

1 Introduction
Allenylmetal reagents [1], including allenylzincs [2], are versatile organometallic species that have been used intensively in organic synthesis. For more than a decade, work from our laboratories [3] and others [1,2] have demonstrated that α-silyl–substituted allenylzincs are a particularly useful substrate class of this family. Typically, allenylzinc reagents exist in a solution as mixture of two rapidly interconverting metallotropic forms: the propargylic and the allenic one. When opposed to carbonyl derivatives (i.e., aldehydes, ketones, or imines), each form has the potential to undergo electrophilic substitution through an SE2′ mechanism providing either allenes (from the propargylic form) or alkynes (from the allenic form). Lack of regioselectivity is thus a common drawback for such condensations [2].
A key feature of α-silyl–substituted allenylzinc reagents is that the organosilyl group geminal to the carbon–zinc bond stabilizes the allenic form (α-silicon effect) and thereby shifts the propargyl/allenyl metallotropic equilibrium toward the latter form (Scheme 1) [4]. Hence, condensation with carbonyl derivatives occurs with exquisite regioselectivity and affords exclusively the propargylic adducts [3,5]. Furthermore, in cases where two stereogenic centers are created during the condensation event, excellent levels of diastereocontrol are usually observed. In addition, their high functional-group tolerance and the fact that they lead to silylated alkynes that can be easily protodesilylated to unmask a terminal alkyne that then serves as handle for further synthetic elaboration are also important qualities of these propargylating reagents [6].

Condensation of aldehydes with α-silyl–substituted propargyl/allenyzinc reagents.
α-Substituted allenylmetals can be conveniently accessed by the 1,2-metalate rearrangement of alkynylogous carbenoids. Commonly, migration of alkyl groups is involved and α-alkyl–substituted allenylmetals are obtained [7–10]. However, the approach is also suitable to access α-silyl allenylmetals, but this prospect has remained so far virtually unexplored [10b,11]. In seminal studies, Harada et al. demonstrated that zincate complexes II, obtained from bromopropargyl mesylates I by halogen–metal exchange with (Me2PhSi)3ZnLi, provided α-silyl allenylzinc intermediates III after 1,2-migration of the organosilyl group with displacement of the propargylic leaving group through an SNi mechanism (Scheme 2, top) [10b,12]. Inspired by these results and as part of our longstanding interest in ring-opening reactions of acetylenic epoxides [13] and aziridines [14], we reasoned that implementation of a similar approach with lithium alkynyl(disilyl)zincates V derived from those substrates and disilylzinc reagents would lead to δ-heteroatom substituted α-silyl allenylzinc intermediates VI that could offer an expeditious stereoselective access to valuable 1,3-diols (VII, X = O) or 1,3-aminoalcohols (VII, X = NR) on condensation with aldehydes (Scheme 2, bottom). In this article, we describe our research endeavors in this area combining an overview of previous work on acetylenic epoxides [10e] and recent results on the ring-opening of N-tert-butanesulfinylaziridines and N-tert-butanesulfonylaziridines.

Formation of α-silyl–substituted propargyl/allenyzinc reagents by 1,2-metalate rearrangement of alkynylogous carbenoids.
2 1,2-Rearrangement of alkynyl(disilyl)zincates derived from acetylenic epoxides
Our first foray in this field was the result of our studies on the rearrangement of lithiated ethynylepoxides by reaction with dialkylzinc reagents [10e]. As part of this work, it was found that lithiated acetylenic epoxides also underwent transmetalation with (Me2PhSi)2Zn at −60 °C to afford lithium alkynyl(disilyl)zincates 2 (Scheme 3). 1,2-Silyl migration inducing opening of the oxirane ring occurred on warming the reaction mixture to −20 °C and afforded the corresponding α-silyl allenylzinc intermediates. After acidic aqueous workup, a mixture of homopropargylic (3) and allenic (3′) alcohols was obtained. The 3/3′ ratio was found to be highly dependent on the quench conditions and difficult to control. Ultimately, it was possible to find conditions to prepare regioselectively homopropargylic alcohols 3 using MeOH (0 °C) to quench the reaction mixture after rearrangement. This optimized protocol was suitable for di- and tri-substituted acetylenic epoxides having either alkyl or aryl substituents and the corresponding alcohols 3 were obtained in 64–86% yield with good to excellent regioselectivity (3/3′ > 84:16).

