

1 Introduction
Catalytic oxidative dehydrogenation (ODH) of light alkanes and particularly of propane is an interesting route to alkenes, which are useful intermediates for the petrochemical and energy industries [1].
A great variety of catalysts with various compositions have been used in the production of propene from propane by ODH [2–8]. Among them, vanadium-containing catalysts are probably the most broadly studied [1,9–11]. Vanadia dispersed on a variety of supports (SiO2, TiO2, ZrO2, Al2O3 and, more recently, graphene) have been indeed reported as catalysts for ODH processes [1,10,12–14]. VMgO mixed oxides, in particular, are among the most active and selective catalysts for the ODH of propane [5,15–19]. The catalytic activity of these materials is closely related to the V content and the composition of the vanadium oxide phase, which can be tuned during preparation, whereas the selectivity depends on the nature of the vanadium species at the catalyst surface. Consequently, the development of effective catalysts for propane ODH strongly depends on the preparation method [9,11].
Various methods can be used to prepare metallic vanadates [9,11]. An attractive route to prepare vanadium mixed oxides with highly dispersed vanadium species is the thermal decomposition of layered double hydroxide (LDH) precursors containing the cations to be present in the final mixed oxide materials.
LDH belongs to a large class of synthetic 2D nanostructured anionic clays, having the general formula [MII1 − xMIIIx(OH)2]x+An−x/n·nH2O, where MII and MIII stand for divalent and trivalent cations, respectively, and An− is a charge-balancing anion. The structure is composed of positively charged brucite-like Mg(OH)2 layers in which a fraction of divalent cations is partially substituted by trivalent cations introducing a positive charge balanced by exchangeable anions located in the interlayer region, where crystallization water molecules are also found [20].
LDHs provide high versatility in their composition because of the different nature of the metallic layer components and the balancing anions. This makes them very interesting mixed oxide catalyst precursors. Transition metal-containing LDH-derived mixed oxides with tuned redox and acid–base properties are remarkable oxidation catalysts [21], and those containing vanadium show particular activities and selectivities in the ODH reaction. For instance, Rives et al. [18] have reported the preparation of VMgAlO mixed oxides by calcination of a vanadate-exchanged MgAl–LDH obtained by coprecipitation of the precursor metallic salts. The activity of these LDH-derived catalysts was higher compared with that of the mixed oxides obtained by the impregnation of a MgO support with an aqueous vanadate salt solution.
Compared with the VMgO system, the properties of the VZnO mixed oxides in ODH have been studied to a lesser extent. To the best of our knowledge, no studies regarding VZnO mixed oxides obtained from anion-exchanged LDH precursors have been reported so far, although the substitution of Zn2+ for Mg2+ leads to different acid–base properties largely involved in the reaction.
Herein, we report the catalytic performance in propane ODH of a VZnAlO mixed oxide obtained via the calcination of a decavanadate-exchanged ZnAl–LDH. The ZnAl–LDH with nitrate as the interlayer anion was first prepared using the sol–gel method, then submitted to exchange with the decavanadate anion, and finally calcined to yield the mixed oxide catalyst. The catalytic performance of these materials was evaluated in propane ODH and compared with that of the VZn(Al)O catalysts prepared by conventional impregnation of either ZnO or ZnAlO mixed oxide supports with a decavanadate aqueous solution. By using these two methods, catalysts with different phase composition, acid–base properties, and reducibilities were obtained, as revealed by powder X-ray diffraction (PXRD), Fourier transform infrared (FTIR) spectroscopy monitoring of pyridine adsorption, temperature-programmed desorption (TPD) of CO2, and temperature-programmed reduction (TPR) experiments, respectively, which ultimately led to the different catalytic behavior in propane ODH.
2 Experimental section
2.1 Preparation of the catalysts
All chemicals were from Fluka and used without any further purification. Nitrate-containing ZnAl–LDH with nominal Zn/Al molar ratios of 2 or 3 were prepared using the sol–gel method as follows: 0.05 mol of zinc acetylacetonate Zn(C5H7O2)2 (98%) was dissolved under reflux (at 80 °C) in 100 mL of ethanol with vigorous stirring, and then 8.5 mL of HNO3 (60%) was added. A second solution containing a suitable amount of aluminum acetylacetonate Al(C5H7O2)3 (99%) (0.025 or 0.0167 mol) in 80 mL of a 1:1 acetone/ethanol mixture was then added dropwise to the former one under reflux at 80 °C with constant stirring. The pH was adjusted to 10 by addition of aqueous NH3 (33%). The suspension was further refluxed at 80 °C for 24 h and the gel formed was washed several times with ethanol, then with water at 80 °C to remove the unreacted organosalts, centrifuged, and dried overnight at 80 °C, yielding the samples ZnAlNx differing in the nominal Zn/Al molar ratio (x = 2 or 3).
The polyoxovanadate-containing samples were prepared by ion exchange from parent nitrate-containing ZnAl–LDH samples. One hundred milliliters of an aqueous solution of NH4VO3 (Panreac, 99%) was added to the gel suspension of precursors ZnAlN2 or ZnAlN3. The amount of NH4VO3 added was that necessary to yield a concentration of [V10O28]6− species 50% larger than that required to balance the positive charge of the brucite-type layer of the LDH. The pH of the mixture was adjusted to 4.5 by slow addition of aqueous HNO3 (30%), and the solution was stirred for 48 h under nitrogen. The solids were finally filtered, washed with decarbonated water, and dried overnight at 80 °C. The samples were named as ZnAlVx.
The LDH-derived mixed oxide catalysts were prepared by the calcination of the dried ZnAlVx precursors in air at 500 °C for 5 or 24 h, at a heating rate of 10 °C/min, yielding the mixed oxides ZnAlVO–LDHx,y (y = calcination time in hours). Thermal analyses have shown that this calcination temperature is high enough to yield thermodynamically stable phases.
For the sake of comparison, two additional samples were prepared by conventional wet impregnation of either commercial ZnO (Panreac, 99%; specific surface area = 10 m2 g−1) or ZnAlN2 LDH calcined at 500 °C with an aqueous solution containing the required amount of NH4VO3 necessary to reach a V/(Zn + Al) molar ratio of ca. 0.6. After impregnation, the solids were filtered, dried overnight at 80 °C, and finally calcined at 500 °C for 5 or 24 h. The dried samples were named as ZnV-I and ZnAlV-I, respectively. The calcined catalysts were named as ZnVO-I,y and ZnAlVO-I,y, respectively (where I stands for “impregnation” and y is the calcination time in hours).
