

1 Introduction
Noxious gases are one of the most important pollutants [1,2], released by the burning of fossil fuels including carbon monoxide (CO), nitrogen dioxide (NO2), sulphur dioxide (SO2), etc. NO2 gas can be converted to nitric acid and nitrous in the atmosphere causing acid rain [3]. Therefore, discovery of a portable, highly sensitive, durable, and low cost NO2 sensor is of great importance. Since one of the places where NO2 sensors should be used is exhaust systems, high-temperature durability is necessary. With the advent of nanotechnology, different nanostructures have attracted much more attention as sensor devices because of their high surface/volume ratio and exceptional electronic properties [4–14]. Carbon nanotubes (CNTs) and graphene are two kinds of nanostructures which have been extensively investigated as gas sensors [15–21]. The common gas sensing mechanism is based on the change of the adsorbent electrical conductance because of its interaction with the gas molecules [19,22–31]. This change can be converted to an electrical noise.
It has been indicated that exposure to NO2 gas could change the electrical conductivity of CNTs [32]. Chang et al. have investigated the adsorption of NO2 gas on CNTs showing that this gas interacts with the tube surface via the physisorption process, affecting the electrical conductivity [33]. It has been claimed that pristine graphene cannot detect NO2 gas and a structural manipulation is needed to overcome this problem [34,35]. For instance, Dia et al. have indicated that Al doping makes the electrical conductivity of graphene much more sensitive to NO2 gas [34]. Graphene has an atom-thick two-dimensional conjugated layer, large specific surface area, high charge mobility and high conductivity [36]. It has no band gap which limits its applications especially in electronic devices [37]. Thus, several research studies have been conducted on nanographenes which are a part of graphene whose dangling atoms are saturated with hydrogen atoms [38–40]. Because of geometric confinement, nanographene introduces a band gap which makes it appropriate for electronic applications such as in field emission transistors [41]. Hexa-peri-hexabenzocoronene (HBC, C42H18) is a popular nanographene and much attention has been focused on its applications because a wide variety of techniques are available for its preparation [42,43]. Herein, we investigate the electronic sensitivity of pristine HBC nanographene to NO2 molecules using density functional theory (DFT) calculations. Also, the selectivity is explored by adsorption of N2, O2, H2, CO2, and H2O gases.
2 Computational methods
The geometries of pristine HBC and NO2/HBC complexes have been optimized at the B3LYP level of theory with a Grimme [44] empirical dispersion correction (D). The 6-31G* basis set was used for all calculations. With respect to the previous recommendations [45], the B3LYP/6-31G(d) method was employed to calculate the nucleus independent chemical shift (NICS) values, based on the gauge independent atomic orbital (GIAO) approach [46]. The NICS is a descriptor of local aromaticity which has been introduced by Schleyer et al. [47]. The NICS is predicted as a negative value of the absolute shielding computed in the centre of a definite ring, NICS(0), or 1 Å above the centre, NICS (1). Also, molecular electrostatic potential (MEP), density of states (DOS), natural bond orbital (NBO), and frontier molecular orbital (FMO) analyses were performed at the same level of theory. The B3LYP is a reliable functional which has been normally employed in the investigation of nanostructures [48–57]. Vibrational frequencies were computed for all optimized structures at the same level to verify that all structures are in true local minima. We employed the GaussSum program to design the DOS diagrams [58].
The adsorption energy is predicted as
| (1) |
3 Results and discussion
3.1 The HBC specifications
The optimized structure of HBC nanographene is shown in Fig. 1, indicating that five kinds of CC bonds can be found (1–6) with the bond length in the range of 1.37–1.44 Å, in good agreement with the experimental values [60]. In comparison to the benzene molecule whose CH vibrational stretching appears at 3214 cm−1, the vibrational modes of CH bonds in the HBC are stronger with values in the range of 3224–3275 cm−1. This indicates the more aromatic character of the peripheral hexagonal rings of the HBC compared to the benzene molecule. The NICS (0) value is calculated to be about −9.9 ppm for these rings which is slightly more negative than that of benzene (∼9.7 ppm at B3LYP/6-31G*). The central ring of HBC is considerably aromatic (NICS (0) ∼ −13.7 ppm, and NICS (1) ∼ −15.0 ppm). The mid rings are nonaromatic because of a small positive NICS (0) value of about 0.6 ppm. This pattern is in good agreement with Clar's isolated sextet rule which states that the Kekulé resonance building with the largest number of disjoint aromatic π-sextets is the most important for characterization of the properties of polycyclic aromatic hydrocarbons [61].