Rearrangement of lithium alkynyl(disilyl)zincates.
The possibility to use this chemistry to access 1,3-diols was also demonstrated. Condensation of propionaldehyde with the α-silyl allenylzinc intermediate 2a′ (obtained from epoxide 1a) was achieved at −40 °C and led to diol 4 [15] in a remarkable 78% yield with excellent regio- and stereoselectivity (Scheme 4). Trapping with acetone was less efficient and tertiary alcohol 5 was obtained in a lower 42% yield, still with excellent stereoselectivity. Interestingly, in the latter case, the presence of allenic regioisomers was also detected to some extent.

Preparation of 1,3-diols 4 and 5.
3 1,2-Rearrangement of alkynyl(disilyl)zincates derived from acetylenic aziridines
Following on these results, we considered the rearrangement of acetylenic aziridines and focused on N-tert-butanesulfinylethynylaziridines (6) and N-tert-butanesulfonylethynylaziridines (7) (Scheme 5). Model substrates 6a–c were prepared as pure stereoisomers in two steps from N-tert-butanesulfinylimines, according to a procedure previously reported by our group [14,16]. Oxidation of 6a–c with meta-Chloroperoxybenzoic acid (m-CPBA) afforded the corresponding N-tert-butanesulfonylethynylaziridines 7a–c in excellent yields.

Preparation of N-tert-butanesulfonyl(ethynyl)aziridines 7a–c.
First, the lithiation step was examined (Table 1). To this end, substrate 6a (representative of N-tert-butanesulfinylethynylaziridines) was treated with nBuLi in THF at −80 °C for 15 min and the reaction mixture was then quenched with MeOD. After workup, [2H]-6a was obtained in low 25% yield along with 71% of unreacted 6a (entry 1). To drive the metalation to completion, the reaction time was then increased to 1 h: [2H]-6a was obtained in 57% yield along with 7% of 6a (entry 2). Hence, although the deuteration grade was significantly improved (89%), the overall lithiation yield remained modest because around 30% of the starting material had been consumed unproductively to yield unidentified material. Sulfoxide 8 was also isolated from this reaction in 5% yield, suggesting that substitution at the sulfur atom of the tert-butanesulfinyl moiety was at the origin of the starting material degradation. To minimize this possible side reaction, changing nBuLi for other alkyllithium reagents was considered. When tBuLi was used (entry 3), sulfoxide 9 was isolated in a higher 16% yield and some starting material was lost. In contrast, degradation did not occur using MeLi or PhLi (entries 4 and 5). Yet, complete metalation was not achieved (65–68% deuteration grades) even after prolonged reaction times (3 h).
Deprotonation studies of 6a.
| Entry | Base | Conditions | [2H]-6a (%)a | 6aa (%) | tBuS(O)Ra (%) |
| 1 | nBuLi | −80 °C, 15 min | 25 | 71 | 8: –b |
| 2 | nBuLi | −80 °C, 1 h | 57 | 7 | 8: 5 |
| 3 | tBuLi | −80 °C, 1 h | 52 | 11 | 9: 16 |
| 4 | MeLi | −80 °C, 3 h | 62 | 34 | 10: –b |
| 5 | PhLi | −80 °C, 3 h | 63 | 30 | 11: 7 |
| 6 | LDA | −80 °C, 1 h | 47 | 39 | – |
| 7 | LDA | HMPAc, −30 °C, 2 h | 81 | 9 | – |
| 8 | nBu3ZnLid | 0 °C, 2 h | 89 | 11 | 8: –b |
a Yield determined by 1H NMR spectroscopy using biphenyl as an internal standard.
b Product not detected.
c 2.0 equiv.
d 2.2 equiv.