2.2 Characterization of the catalysts
Zn, Al, and V element chemical analyses were carried out by atomic absorption spectroscopy using an ELL-240 Mark 2 instrument after dissolving the dried samples in HNO3.
The PXRD patterns were recorded using a Siemens D-500 diffractometer with the Cu Kα radiation (λ = 1.541 Å). The data were recorded in the 5–70° 2θ range with an angular step of 0.05° at 1.5 s per step, which resulted in a scan rate of 2°/min.
The specific surface areas were determined from the full nitrogen adsorption–desorption isotherms measured at −196 °C using a Micromeritics Gemini instrument on samples previously treated in situ at 150 °C for 2 h in flowing nitrogen.
The FTIR spectra were obtained using a Perking Elmer FTIR 1600 spectrometer in the 4000–400 cm−1 range, averaging 50 scans with a nominal resolution of 4 cm−1 to improve the signal-to-noise ratio. The samples were dispersed in solid KBr.
Thermogravimetric (TG) and differential thermal analysis (DTA) curves of the decavanadate-exchanged samples were recorded using a Perkin Elmer DTA/TG-7 apparatus, at a heating rate of 10 °C/min.
Surface acidity properties were determined by FTIR monitoring of adsorption of pyridine (Py) in a Perkin–Elmer 16PC spectrometer using self-supported discs. The samples were degassed in situ in a special cell with CaF2 windows, at 400 °C for 2 h at 10−4 N/m2, before the adsorption experiments. After degassing, the probe gas was adsorbed at room temperature and the spectrum recorded at increasing temperatures, from room temperature to 400 °C.
TPD of CO2 was conducted using a Micromeritics Autochem model 2910 instrument. The TPD program performed was as follows: ca. 75 mg of sample previously calcined at 500 °C for either 5 or 24 h was pretreated in situ at 150 °C for 60 min to remove the physisorbed species on the surface. The CO2 adsorption was performed with a flowing gas mixture containing 2.5% CO2 balanced with He (30 mL/min) at 80 °C for 30 min. After adsorption, a flow of He (30 mL/min) was passed through the sample at 80 °C for 30 min to remove the physisorbed CO2. Then, the analysis was performed in flowing He (30 mL/min) from 30 to 900 °C (10 °C/min). The number of basic sites was calculated from the area under the CO2 peak. The calibration of the instrument was done using a known amount of CaCO3. Water was trapped on magnesium perchlorate.
TPR studies were carried out in a TPR/TPD 2900 Micromeritics instrument, using 5 vol % H2 in Ar (from L'Air Liquide, Spain) at a heating rate of 10 °C/min, and using CuO (from Merck) for calibration.
2.3 Catalytic tests
The catalytic ODH of propane was carried out in a fixed-bed quartz tube downflow reactor operated at atmospheric pressure. The internal diameter of the reactor tube was 10 mm. A mass of 200 mg of catalyst powder (W), diluted with quartz in a 1:1 mass ratio to prevent the formation of hot spots, was used. The catalyst bed was supported by quartz wool. The axial temperature profile was measured using an electronic thermometer placed in a thermowell centered in the catalyst bed. The reactor temperature was controlled using a chromel–alumel thermocouple attached to the exterior of the reactor. Quartz chips were used to fill the dead volumes before and after the catalyst bed, to minimize potential gas-phase reactions at higher reaction temperatures. The gas mixture (8% O2, 8% C3H8, and 84% He, v/v) was fed into the reactor at a total flow rate (F) of 250 cm3/min. The contact time (W/F) for all the experiments was fixed at 8 × 10−4 min gcat/cm3. The reaction temperature was varied between 325 and 425 °C. Under these conditions, the homogeneous conversion of propane was negligible. The system was allowed to stabilize for about 30 min at the reaction temperature before the first product analysis was made. Each run was carried out over a period of 2–3 h during which four analyses were done. The reaction products were analyzed in a Trace GC Gas-Chromatograph (CE Instruments) equipped with a thermal conductivity detector using a 60/80 Carboxen 1000 column (15 ft × 1/8 in.) and a flame ionization detector using an alumina column (50 m × 0.53 mm). Propene, CO, and CO2 were the major products formed under the reaction conditions chosen. Cracking products (methane and ethylene) were also identified in some cases. The propane conversion and product selectivities were expressed as mol % on a carbon atom basis. The carbon balance was satisfactory in all the runs within an error margin of ±3%.
3 Results and discussion
3.1 Characterization of the catalysts
Elemental chemical analysis data for the LDH precursors are given in Table 1. The Zn/Al atomic ratios of 1.2 and 1.7 for the solid samples differ markedly from the nominal values of 2 and 3, respectively. The Zn/Al molar ratio for a pure LDH phase is usually in the range 2–4 [20], the Al(OH)3 side phase being formed for Zn/Al molar ratios less than 2. Although the Al(OH)3 phase can be identified by X-ray diffraction, it is usually amorphous and difficult to detect. The samples of this study with Zn/Al ratios of 1.2 and 1.7 probably contain amorphous Al(OH)3 extraphase, in agreement with the X-ray diffraction patterns, where only peaks of the LDH phase were detected (Fig. 1A).
Elemental chemical analysis of the parent ZnAlNx and ZnAlVx LDH materials and the dried impregnated samples.
| Sample | Chemical composition (at. %) | Molar ratios | ||||
| Zn | Al | V | Zn/Al | V/Zn | V/(Zn + Al) | |
| ZnAlN2 | 53.7 | 46.3 | – | 1.2 (2)a | – | – |
| ZnAlN3 | 62.9 | 37.1 | – | 1.7 (3)a | – | – |
| ZnAlV2 | 23.2 | 37.6 | 39.2 | 0.6 | 1.7 | 0.6 |
| ZnAlV3 | 22.4 | 31.8 | 45.8 | 0.7 | 2.0 | 0.8 |
| ZnAlV-I | 26.7 | 46.9 | 26.4 | 0.6 | 1.0 | 0.4 |
| ZnV-I | 60.0 | – | 40.0 | – | 0.7 | 0.7 |
a Nominal values in brackets.