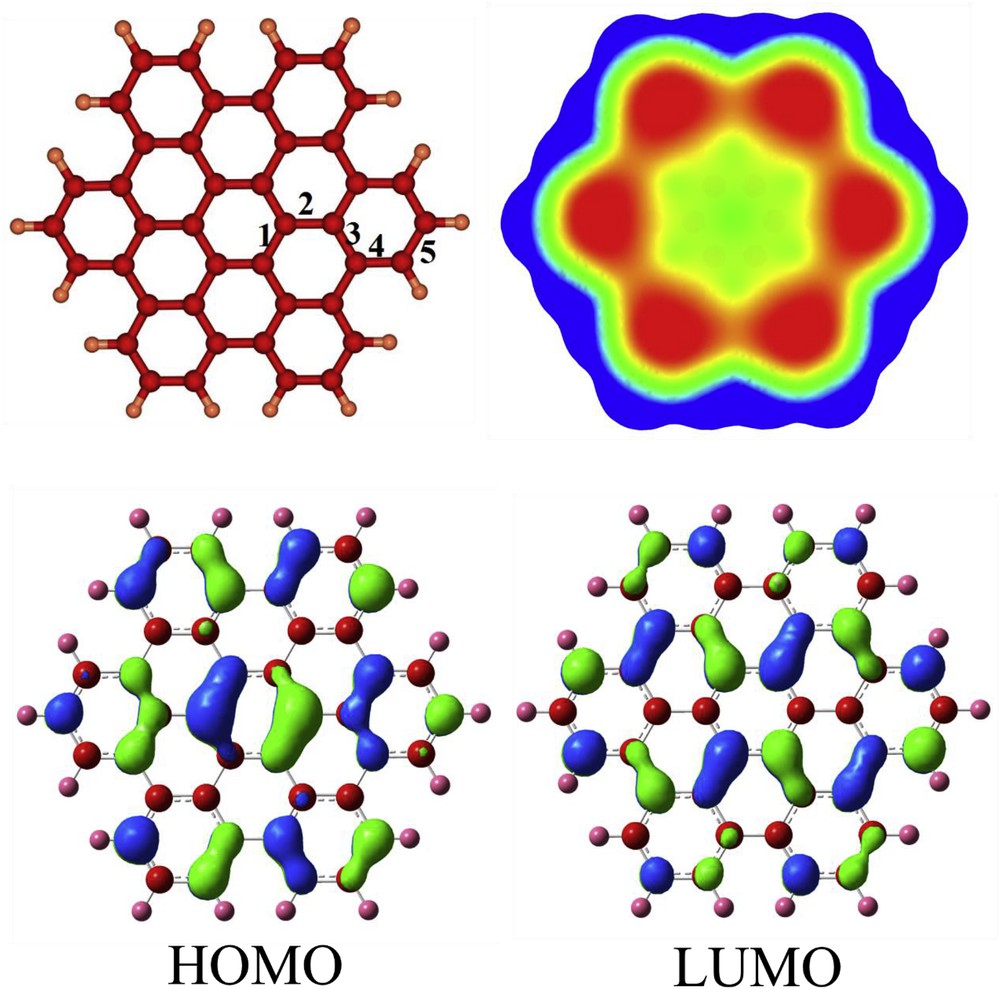
The optimized structure of HBC, its molecular electrostatic potential map, and HOMO and LUMO profiles. Colour ranges, in a.u.: blue, more positive than 0.015; green, between 0.015 and 0; yellow, between 0 and −0.015; red, more negative than −0.015.