Following these failures, we contemplated deprotonation of 6a with LDA. On reaction at −80 °C for 1 h, only partial metalation was obtained, but without any significant starting material degradation (entry 6). Finally, adding HMPA (2.0 equiv) as additive and performing the reaction at −30 °C allowed for convenient lithiation of 6a as [2H]-6a was obtained in 81% yield (entry 7). An additional solution for the metalation of 6a came out of serendipity. Although studying the ring-opening of ethynylaziridines with triorganozincates [17], we found that [2H]-6a could be obtained in 89% yield by treatment with 2.2 equiv nBu3ZnLi at 0 °C for 2 h followed by quenching with MeOD (entry 8) [18].
Unlike for N-tert-butanesulfinylethynylaziridine 6a, lithiation of 7a (representative of N-tert-butanesulfonylaziridines) was not problematic and occurred readily at −80 °C on treatment with nBuLi (1.1 equiv). This was confirmed by the isolation of [2H]-7a in 89% yield after addition of MeOD to the reaction mixture and aqueous workup (Scheme 6).
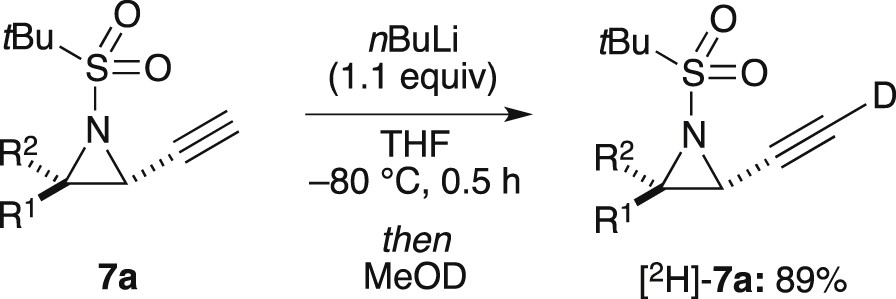
Deprotonation of 7a.
With suitable metalation procedures in hand for 6 and 7, the 1,2-metalate rearrangement of the corresponding alkynyl(disilyl)zincates (V) arising from reaction with (Me2PhSi)2Zn was investigated. N-tert-butanesulfinylethynylaziridines 6 were considered first (Table 2).
Rearrangement of N-tert-butanesulfinylethynylaziridines 6.
| Entry | 6 | Base | (Me2PhSi)2Zn [equiv], T | Product | 12/12′a | 12′(dr)a | Yieldb (%) |
| 1 | 6a | LDA/HMPA | 1.1, −30 °C | 12a/12a′ | ncc | ncc | <5 |
| 2 | 6a | LDA/HMPA | 2.2, −30 °C | 12a/12a′ | <2:98 | 68:32 | 57 |
| 3 | 6a | (nBu)3ZnLid | 2.2, 0 °C | 12a/12a′ | 66:34 | 91:9 | 63 |
| 4 | 6a | nBuLi | 1.5, 0 °C | 12a/12a′ | 84:16 | 33:67 | 60 |
| 5e | 6a | nBuLi | 1.5, 0 °C | 12a/12a′ | <2:98 | 77:23 | 64 |
| 6f | 6a | nBuLi | 1.5, 0 °C | 12a/12a′ | 90:10 | 54:46 | 65 |
| 7 | 6b | nBuLi | 1.5, 0 °C | 12b/12b′ | <2:98 | 97:3 | 75 |
| 8 | 6c | nBuLi | 1.5, 0 °C | 12c/12c′ | – | – | –g |
a Measured by 1H NMR spectroscopy before purification.
b Combined isolated yield of 12 and 12′.
c Not calculated.
d 2.2 equiv.
e Quenched at −20 °C.
f Quenched with 1 M HCl at −30 °C.
g The starting material was recovered.