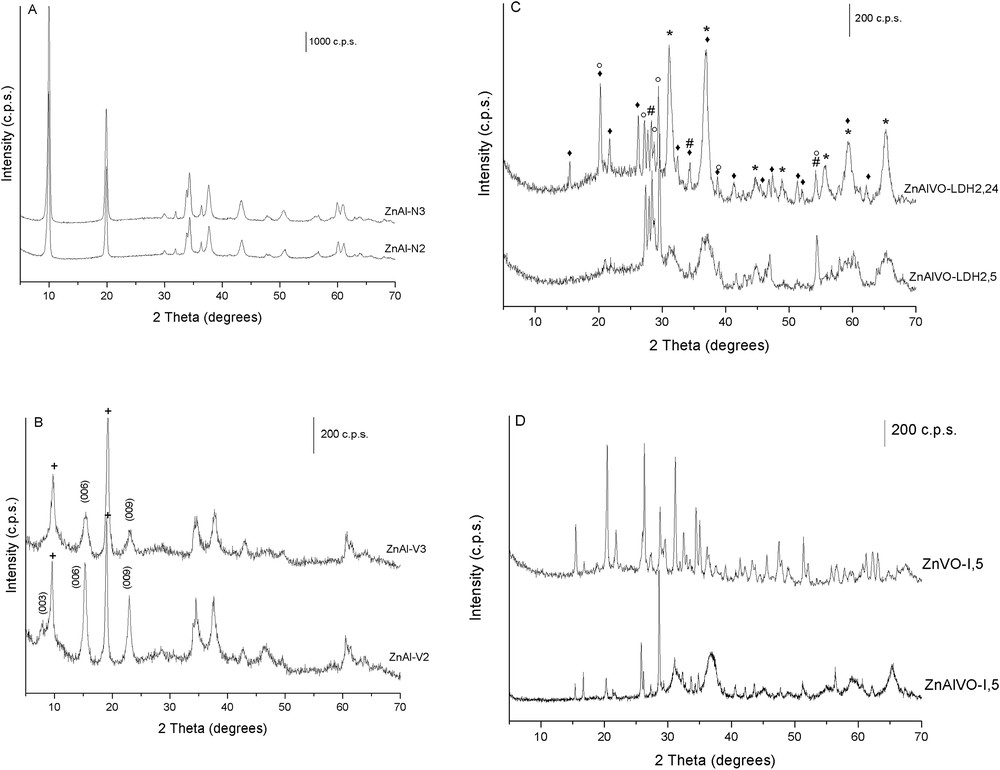
PXRD of the (A) ZnAlNx LDH precursors; (B) ZnAlVx decavanadate-exchanged LDHs; (C) LDH-derived ZnAlVO mixed oxides, and (D) Zn(Al)VO-I mixed oxides. (+) [V4O12]4− interlayer species; (*) ZnAl2O4; (♦) V2O5; (○) ZnV2O6; (#) Zn2V2O7.
After the decavanadate exchange, the Zn/Al ratio dramatically decreases. During the exchange process at pH 4.5 to form the decavanadate anion, Zn2+ cations are probably removed by partial dissolution of the layers, whereas precipitated aluminum hydroxides should still be present. This behavior would account for the abnormally low Zn/Al ratios observed [22]. Despite the different Zn/Al molar ratios in the ZnAlNx LDH precursors, both ZnAlVx LDH samples show similar Zn/Al ratios. The vanadium content is slightly higher for the ZnAlV3 sample. An increase in the vanadium content may be expected when the Zn/Al ratio decreases to balance the higher positive charge in the layers. However, the values found here strongly suggest the presence of different vanadium polyoxometalates (POMs) in the interlayer and the existence of a coprecipitated aluminum-containing side phase.
Regarding the impregnated materials, the V content of the ZnV-I sample is similar to that of the ZnAlVx LDH samples, whereas that of the ZnAlV-I sample is lower. However, the V surface density calculated for the impregnated oxides, allowing to compare supported catalysts with different surface areas, is much higher for the ZnV-I sample (945 V nm−2) than that for ZnAlV-I sample (42 V nm−2).
The PXRD patterns of vanadium-containing ZnAlV2 and ZnAlV3 samples (Fig. 1B) show the typical reflections of layered compounds. The presence of various layered phases can be concluded for both samples because of the presence of two different series of harmonics in the diffractograms. For instance, the PXRD pattern of the ZnAlV2 sample shows (00l) harmonics at d values of 11.6, 5.8, and 3.8 Å, corresponding to the diffraction by (003), (006), and (009) planes, respectively. The positions of the planes are in agreement with the intercalation of decavanadate species, [V10O28]6−, in the interlayer of the LDH oriented with its six-octahedra plane parallel to the brucite-like layers [24].
The (003) diffraction peak is generally the most intense for hydrotalcite-like compounds, but in these diffractograms the second (006) harmonic is the most intense one. This effect has been attributed to the formation of a new compound resulting from the reaction between the basic LDH layers and the acidic POMs [25–27]. However, this phenomenon, also observed in several LDHs intercalated with heavy molecular species, is attributed to the increase in the electron density in the midplane of the interlayers because of the presence of the heavy metal [28]. A weak peak at 10.8 Å in the PXRD patterns of similar samples has already been attributed to the Zn2+ and Al3+ salts of the POM anion [23]. Furthermore, the sharp reflections at 9.6 and 4.7 Å suggest the presence of intercalated metavanadate species, [V4O12]4−, formed at higher pH (>5.5) induced by the intrinsic basicity of the aqueous solution containing the dispersed ZnAlNx LDH [22].
The calcination of ZnAlVx LDH precursors at 500 °C in static air during 5 or 24 h results in different mixed oxide materials. As representative ones, the PXRD patterns of the ZnAlVO–LDH2,5 and ZnAlVO–LDH2,24 samples are depicted in Fig. 1c, and the crystalline phases identified in all the samples are reported in Table 2. The set of the maxima at 2θ = 25–30° for the samples calcined for 5 h has been assigned to a mixture of zinc vanadates, that is, zinc pyro- (Zn2V2O7; JCPDS 52-1893) and meta-vanadate (ZnV2O6; JCPDS 75-1392). The presence of additional V-containing extraphases in these samples may be expected because of the V/Zn atomic ratios greater than 2 (Table 1). Additional peaks corresponding to ZnO were not found. The calcination for 24 h improves the crystallinity and new lines develop, whereas those corresponding to ZnV2O6 and Zn2V2O7 are still present. Maxima corresponding to a ZnAl spinel-type phase (gahnite syn; JCPDS 5-0669) and V2O5 (JCPDS 74-1595) are also identified. Both ZnAlVO–LDH3,5 and ZnAlVO–LDH3,24 samples contain the same crystalline phases, their crystallinity being also enhanced by increasing the calcination time. This is consistent with the decrease in the specific surface area of the catalysts as the calcination time increases from 5 to 24 h (Table 2).