Two kinds of hydrogen atoms are distinguished in the 1H NMR spectrum including para and meta ones at 7.9 and 8.8 ppm, respectively. It has been demonstrated that the adjacent ring current can affect the meta hydrogens, and more deshield them [62]. The HOMO and LUMO levels of pristine HBC are localized at −5.23 and −1.63 eV, respectively (Table 1). Therefore, the Eg is about 3.60 eV, indicating a semiconductor character. Fig. 1 specifies that HOMO is more located on the bonds of central and marginal hexagons, while the LUMO on the bonds of mid-hexagonal rings. This pattern is in consistence with Clar's isolated sextet rule as mentioned before.
The adsorption energies in kJ/mol for different configurations of NO2 adsorption on the HBC. Energies of the HOMO, LUMO and Eg in eV. The ΔEg indicates the change of Eg of HBC after the NO2 adsorption and Q is the NBO charge on the NO2 molecule which is adsorbed on the HBC. See Figs. 1 and 2. Calculations are at the B3LYP level with the 6-31G* basis set.
| Structure | Ead | EHOMO | ELUMO | Eg | %ΔEg | Q |
| HBC | – | −5.23 | −1.63 | 3.60 | – | – |
| A | −13.2 | −5.26 | −3.92 | 1.35 | −62.6 | −0.013 |
| B | −11.4 | −5.22 | −3.97 | 1.26 | −65.1 | −0.011 |
| C | −10.1 | −5.23 | −3.94 | 1.29 | −64.2 | −0.010 |
3.2 NO2 adsorption on the pristine HBC
In order to find the local minima for NO2/HBC complexes, several potential initial geometries including single (O or N), double (OO or NO) and triple ONO bonded atoms close to the C atoms or bridge sites or on the hexagonal rings were examined. Previously, three major possible configurations have been suggested for NO2/CNT complexes [63], including that the molecule is approached to the surface via a N atom (nitro configuration), bonded by one O atom (nitrite configuration) and bonded with both O atoms (cycloaddition configuration). We have predicted one nitro and two cycloaddition configurations (Fig. 2) after relax optimization without any symmetry constrain. Both of the cycloadditions on the HBC give rise to adsorption energies which are higher (less negative) than that of the nitro configuration (Table 1).
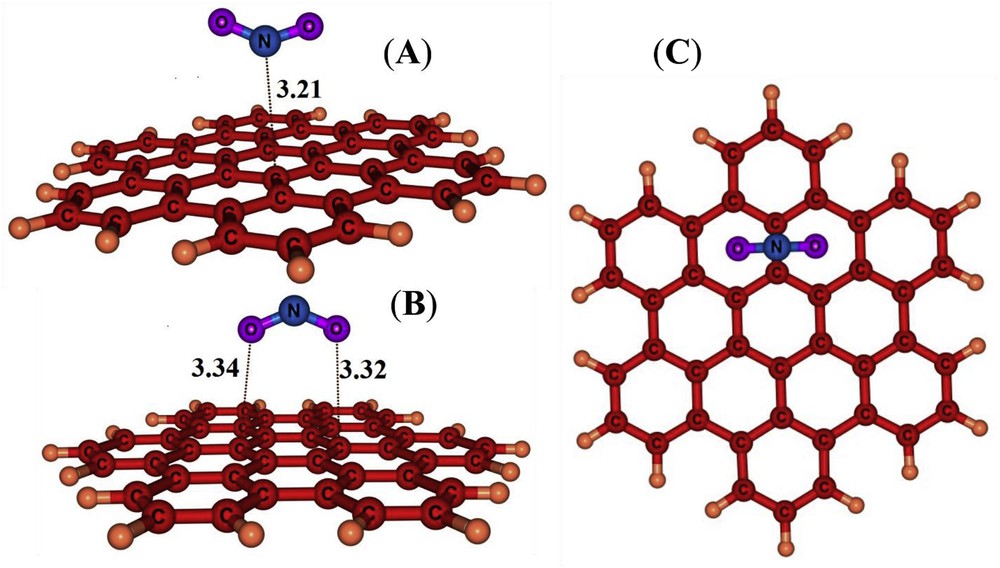
Optimized structures of NO2/HBC complexes. Distances are in Å.