Trace amounts of rearrangement products were obtained with the initial set of conditions involving LDA as base, addition of 1.1 equiv (Me2PhSi)2Zn at −60 °C, and warming the reaction mixture to −30 °C (Table 2, entry 1). After addition of MeOH (at −30 °C) and work up, around 80% of the starting aziridine 6a was recovered. To our delight, when a larger amount of (Me2PhSi)2Zn was introduced (2.2 equiv), complete ring opening of 6a was observed and δ-amino allenylsilane 12a′ was isolated in 57% yield as a 68:32 mixture of diastereomers (entry 2). 1,2-Rearrangement also occurred at 0 °C with the zincate obtained using nBu3ZnLi for the metalation step (entry 3). In this case, a mixture of 12a and 12a′ in a 12a/12a′ = 66:34 ratio was isolated in 63% yield. Quite unexpectedly, given the apparent moderate efficiency of the lithiation step (see above), similar yields (60–65%) in rearranged products were obtained when nBuLi was used as base (entries 4–6). Here, mixtures of 12a/12a′ were isolated in regio- and stereoselectivities that were difficult to control and varied with the workup conditions [19]. Importantly, these substantial disparities provided good evidence for the intermediate formation of a δ-amino α-silyl allenylzinc intermediate VI and, in-line with the behavior of the related δ-oxy α-silyl allenylzinc reagents obtained from ethynylepoxides, are likely the result of the difficulty to control the selectivity of the protodemetalation reaction.
The 1,2-metalate rearrangement also occurred efficiently in the case of ethynylaziridine 6b having a secondary alkyl substituent (entry 7). Interestingly, with the protocol relying on nBuLi for the metalation step, the exclusive formation of δ-amino allenylsilane 12b′ (97:3 dr) was obtained in 75% yield. On the contrary, ring-opening by 1,2-metalate rearrangement did not occur for the alkynyl(disilyl)zincate derived from cis-configured ethynylaziridine 6c and the starting material was recovered (entry 8) [20].
N-tert-Butanesulfonylethynylaziridines 7 were considered next (Table 3). The rearrangement of the alkynyl(disilyl)zincate derived from lithiated 7a and (Me2PhSi)2Zn (1.5 equiv) occurred fully at −30 °C and a mixture of products 13a and 13a′ (13a/13a′ = 78:22) was isolated after methanolysis and workup in 54% yield (entry 1). Performing the rearrangement at 0 °C led to a better 70% yield (entry 2).
Rearrangement of N-tert-butanesulfonylethynylaziridines 7.
| Entry | 7 | T | Product | 13/13'a | 13′(dr)a | Yieldb (%) |
| 1 | 7a | −30 °C | 13a/13a′ | 78:22 | 90:10 | 54 |
| 2 | 7a | 0 °C | 13a/13a′ | 93:7 | nc | 70 |
| 3c | 7b | −30 °C | 13b/13b′ | 98:2 | nc | 70 |
| 4 | 7c | −30 °C | 13a/13a′ | 87:13 | 74:16 | 80 |
a Measured by 1H NMR spectroscopy before purification.
b Combined isolated yield of 13 and 13′.
c Quench was performed at −80 °C.
In the case of substrate 7b having a secondary alkyl substituent, the yield in rearranged products 13b and 13b′ was already good when the rearrangement was carried out at −30 °C (entry 3). Here, in sharp contrast with N-tert-butanesulfinyl substrates, no significant reactivity difference was noted between trans and cis configured ethynylaziridines. Thus, starting from cis-configured 7c, the ring-opening products 13a and 13a′ were isolated in 80% combined yield after rearrangement at −30 °C and treatment with MeOH (entry 4). Again, evidencing the formation of δ-amino α-silyl allenylzinc intermediates (VI), the regio- and stereoselectivity of the process was, for all the substrates, somewhat erratic and dependent on the workup conditions. Nevertheless, quenching the reactions with MeOH before aqueous workup favored in general the formation of homopropargylamines 13, as indicated in Table 3.