Crystalline phases and specific surface areas of the calcined catalysts.
| Sample | SBET (m2 g−1) | Crystalline phasesa | |
| Zn-vanadate | Other | ||
| ZnAlVO–LDH2,5 | 11 | meta; pyro | V2O5; ZnAl2O4 |
| ZnAlVO–LDH2,24 | 6 | meta; pyro | V2O5; ZnAl2O4 |
| ZnAlVO–LDH3,5 | 10 | meta; pyro | V2O5; ZnAl2O4 |
| ZnAlVO–LDH3,24 | 7 | meta; pyro | V2O5; ZnAl2O4 |
| ZnVO-I,5 | 3 | ortho; meta; pyro | ZnO; V2O5 |
| ZnAlVO-I,5 | 55 | ortho; pyro | ZnAl2O4; ZnO; V2O5 |
| ZnAlVO-I,24 | 55 | ortho; pyro | ZnAl2O4; ZnO; V2O5 |
a Detected by PXRD.
The PXRD pattern of the mixed oxide obtained by the impregnation of commercial ZnO (ZnVO-I,5 sample) shows a different profile than those of the oxides derived from the LDH precursors (Fig. 1d). It is more complex showing the presence of a mixture of meta-, pyro-, and ortho-vanadate phases, as well as V2O5 and ZnO (JCPDS 36-1451). On the other hand, the sample obtained by the impregnation of the LDH-derived ZnAl mixed oxide (ZnAlVO-I,5) shows diffraction lines corresponding to a mixture of pyro- and ortho-vanadate phases, the latter being absent in the ZnAlVO–LDHx,y mixed oxides (Fig. 1d). Other phases observed in the ZnAlVO-I,y samples are gahnite (ZnAl2O4), V2O5, and ZnO. However, in this case, peaks corresponding to the ZnAl2O4 spinel phase are markedly weaker and the crystallinity is less affected by the calcination time compared with the LDH-derived mixed oxides.
The FTIR spectra of representative samples intercalated with nitrate and decavanadate are shown in Fig. 2. Concerning the ZnAlN2 sample, the broad absorption between 3600 and 3300 cm−1 is due to the ν(OH) mode of the hydroxyl groups from the layers and to the antisymmetric and symmetric OH stretching modes of the interlayer water molecules. The band close to 1620 cm−1 is due to δ(H2O). The very intense absorption band close to 1370 cm−1 is attributed to the antisymmetric NO stretching mode of interlayer nitrate species, which are also responsible for the sharp, very weak absorptions at 1760 cm−1 (ν1 + ν4) and 827 cm−1 (ν2) [29]. The absorptions below 800 cm−1 are attributed to the lattice vibrations involving metal–oxygen stretching modes. The FTIR spectra of the polyoxovanadate-containing samples show new bands in the 1000–500 cm−1 range, rather broad probably because of the coexistence of two different polyoxovanadate species, as already suggested by the elemental chemical analysis and XRD data. As an example, the spectrum of the ZnAlV2 sample is included in Fig. 2. The band at 955 cm−1 is attributed to the stretching mode of terminal VO species, typical of decavanadate species in the interlayer space [22]. A large broadening of the ν(OH) mode extending from 3600 to 2500 cm−1 is also observed due to the presence of hydroxyls of the layers interacting with different intercalated species, and to differently bonded water molecules. Remarkably, the absence of the absorption band at around 1370 cm−1 associated with the antisymmetric NO stretching mode of interlayer nitrate species confirms that nitrate was completely exchanged by the polyoxovanadate in the interlayer space. For the calcined ZnAlVO–LDH2,24 sample, the doublet just below 1000 cm−1 is due to the ν(VO) and νs(VO4) modes, whereas the band and the shoulder at 666 and 712 cm−1 are due to the νas(VO4) and νas(VOV) modes, respectively. The sharp band at 1018 cm−1 is associated with the crystalline V2O5, in line with the PXRD data.
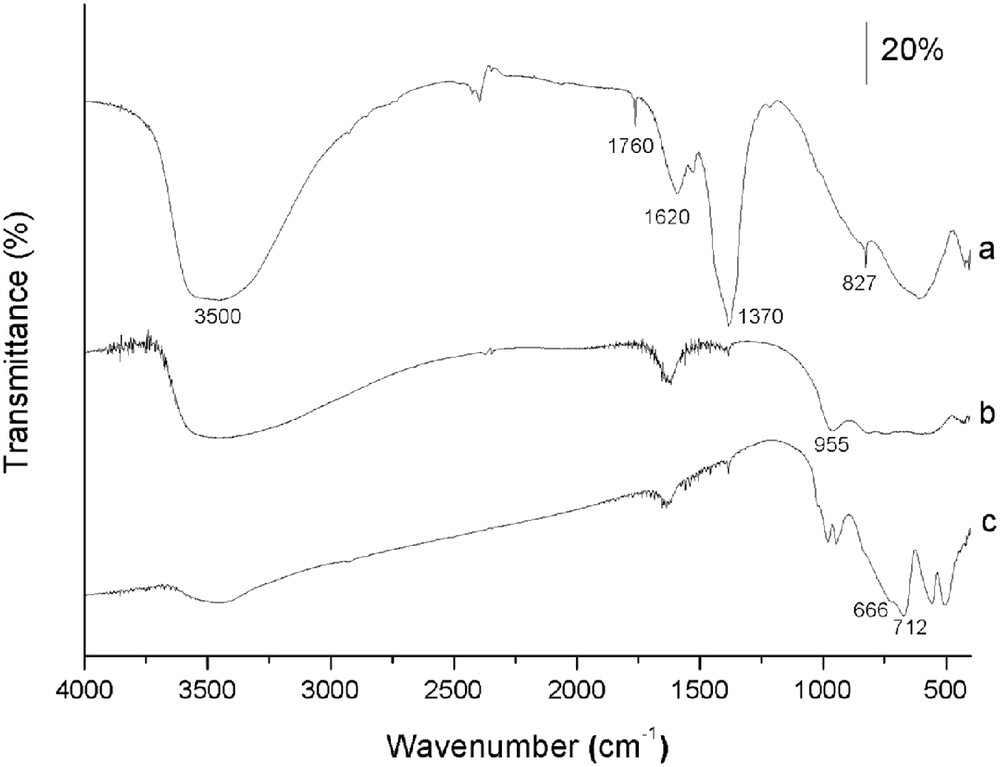
FTIR spectra of the (a) ZnAlN2, (b) ZnAlV2, and (c) ZnAlVO–LDH2,24 samples.