The nitro complex is more compatible with the electron withdrawing nature of the NO2 molecule. An NBO charge transfer of about 0.013 e is predicted from HBC to the NO2 molecule. The adsorption energy is about −13.2 kJ/mol with the nearest NC distance of about 3.21 Å (Fig. 2). However, the nitro configuration favours the interaction between the electropositive N atom and the electron rich surface of the nanographene, indicating that the NO2 acts as an electron accepter. The symmetric vibrational frequency of ONO is decreased from 1441 cm−1 in the isolated NO2 molecule to 1413 cm−1 in the nitro configuration because of a large symmetry broken of the NO2 molecule upon its interaction with the HBC.
3.3 Electronic sensitivity
Herein, our main objective is investigating the electronic sensitivity of the pristine HBC to NO2 gas. To this aim, a simple and widespread approach was used which relates the change of the Eg to the population of conduction electrons of a semiconductor as follows [64]:
| (2) |
Table 1 indicates that for all of the cycloaddition and nitro configurations, the electronic properties of HBC (HOMO, LUMO and Eg) are considerably changed after the adsorption of the NO2 molecule. Especially, the LUMO level significantly stabilized by shifting from −1.63 to lower energies. Approximately, the pattern of the electronic property change is similar for all configurations in which the HOMO is not affected, unlike the LUMO. This can be understood by the fact that although the NO2 molecule acts as an electron withdrawing agent via charge induction, it shares its lone pair directly with the LUMO of the sheet. The partial DOS plot and LUMO profile of the nitro complex (Fig. 3) show that the shape of the LUMO is significantly changed after the NO2 adsorption so that it shifts from the HBC surface on the NO2 molecule. This is in agreement with the large shift in the energy of the LUMO level. But the HOMO is still located on the sheet (Fig. 3) in good agreement with its insignificant energy change. The partial DOS plot of the nitro configuration indicates that some new states appeared through the Eg of HBC after the NO2 adsorption and the lowest state is made by the NO2 molecule.
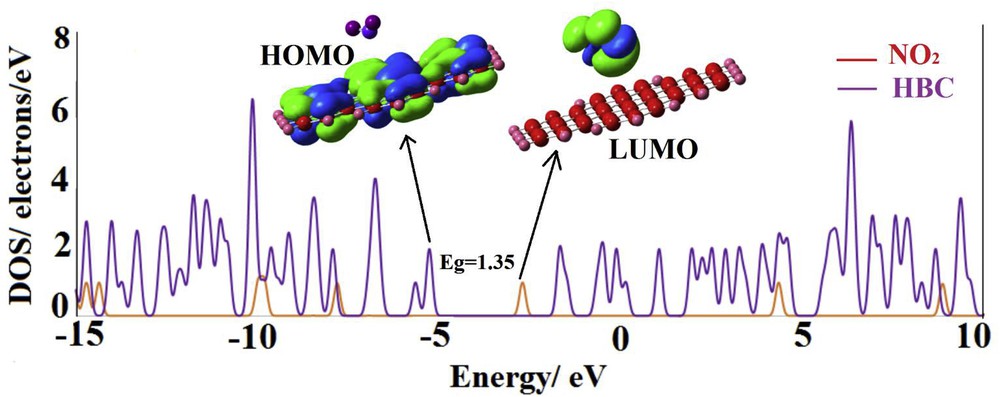
The partial density of states of the nitro complex (Fig. 2), and its HOMO and LUMO profiles.
Nevertheless, the significant stabilization of the conduction level (LUMO) energetically facilitates the electron transfer from the HOMO level to this level by reducing the Eg. The Eg is sharply reduced from 3.60 eV in the pristine HBC to 1.35 eV in the nitro complex, indicating a change by about 62.6%. This large change significantly increases the electrical conductivity based on Eq. 2. Therefore, the HBC is transformed from a semiconductor to a semi-metal compound in the presence of the NO2 molecule. It can be decided that the HBC may be used in NO2 sensor devices.