Finally, the reactivity against aldehydes of the δ-amino α-silyl allenylzinc intermediates (VI) obtained from ethynylaziridines was investigated. Despite extensive experimentation, condensation was not observed to significant extents (<10–15%) between the allenylzinc species obtained from N-sulfinylated ethynylaziridines 6a or 6b and neither propionaldehyde nor acetone. The allenylzinc intermediates arising from N-sulfonylated ethynylaziridines 7a or 7b were slightly more prone to react but still satisfactory results were not achieved. The most promising reaction was that of 7b that afforded 1,3-aminodiol 14 as a single stereoisomer in 30% yield [21], by sequential ring opening by 1,2-metalate rearrangement and condensation with benzaldehyde (Scheme 7).

Preparation of 1,3-aminoalcohol 14.
4 Conclusion
To sum up, lithium alkynyl(disilyl)zincates derived from ethynylepoxides and ethynylaziridines undergo 1,2-migration of the organosilyl group with ring opening of the oxirane or aziridine ring by SNi displacement. This protocol, initially developed for the rearrangement of ethynylepoxides, was successfully extended to aziridines and thus offers a straightforward approach to both δ-oxy- and δ-amino α-silyl allenylzinc intermediates. δ-Oxy α-silyl allenylzinc intermediates could undergo condensation with aldehydes or ketones to provide 1,3-diols in good yields and stereoselectivities. In contrast, the δ-amino α-silyl allenylzinc intermediates only reacted sluggishly. Understanding the reasons for this lack of reactivity and finding suitable conditions allowing for the preparation of 1,3-aminoalcohols with satisfactory yields using this chemistry are currently our research objectives in this promising field.
5 Experimental section
5.1 General methods
Experiments involving organometallic compounds were carried out in dried glassware under a positive pressure of dry argon or nitrogen using Schlenk techniques. THF was dried over Na/benzophenone and distilled before use. Commercial nBuLi (Sigma–Aldrich or Acros) was titrated with Sufferts's reagent [22]. Commercial ZnBr2 was melted under nitrogen and immediately after cooling to room temperature dissolved in dry THF. Chloro(dimethyl)phenylsilane (Sigma–Aldrich) and all other reagents were of commercial quality and were used without purification. Flash column chromatography was performed using the indicated solvents on silica gel 60 (15–40 or 40–63 μm particle size) by Merck. 1H NMR and 13C NMR spectra were recorded with a Bruker AV 400. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to the residual solvent resonance as the internal standard (CHCl3: δ = 7.26 ppm for 1H NMR and CDCl3: δ = 77.16 ppm for 13C NMR). Data are reported as follows: chemical shift, multiplicity (br s = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet), coupling constant, and integration. Infrared (IR) spectra were recorded on a Bruker Tensor 27 ATR diamond spectrophotometer equipped with an ATR unit. High-resolution mass spectra (HRMS) were obtained using a Bruker micrOTOF instrument equipped with ESI source. Product characterization data for compounds 5 [10e] and 12′a–c [14,19b] have been previously reported.
5.1.1 Preparation of PhMe2SiLi (∼0.40 M in THF)
Under argon, Li (557 mg, 80.0 mmol) was washed with dry THF (3 × 5 mL). TMSCl (5.1 mL, 40.0 mmol) and THF (20 mL) were added. After 15 min of sonication, Li was washed with dry THF (4 × 5 mL) and then taken up in dry THF (40 mL). After an additional 5 min of sonication, PhMe2SiCl (3.3 mL, 20.0 mmol) was added and the resulting mixture was sonicated for 2 h at 0 °C to give a dark red THF solution of PhMe2SiLi (∼0.40 M). The halogen–lithium exchange was assumed to be effective at 80% [23]. The solution was stored at −18 °C and used within 2 weeks.
5.1.2 Preparation of (PhMe2Si)2Zn (∼0.16 M in THF)
To a stirred THF solution of PhMe2SiLi (∼0.40 M, 3.75 mL, 1.50 mmol) was added dropwise a THF solution of ZnBr2 (1.0 M, 0.75 mL, 0.75 mmol) at −20 °C giving a dark green solution within 10 min.