The TG and DTA curves of the decavanadate-exchanged samples are displayed in Fig. 3. They show a nearly gradual mass loss from 65 to 600 °C, with three endothermic events. The first intense endothermic peak at 155 °C is attributed to the removal of physisorbed and interlayer water molecules. The second endothermic signal, at 295 °C, is generally attributed to the dehydroxylation of the brucite-like layers. Finally, the less intense endothermic peak, recorded at 630 °C, corresponds to the depolymerization of the POM species [27]. The total mass loss found for the decavanadate-exchanged ZnAl LDH sample corresponds only to water removal by dehydration and dehydroxylation of the layers, because upon thermal decomposition, polyoxovanadate anions do not give rise to gaseous phases. A total mass loss less than 20–25% has usually been reported for this kind of samples, close to the theoretical mass loss of 24.6% for a pure decavanadate-exchanged hydrotalcite [22]. In our case, the ZnAlV2 and ZnAlV3 samples exhibited total mass losses of 10.5% and 21.2%, respectively. The ZnAlV3 sample contains a larger amount of oxovanadate species as shown by elemental chemical analysis (Table 1), which is also in agreement with the higher intensity of the endothermic peak in the DTA profile at 630 °C.
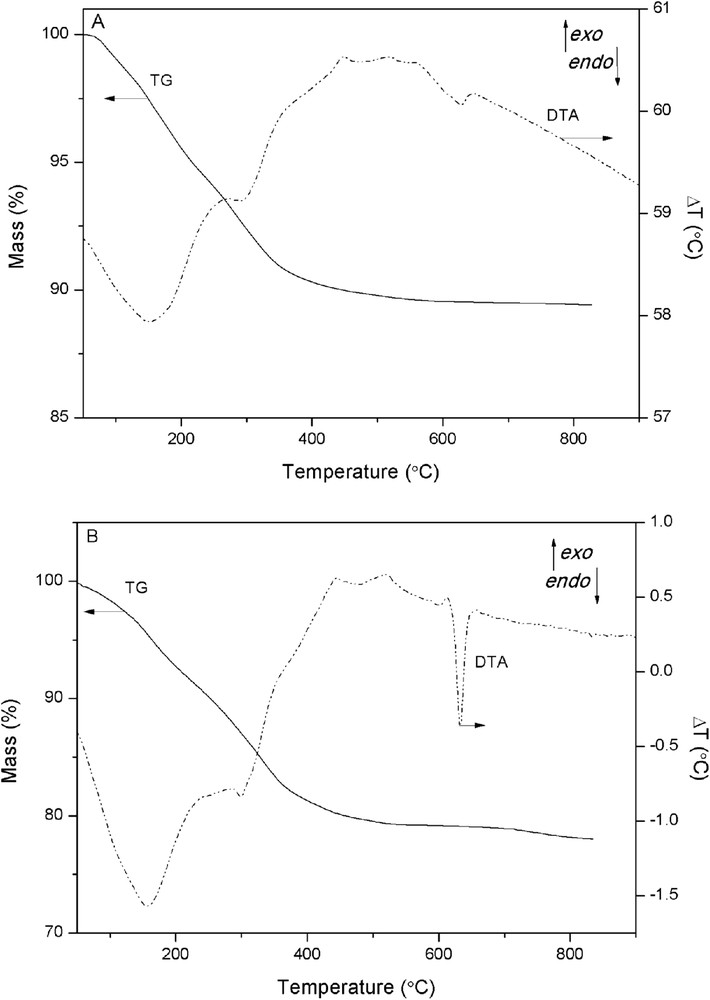
TG and DTA curves of the ZnAlV2 (A) and ZnAlV3 (B) samples.
The surface acidity of the calcined samples was studied by FTIR spectroscopy monitoring of the adsorption of pyridine (Fig. 4). The bands at 1607, 1575, and 1447 cm−1 recorded for the ZnAlVO–LDH3,24 sample (Fig. 4a) are ascribed to the ν(8a), ν(19a), and ν(19b) modes, respectively, of the pyridine adsorbed on surface Lewis acid sites resulting probably from the coordinatively unsaturated V5+ and Al3+ cations. The intensities of these bands slightly decrease and shift toward higher wavenumbers, that is, from 1607 to 1613 cm−1 for ν(8a) and from 1447 to 1449 cm−1 for ν(19b), when the sample is outgassed at increasing temperature. They are still observed after outgassing at 300 °C, indicating that these are strong Lewis acid sites. Other bands corresponding to the ν(8a) and ν(19b) modes of protonated pyridine are observed at 1537 and 1638 cm−1, indicating the presence of surface Brønsted acid sites. These bands totally vanish when the sample is outgassed at 300 °C, demonstrating that the Lewis-type acid sites are stronger than the Brønsted-type ones. These results differ from those reported by Blanco et al. [18] for VMgAlO mixed oxides derived from decavanadate-intercalated MgAl–LDH precursors, where only surface Lewis acid sites were observed. Blasco et al. [16] also observed only surface Lewis acid sites for vanadium supported on MgO and MgAl mixed oxide catalysts and weak Brønsted acid sites for vanadium supported on alumina catalyst. The same types of Lewis and Brønsted acid sites are found in our sample calcined for a shorter period of time, that is, ZnAlVO–LDH3,5, but they are of weaker strength because outgassing at 200 °C totally removes the adsorbed pyridine (Fig. 4B). Moreover, this sample presents a band at 1595 cm−1 after evacuation at room temperature attributed to hydrogen-bonded pyridine on the surface, similar to that observed by Connell and Dumesic [30] for pure alumina. These bands are easily removed after evacuation at 200 °C. Thus, ZnAlVO mixed oxides derived from decavanadate-exchanged ZnAl–LDH show Lewis and Brønsted acid sites whose strength increases with the calcination time from 5 to 24 h.