3.4 Effect of density functional
Several studies have been published indicating that the electronic properties of materials may be depended on the density functional especially on the percentage of Hartree–Fock (HF) exact exchange [70,71]. Thus, herein, we explored the effect of density functional on the sensitivity and electronic properties of HBC. To this aim, we performed single point calculations on the nitro complex and the HBC by using four Minnesota 06 density functionals including M06-HF, M06-2X, M06 and M06-L [72,73] with a %HF of 100, 54, 27, and 0, respectively. Table 2 demonstrates that the HOMO level of the HBC is stabilized, increasing the %HF exchange and the LUMO becomes more unstable. Consequently, the Eg is significantly affected and enlarged by increasing the HF exchange. Also, by increasing the %HF exchange, the sensitivity is decreased due to increasing the Eg of the base material (HBC). A review of the literature [74,75] indicates that the M06-2X and M06-HF functionals with high %HF significantly overestimate the Eg value and are not reliable methods for the electronic property evaluation.
The adsorption energies in kJ/mol for the nitro configuration (complex A, Fig. 2). Energies of the HOMO, LUMO and Eg in eV. The ΔEg indicates the change of Eg of HBC after the NO2 adsorption. Different density functionals were used with the 6-31G* basis set. The %ΔEg specifies the change of Eg of HBC after the adsorption process.
| Functional | System | EHOMO | ELUMO | Eg | %ΔEg |
| M06-L | HBC | −4.88 | −2.15 | 2.73 | – |
| Complex A | −4.88 | −4.67 | 0.21 | −92.2 | |
| M06 | HBC | −5.52 | −1.54 | 3.97 | – |
| Complex A | −5.51 | −3.87 | 1.63 | −58.8 | |
| M06-2X | HBC | −6.40 | −1.01 | 5.39 | – |
| Complex A | −6.39 | −2.59 | 3.80 | −29.5 | |
| M06-HF | HBC | −7.85 | −0.11 | 7.74 | – |
| Complex A | −7.84 | −1.44 | 6.40 | −17.3 |
3.5 Recovery time
The sensor recovery time is an important factor because without a fast recovery, a sensor may not be able to inform whether a gas leak is occurred or just how much gas has escaped into the atmosphere. Experimentally, the desorption process can be performed by heating the sensor to higher temperatures or by exposure to UV light [76]. The recovery time can be predicted from the following formula of transition theory:
| (3) |
3.6 Selectivity
Herein, we investigate the effect of the potential interfering gas molecules on the adsorption properties and sensitivity of HBC to NO2 molecules. To this aim, the adsorption of some gases including H2, N2, H2O, O2 (triplet), and CO2 on the HBC was investigated at the same level of theory. The results of adsorption energy and the electronic properties are summarized in Table 3. The HBC and also molecules H2, N2, O2, and CO2 are nonpolar species and, therefore, the nature of interaction between the HBC and these molecules is mainly induced dipole-induced dipole attraction. In such interactions a temporary (instantaneous) dipole is occurred on a system and induced a dipole on the adjacent molecule, attracting each other. As can be found in Table 3, this type of interaction is very weak, and these nonpolar molecules cannot impede the NO2 adsorption on the HBC.
Adsorption energy of different molecules on the HBC (Ead, kJ/mol), and the HOMO, LUMO and gap energy (Eg) in between (eV). The %ΔEg specifies the change of Eg of HBC after the adsorption process. The symbol “d” indicates the smallest distance between the molecule and surface of HBC (Figs. 2 and 4).
