (m, 3H), 7.77–7.79 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 26.9, 112.2, 121.4, 124.6, 126.8, 127.1, 128.8, 129.9, 130.1, 134.2, 140.2, 155.1, 166.0. FT-IR (film, cm−1): 3133, 2964, 1596, 1496, 751, 698. HRMS (ESI): calcd for C15H12N2OS2 [M+H]+, 301.0464; found, 301.0435.
4.4.6 (Z)-2-(Thiophen-2-ylthio) acetophenone oxime (3i)
White solid mp 79–81 °C, 88% yield. 1H NMR (600 MHz, CDCl3): δ 4.12 (s, 2H), 6.92 (dd, J = 4.8, 3.0 Hz, 1H), 7.12 (dd, J = 3.6, 1.2 Hz, 1H), 7.33–7.39 (m, 4H), 7.57–7.58 (m, 2H), 8.37 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 32.4, 126.4, 127.5, 128.5, 129.5, 130.5, 132.9, 134.4, 135.4, 155.2. FT-IR (film, cm−1): 3228 (OH), 2913, 1731, 1454, 1417, 1400, 1316, 1301, 1052, 945. HRMS (ESI): calcd for C12H11NOS2 [M+H]+, 250.0355; found, 250.0371.
4.4.7 (Z)-2-[5-(Methylthio)-1,3,4-thiadiazol-2-ylthio]acetophenone oxime (3j)
White solid, mp 160–163 °C, 86% yield. 1H NMR (400 MHz, CDCl3): δ 2.75 (s, 3H), 4.66 (s, 2H), 7.32–7.41 (m, 3H), 7.63–7.71 (m, 2H), 8.73 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 16.5, 27.2, 126.4, 128.8, 129.9, 133.7, 154.3, 163.8, 167.3. FT-IR (film, cm−1): 3256 (OH), 2921, 2821, 1638, 1383, 1303, 1043, 948. HRMS (ESI): calcd for C11H11N3OS3 [M+H]+, 298.0137; found, 298.0167.
4.4.8 (Z)-2-(Phenylsulfonyl)cyclohexanone oxime (7a)
White solid, mp 151–154 °C, 98% yield. 1H NMR (600 MHz, CDCl3): δ 1.33–1.41 (m, 1H), 1.67–1.86 (m, 2H), 1.96–1.98 (m, 1H), 2.10–2.18 (m, 1H), 2.30–2.36 (m, 1H), 2.71–2.74 (m, 1H), 3.22–3.24 (m, 1H), 3.76–3.78 (m, 1H) 7.52–7.55 (m, 2H), 7.61–7.65 (m, 1H), 7.80–7.82 (m, 2H), 7.96 (br s, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 21.2, 22.2, 24.3, 25.6, 65.2, 128.7, 128.9, 133.7, 137.8, 153.8. FT-IR (film, cm−1): 3285, 2941, 2861, 1791, 1447, 1306, 1084. GC–MS (70 eV): m/z (%) 253 (M+, 2) 143 (29), 125 (13), 111 (100), 77 (44). HRMS (ESI): calcd for C12H15NO3S [M+H]+, 254.0845; found, 254.0898.
4.4.9 (Z)-2-(Phenylsulfonyl)acetophenone oxime (7b)
White solid, mp 156–158 °C, 72% yield. 1H NMR (600 MHz, CDCl3): δ 4.73 (s, 2H), 7.33–7.38 (m, 3H), 7.42–7.46 (m, 2H), 7.55–7.62 (m, 3H), 7.82–7.85 (m, 3H, 1H, exchanges with D2O). 13C NMR (125 MHz, CDCl3): δ 52.6, 126.6, 128.4, 128.6, 128.8, 129.9, 133.5, 133.8, 139.3, 147.9. FT-IR (film, cm−1): 3394, 3062, 2924, 1447, 1308, 1157, 1308, 1157, 1134, 1085, 1055, 956, 745, 686. HRMS (ESI): calcd for C14H13NO3S [M+Na]+, 298.0508; found, 298.0501.
Acknowledgments
This work was financially supported by the University of Bari within the framework of the Project “Sviluppo di nuove metodologie di sintesi mediante l'impiego di biocatalizzatori e solventi a basso impatto ambientale” (code: Perna01333214Ricat) and the Interuniversities Consortium CINMPIS. The authors would like to thank Mr. Francesco Messa for his contribution to the experimental section.