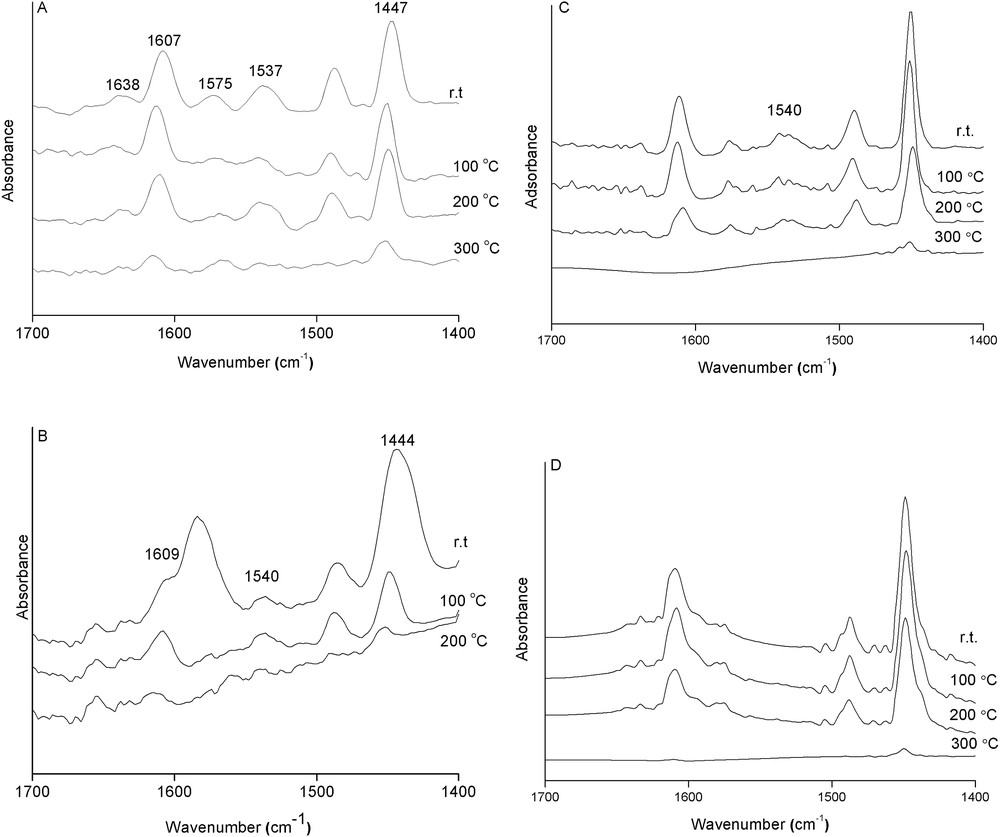
Infrared spectra of pyridine preadsorbed upon desorption at increasing temperatures on the (A) ZnAlVO–LDH3,24; (B) ZnAlVO–LDH3,5; (C) ZnAlVO-I,24; and (D) ZnAlVO-I,5 samples.
Previous studies of pyridine adsorption on calcined ZnAlNO3 and ZnAlCO3 LDHs revealed only the presence of Lewis acid sites [31,32], suggesting that the Brønsted acid sites found in the ZnAlVO mixed oxides of this study are associated with the presence of vanadium oxides.
ZnAlVO-I,y mixed oxides prepared by impregnation show different acid sites depending on the calcination time (Fig. 4c and d). ZnAlVO-I,5 shows only bands due to the pyridine adsorbed on surface Lewis acid sites, whereas ZnAlVO-I,24 shows additional bands corresponding to Brønsted acid sites. In both cases, pyridine is totally desorbed at 300 °C, indicating medium-strength acidity, weaker than that found in LDH-derived mixed oxides.
No pyridine adsorption could be observed for the ZnVO-I sample, therefore exhibiting no acidity.
The nature and strength of the acid sites existing in all the samples can hardly be related to the nature and the relative amount of the crystalline phases. Indeed, samples exhibiting the same crystalline phases, such as ZnAlVO–LDH3,5 and ZnAlVO–LDH3,24, or ZnAlVO-I,5 and ZnAlVO-I,24, show different Lewis and Brønsted acid strengths in the two former samples, or sites of different nature and strength in the two latter samples. The different acidities can be due to the different degrees of crystallinity of the vanadium-containing phases and to their interaction with the aluminum species existing in the samples.
The CO2-TPD profiles of the catalysts are presented in Fig. 5, whereas the amount and the surface density of basic sites estimated from the area under the peaks are tabulated in Table 3. The ZnAlVO-I,5 sample presents a TPD profile with two distinguishable peaks of CO2 desorption at ca. 440 and 634 °C, thus presenting the highest basic strength. The ZnAlVO-I,24 sample shows a single peak at ca. 450 °C suggesting that the strongest basic sites vanish by increasing the calcination time for the impregnated catalysts. The ZnAlVO–LDH2,5 sample shows two broad and weakly intense peaks in the range 250–600 °C accounting for a low amount of basic sites, whereas the ZnAlVO–LDH2,24 sample exhibits a very weak peak at ca. 600 °C accounting for a very low amount of basic sites. This shows that the basicity almost disappears at larger calcination time. The ZnAlVO–LDH3,5 sample exhibits no basicity. As a general trend, one could say that the catalysts prepared by impregnation (ZnAlVO-I) presented higher basic character than those prepared from exchanged Zn/Al LDHs, both in terms of strength and amount of basic sites. This mainly accounts for the higher specific surface area of the former. Indeed, regarding the surface density of basic sites, all the catalysts show low and similar basicity (Table 3).