5.2 Preparation of N-tert-butanesulfonylaziridines 7a–c
5.2.1 Typical procedure for the oxidation of N-tert-butanesulfinylaziridines (preparation of 7a)
To a stirred CH2Cl2 (40 mL) solution of N-tert-butanesulfinylaziridine 6a (1.54 g, 7.22 mmol) at room temperature was added m-CPBA (≤77% assay, 1.96 g, 7.94 mmol). The mixture was stirred for 10 min and an aqueous saturated solution of Na2S2O3 (40 mL) was added. After 30 min of stirring, the layers were separated and the aqueous layer was extracted with AcOEt (×2). The combined organics were washed with an aqueous saturated solution of NaHCO3 (×3), water (×2), and brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (cyclohexane/AcOEt = 90:10) afforded analytically pure 7a (1.32 g, 80%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ = 3.00 (dd, J = 4.3, 2.0 Hz, 1H), 2.87 (dt, J = 8.2, 4.3 Hz, 1H), 2.33 (d, J = 2.0 Hz, 1H), 2.07–1.97 (m, 1H), 1.70–1.59 (m, 3H), 1.48 (s, 9H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ = 78.3, 72.8, 60.6, 49.1, 34.8, 31.4, 24.1 (3C), 20.4, 13.8. IR (neat): 3244, 2967, 2935, 2875, 2361, 1301, 1123, 1018, 881, 729, 669 cm−1. HRMS (ESI): m/z [M + Na] + calcd for C11H19NNaO2S, 252.1029; found, 252.1020.
5.2.2 Preparation of product 7b
The typical procedure was followed with 6b (320 mg, 1.50 mmol). Purification by flash column chromatography on silica gel (cyclohexane/AcOEt = 70:30) afforded analytical pure 7b (340 mg, 99%) as a pale yellow amorphous solid. 1H NMR (400 MHz, CDCl3): δ = 2.99 (dd, J = 4.3, 2.0 Hz, 1H), 2.79 (dd, J = 7.1, 4.3 Hz, 1H), 2.36 (d, J = 2.0 Hz, 1H), 1.94–1.85 (m, 1H), 1.47 (s, 9H), 1.11 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ = 77.9, 73.6, 60.5, 53.8, 32.8, 28.7, 24.2 (3C), 20.2, 19.0. IR (neat): 3245, 2977, 2929, 2361, 1303, 1131, 886, 703, 670 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C11H19NNaO2S, 252.1029; found, 252.1035.
5.2.3 Preparation of product 7c
The typical procedure was followed with 6c (516 mg, 2.42 mmol). Purification by flash column chromatography on silica gel (cyclohexane/AcOEt) afforded analytical pure 7c (500 mg, 90%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ = 3.19 (dd, J = 6.8, 1.9 Hz, 1H), 2.86–2.76 (m, 1H), 2.24 (d, J = 1.9 Hz, 1H), 1.69–1.59 (m, 2H), 1.57–1.48 (m, 2H), 1.48 (s, 9H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ = 72.7, 72.6, 59.6, 41.6, 35.0, 30.5, 24.1 (3C), 19.9, 13.8.