| Structure | d | Ead | EHOMO | ELUMO | Eg | %ΔEg |
| HBC | – | – | −5.55 | −1.95 | 3.60 | – |
| NO2 | 3.20 | −14.1 | −5.55 | −4.48 | 1.06 | −70.5 |
| H2 | 3.39 | −3.7 | −5.55 | −1.95 | 3.60 | −0.0 |
| N2 | 3.33 | −5.5 | −5.56 | −2.13 | 3.42 | −4.9 |
| O2 | 3.31 | −6.3 | −5.55 | −2.18 | 3.38 | −6.2 |
| CO2 | 3.49 | −6.9 | −5.56 | −1.97 | 3.59 | −0.3 |
| H2O | 2.87 | −9.6 | −5.67 | −2.09 | 3.59 | −0.4 |
The electron density, size, shape, polarizability, and orientation of the molecules control the strength of this interaction. The H2 molecule is the smallest one and has a very low polarizability and, therefore, it has the smallest adsorption energy (less negative) of about −3.1 kJ/mol with a distance of 3.41 Å from the HBC surface as shown in Fig. 4. It has been previously indicated that the polarizability of the N2 molecule is somewhat greater than that of the O2 molecule because of its two π-bonds [79], while the more negative adsorption energy (∼−5.9 kJ/mol, Table 3) of the O2 molecule is ascribed to its charge acceptor character compared to the N2 [80]. The CO2 molecule is larger and has higher polarizability and, therefore, more negative adsorption energy compared to the other studied nonpolar molecules. The H2O molecule is a polar molecule with higher dipole induction on the HBC and more negative adsorption energy about −8.6 kJ/mol which is less negative compared to that of the NO2 molecule. However, our calculations (Table 3) indicate that none of these molecules can affect the electronic properties of the HBC, compared to the NO2 molecule. Thus, we think that the HBC can detect NO2 gas in the presence of O2, H2, N2, CO2, and H2O molecules.

The optimized structures of different gas complexes with the HBC. Distances are in Å.
We also repeated all calculations on the most stable structures at the B3LYP level with a larger basis set 6-31+G** and the results are collected in Table 4. This basis set includes the diffuse functions on the N, C and O atoms, and also, polarization functions on the all atoms. The adsorption energy of all molecules is slightly more negative with this basis set compared to the results of 6-31G* one, and also, the structures are changed very slightly. For example, the interaction distance of H2 with HBC is shortened from 3.41 to 3.39 Å (Fig. 4). Table 4 specifies that the larger basis set somewhat stabilizes both of the absolute quantities (HOMO and LUMO energies). While the relative quantity (ΔEg) is still very small for adsorption of O2, H2, N2, CO2, and H2O molecules and is very large (∼−70.5%) for the NO2 molecule. Thus, similar to the 6-31G* basis set, the 6-31+G** basis set indicates that the HBC can identify the NO2 molecule in the presence of O2, H2, N2, CO2, and H2O molecules.
Adsorption energy of different molecules on the HBC (Ead, kJ/mol), and the HOMO, LUMO and gap energy (Eg) in between (eV). The %ΔEg specifies the change of Eg of HBC after the adsorption process. Calculations are at the B3LYP level with the 6-31+G** basis set.
| Structure | Ead | EHOMO | ELUMO | Eg | %ΔEg |
| HBC | – | −5.23 | −1.63 | 3.60 | – |
| H2 | −3.1 | −5.24 | −1.63 | 3.61 | 0.0 |
| N2 | −5.2 | −5.24 | −1.62 | 3.62 | 0.5 |
| O2 | −5.9 | −5.25 | −1.63 | 3.62 | 0.5 |
| CO2 | −6.3 | −5.29 | −1.60 | 3.69 | 2.5 |
| H2O | −8.6 | −5.38 | −1.52 | 3.86 | 7.2 |
4 Conclusions
We investigated the structural properties, reactivity and electronic sensitivity of HBC to NO2 gas by means of DFT calculations. Energetically, different NO2/BNG complexes including one nitro configuration and two cycloaddition configurations are predicted. It was found that, unlike graphene, the electronic properties of HBC are highly sensitive to the presence of NO2 molecules so that after the NO2 adsorption, the HBC is converted from a semiconductor to a semimetal compound. The Eg of HBC is reduced by about 62.6% upon the NO2 adsorption. Thus, it was concluded that the HBC may be used in NO2 sensor devices. Also, a short recovery time of 1.9 ns is predicted for the NO2 desorption process at room temperature. The HBC selectively detects the NO2 molecules in the presence of H2, N2, H2O, O2 (triplet), and CO2 molecules.