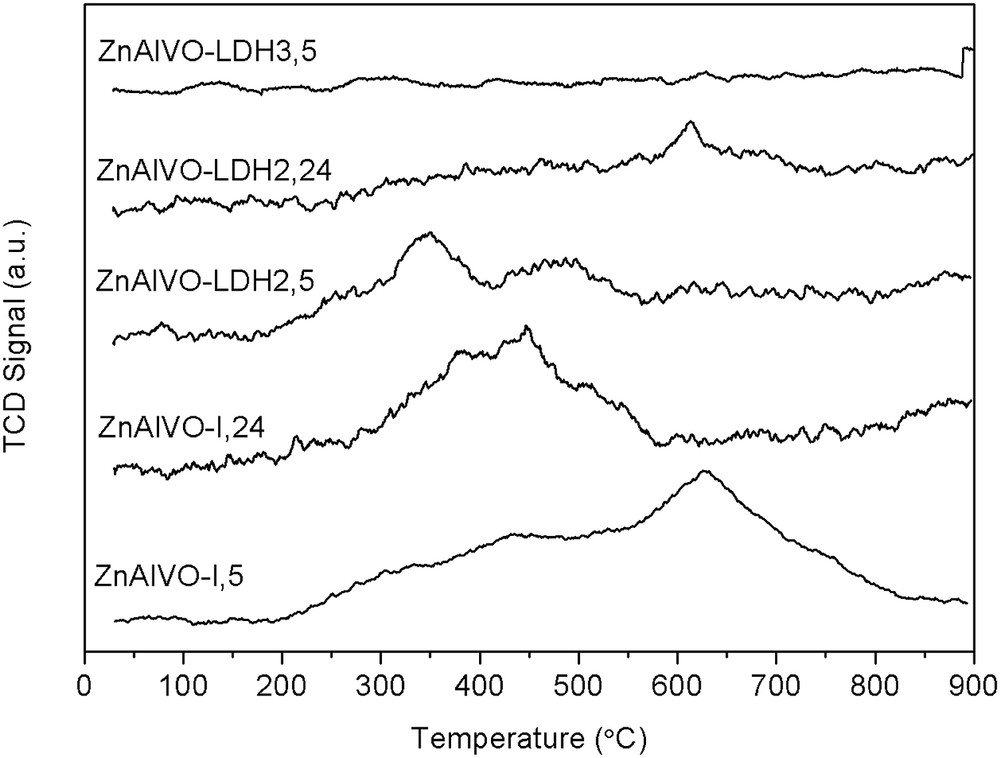
CO2-TPD profiles of the ZnAlVO-I,5, ZnAlVO-I,24, ZnAlVO–LDH2,5, ZnAlVO–LDH2,24, and ZnAlVO–LDH3,5 catalysts.
Basic properties of the catalysts as determined by CO2-TPD.
| Sample | Amount of CO2 (μmol g−1) | Surface density of sites (μmol m−2) |
| ZnAlVO–LDH2,5 | 113 | 10.3 |
| ZnAlVO–LDH2,24 | Not integrated | – |
| ZnAlVO–LDH3,5 | – | – |
| ZnAlVO-I,5 | 390 | 7.0 |
| ZnAlVO-I,24 | 304 | 5.5 |
The H2-TPR patterns of the catalysts are displayed in Fig. 6. ZnAlVO–LDHx,y samples show only one peak with the maximum reduction rate around 520–540 °C, whereas for the samples synthesized by conventional impregnation of ZnO and ZnAlO mixed oxide, that is, ZnVO-I,5 and ZnAlVO-I,5, respectively, two broad peaks are observed. In the case of the ZnVO-I,5 sample, the pattern has been deconvoluted into three peaks with maxima at 472, 546, and 596 °C, whereas the pattern of the ZnAlVO-I,5 sample has been deconvoluted in four peaks with maxima at 444, 474, 580, and 615 °C. This indicates a markedly different reducibility of the existing vanadate phases detected by PXRD, dispersed in a nonuniform manner on the mixed oxide matrix. The lower temperature reduction peaks are attributed to monomeric VOx and oligomeric species in tetrahedral coordination geometry, whereas the higher temperature reduction peaks belong to both polymeric and bulk-like V2O5 species, more difficult to be reduced and whose reduction follows several steps: V2O5 → ⅓ V6O13 → V2O4 → V2O3 [17,33]. An amount of vanadium oxide species more reducible than those existing in the ZnAlVO–LDHx,y samples was indeed observed below 500 °C in the ZnVO-I,5 and ZnAlVO-I,5 samples. The latter contains a higher proportion of more reducible vanadium sites than the former. The onset reduction temperatures for the catalysts prepared from LDH precursors of ca. 450 °C are rather similar, indicating the comparable reduction behavior of the near surface region of these oxides. The single-peak reduction pattern, identified as a one-step transition from V5+ to V3+ in the catalysts prepared from LDH precursors, was also observed by other authors [34,35]. The shift in the temperature for the maximum hydrogen consumption toward higher values at higher vanadium loading was equally noticed [34,36]. Notably, almost quantitative reduction of V5+ to V3+ species was observed in all the cases, the H2 consumption being higher than 90% (Table 4).
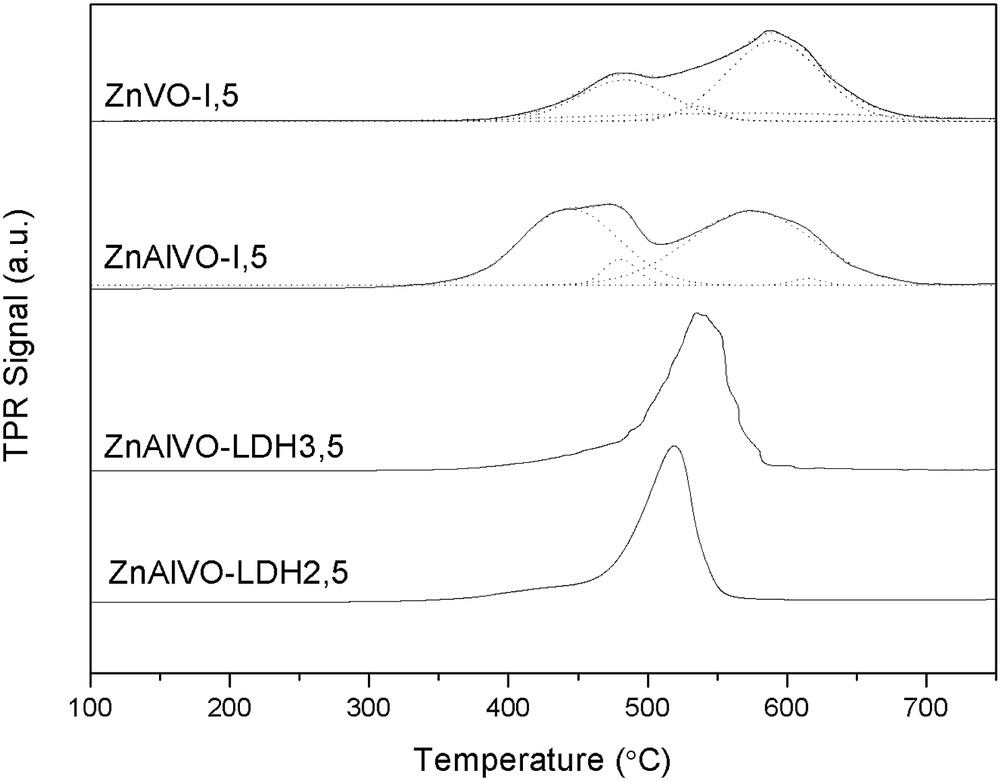
TPR profiles of the ZnVO-I,5, ZnAlVO-I,5, ZnAlVO–LDH2,5, and ZnAlVO–LDH3,5 catalysts.