5.3 Typical procedure for the rearrangement of ethynylepoxides and ethynylaziridines (preparation of 13a (Table 3, entry 2))
nBuLi (2.40 M in hexanes, 0.23 mL, 0.55 mmol) was added dropwise at −80 °C to a THF (5 mL) solution of ethynylaziridine 7a (115 mg, 0.50 mmol). The mixture was stirred for 1 h at this temperature, then warmed to −60 °C, and added via cannula to a THF solution of (PhMe2Si)2Zn (0.16 M, 4.7 mL, 0.75 mmol) at −60 °C. The reaction mixture was then warmed to 0 °C and stirred for 2 h. MeOH (1 mL) was added and the mixture was left to warm to room temperature. Et2O (10 mL) was then added and the layers were separated. The aqueous layer was extracted with Et2O (×2), and the combined organics were washed with water and brine, dried over MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (cyclohexane/AcOEt = 90:10) afforded analytical pure 13a (127 mg, 70%) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ = 7.62–7.60 (m, 2H), 7.44–7.38 (m, 3H), 4.01 (d, J = 9.9 Hz, 1H, NH), 3.55–3.43 (m, 1H), 2.68 (AB system, J = 17.1, 5.7 Hz, 1H), 2.56 (AB system, J = 17.1, 4.4 Hz, 1H), 1.76–1.65 (m, 2H), 1.46–1.34 (m, 2H), 1.40 (s, 9H), 0.99 (t, J = 7.5 Hz, 3H), 0.41 (s, 6H). 13C NMR (100 MHz, CDCl3): δ = 137.2, 133.7 (2C), 129.5, 128.0 (2C), 104.2, 86.3, 60.0, 54.7, 28.4, 26.9, 24.4 (3C), 10.6, −0.67, −0.69 (one C missing). HRMS (ESI): m/z [M + Na]+ calcd for C19H31NNaO2SSi, 388.1737; found, 388.1728.
5.3.1 Preparation of product 12a (Table 2, entry 6)
The typical procedure was followed with 6a (107 mg, 0.50 mmol) but using an aqueous solution of HCl (1.0 M) for hydrolysis instead of MeOH. Purification by flash column chromatography on silica gel (cyclohexane/AcOEt = 90:10) afforded 12a contaminated with 12a′ (114 mg, 12a/12a′ = 90:10, 65%). Data for 12a: 1H NMR (400 MHz, CDCl3): δ = 7.64–7.58 (m, 2H), 7.40–7.30 (m, 3H), 3.57 (d, J = 8.1 Hz, 1H, NH), 3.39 (m, 1H), 2.72 (ABX system, J = 16.9, 5.8, 0.7 Hz, 1H), 2.56 (ABX system, J = 16.9, 4.7, 0.7 Hz, 1H), 1.65–1.51 (m, 2H), 1.48–1.30 (m, 2H), 1.19 (s, 9H), 0.92 (t, J = 7.4 Hz, 3H), 0.40 (s, 6H). 13C NMR (100 MHz, CDCl3): δ = 137.3, 133.7 (2C), 129.5, 128.0 (2C), 104.8, 86.3, 56.0, 54.5, 37.2, 28.2, 22.8 (3C), 19.1, 14.0, −0.61, −0.65. IR (neat): 3209, 3069, 2957, 2870, 2173, 1939, 1457, 1248, 114, 1051, 835, 814, 779, 730, 698 cm−1.
5.3.2 Preparation of product 13b (Table 3, entry 3)
The typical procedure was followed with 7b (115 mg, 0.50 mmol) but methanolysis was carried out at −80 °C. Purification by flash column chromatography on silica gel (cyclohexane/AcOEt = 90:10) afforded analytical pure 13b (127 mg, 70%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ = 7.62–7.60 (m, 2H), 7.39–7.35 (m, 3H), 3.92 (d, J = 10.0 Hz, 1H, NH), 3.36 (dddd, J = 10.1, 6.5, 5.7, 4.6 Hz, 1H), 2.68 (AB system, J = 17.3, 5.7 Hz, 1H), 2.60 (AB system, J = 17.3, 4.7 Hz, 1H), 2.05 (dq, J = 13.5, 6.8 Hz, 1H), 1.40 (s, 9H), 1.02 (d, J = 6.8 Hz, 3H), 1.00 (d, J = 6.8 Hz, 3H), 0.40 (s, 6H). 13C NMR (100 MHz, CDCl3): δ = 137.2, 133.7 (2C), 129.5, 128.0 (2C), 104.6, 86.3, 60.2, 58.4, 32.2, 24.9, 24.5 (3C), 19.1 (2C), −0.7 (2C). IR (neat): 3275, 2959, 2360, 2341, 1939, 1305, 1122, 814 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C19H31NNaO2SSi, 388.1737; found, 388.1722.
5.4 Typical procedure for sequential rearrangement–condensation with aldehydes (preparation of 4)
nBuLi (2.40 M in hexanes, 0.23 mL, 0.55 mmol) was added dropwise at −80 °C to a THF (5 mL) solution of ethynylepoxide 1a (69 mg, 0.50 mmol). The mixture was stirred for 1 h at this temperature, then warmed to −60 °C and added via cannula to a THF solution of (PhMe2Si)2Zn (0.16 M, 4.7 mL, 0.75 mmol) at −60 °C. The reaction mixture was warmed to −20 °C for 1.5 h and then to 0 °C for 45 min before being cooled to −80 °C. Propionaldehyde (180 μL, 2.5 mmol) was added at this temperature and the mixture was warmed to −40 °C and stirred for 2.5 h. After warming to room temperature, aqueous HCl (1.0 M, 10 mL) and Et2O (10 mL) were added and the layers were separated. The aqueous layer was extracted with Et2O (×2) and the combined organics were washed with water and brine, dried over MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (n-pentane/Et2O = 85:15) afforded analytical pure 4 (129 mg, 78%, 95:5 dr) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ = 7.66–7.61 (m, 2H), 7.43–7.35 (m, 3H), 3.97 (dd, J = 8.7, 3.1 Hz, 1H), 3.84 (m, 1H), 2.74 (dd, J = 8.7, 2.1 Hz, 1H), 1.75–1.25 (m, 7H), 1.00 (t, J = 7.4 Hz, 3H), 0.94 (t, J = 7.4 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H), 0.40 (s, 6H). 13C NMR (100 MHz, CDCl3): δ = 137.3, 133.7 (2C), 129.6, 128.0 (2C), 105.7, 88.4, 72.6, 71.8, 44.2, 43.1, 29.0, 22.9, 20.6, 12.4, 11.9, 10.7, −0.6 (2C). IR (neat): 3393, 3070, 2961, 2929, 2876, 2169, 1719, 1462, 1428, 1250, 115, 837, 817, 780, 731, 700 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C20H32NaO2Si, 355.2064; found, 355.2066.
5.4.1 Preparation of 1,3-aminoalcohol 14
The typical procedure was followed with 7b (115 mg, 0.50 mmol) and benzaldehyde (254 μL, 2.5 mmol). Purification by flash column chromatography on silica gel (n-pentane/Et2O = 90:10–70:30) afforded analytical pure 14 (71 mg, 30%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ = 7.53–7.51 (m, 2H), 7.42–7.28 (m, 8H), 4.98 (m, 1H), 4.0 (d, J = 10.2 Hz, 1H, NH), 3.43 (ddd, J = 10.2, 7.4, 2.9 Hz, 1H), 3.14 (d, J = 5.5 Hz, 1H, OH), 2.94 (dd, J = 7.4, 4.2 Hz, 1H), 2.28 (septd, J = 6.9, 2.9 Hz, 1H), 1.33 (s, 9H), 0.97 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H), 0.28 (s, 3H), 0.27 (s, 3H). 13C NMR (100 MHz, CDCl3): δ = 141.5, 136.9, 133.7 (2C), 129.5, 128.3 (2C), 128.0 (2C), 127.8, 126.5 (2C), 105.1, 90.0, 71.8, 60.7, 59.5, 48.1, 31.0, 24.6 (3C), 21.6, 17.2, −0.8, −0.9. IR (neat): 3490, 3278, 3060, 2964, 2170, 1427, 1297, 1115, 1018, 907, 819, 727 cm−1. HRMS (ESI) m/z [M + Na]+ calcd for C26H37NNaO3SSi, 494.2156; found, 494.2166.
Acknowledgments
The authors wish to thank the MESR for a grant to V.N.B. and Omar Khaled from Institut Parisien de Chimie Moléculaire (IPCM) for HRMS analysis.