H2 consumption in TPR experiments of the different catalysts.
| Sample | ZnAlVO–LDH2,5 | ZnAlVO–LDH3,5 | ZnAlVO-I,5 | ZnVO-I,5 |
| H2 consumption (%)a | 92 | 96 | 91 | 92 |
a Calculated assuming the reduction of V5+ to V3+ species.
3.2 Catalytic studies
The results of the ODH of propane over ZnAlVO–LDHx,y, ZnAlVO-I,y, and ZnVO-I,y catalysts are summarized in Table 5. Taking into account the various physicochemical properties of the different families of mixed oxides, we will particularly focus on the influence of the preparation method, the composition, and the calcination time on their catalytic performances.
Oxidative dehydrogenation of propane on Zn(Al)VO mixed oxides.a
| Catalyst | T (°C) | Propaneb conversion (%) | Rate of propane conversion (mmol m−2 h−1) | Selectivity (%) | Propene yield (%) | Eact (kJ mol−1) | ||
| C3H6 | CO2 | CO | ||||||
| ZnAlVO–LDH2,5 | 325 | 1.0 | 0.23 | 69.4 | 10.1 | 20.5 | 0.7 | 97 |
| 375 | 4.5 | 1.02 | 52.8 | 13.6 | 33.6 | 2.4 | ||
| 425 | 16.3 | 3.70 | 35.7 | 17.4 | 46.9 | 5.8 | ||
| ZnAlVO–LDH2,24 | 325 | 1.8 | 0.75 | 60.9 | 12.2 | 26.9 | 1.1 | 80 |
| 375 | 6.1 | 2.54 | 47.6 | 15.8 | 36.6 | 2.9 | ||
| 425 | 18.2 | 7.57 | 33.3c | 22.4c | 41.2c | 6.1 | ||
| ZnAlVO–LDH3,5 | 325 | 1.2 | 0.30 | 68.3 | 12.0 | 19.7 | 0.8 | 93 |
| 375 | 5.6 | 1.40 | 51.9 | 17.1 | 31.0 | 2.9 | ||
| 425 | 17.4 | 4.34 | 34.1 | 27.2 | 38.7 | 5.9 | ||
| ZnAlVO–LDH3,24 | 325 | 2.2 | 0.78 | 63.9 | 17.0 | 19.1 | 1.4 | 79 |
| 375 | 7.3 | 2.60 | 49.9 | 22.1 | 28.1 | 3.6 | ||
| 425 | 21.5 | 7.67 | 30.3c | 26.8c | 37.4c | 6.5 | ||
| ZnVO-I,5 | 325 | – | – | – | – | – | – | – |
| 375 | – | – | – | – | – | – | ||
| 425 | 0.7 | 0.58 | 54.4 | 26.9 | 18.7 | 0.4 | ||
| ZnAlVO-I,5 | 325 | 0.6 | 0.03 | 79.2 | 10.4 | 10.4 | 0.5 | 99 |
| 375 | 3.4 | 0.15 | 51.6 | 17.1 | 31.3 | 1.8 | ||
| 425 | 10.4 | 0.47 | 40.3 | 19.6 | 40.1 | 4.2 | ||
| ZnAlVO-I,24 | 325 | 0.9 | 0.04 | 88.6 | 5.0 | 6.4 | 0.8 | 95 |
| 375 | 4.4 | 0.20 | 62.1 | 15.7 | 22.2 | 2.7 | ||
| 425 | 13.8 | 0.63 | 30.4 | 21.2 | 48.4 | 4.2 |
a Propane/oxygen/helium feed (vol %): 8/8/84.
b Oxygen conversion was in all cases largely below 100%.
c Cracking products in balance.
3.2.1 Influence of the preparation method
The catalysts prepared by calcination of LDH precursors show higher propane conversions and much higher intrinsic activities in the temperature range studied compared with the catalysts prepared by impregnation of NH4VO3 on commercial ZnO or ZnAlO mixed oxide. Indeed, the latter shows lower activities, although with slightly different behaviors. Thus, the ZnVO-I,5 catalyst, calcined for 5 h, presents only very low activity at 425 °C and is not active when calcined for 24 h. On the contrary, the catalysts prepared by impregnation of the ZnAlO mixed oxide support (ZnAlVO-I,y) are more active than ZnVO-I,5 leading to propane conversion starting from 325 °C, which then increases with the reaction temperature. However, the conversion does not exceed 14% at 425 °C for the impregnated catalysts, value that is substantially lower than that of the best LDH-derived catalyst at the same reaction temperature, that is, ca. 22% (Table 5). Notably, the intrinsic rate of propane conversion is ca. 10 times higher for the LDH-derived catalysts compared to the impregnated ones.
The selectivity to propene over both families of catalysts, that is, ZnAlVO–LDH and ZnAlVO-I, decreases as the reaction temperature increases. This is a common feature of most of the ODH catalysts because of the increasing contribution of the total oxidation processes, that is, consecutive propene oxidation and parallel propane combustion. Therefore, all these catalysts show increasing CO2 and CO selectivities with temperature, the selectivity to CO being always higher than that to CO2 (Table 5). It is noteworthy that CO is formed mainly by consecutive propene oxidation, whereas CO2 is produced from propane combustion via parallel oxidation reactions [14,37]. Thus, propene oxidation to CO is predominant on both ZnAlVO–LDH and ZnAlVO-I families of catalysts, likely due to their acid properties evidenced by pyridine adsorption. On the ZnVO-I catalyst, which exhibits no acidity, CO2 was predominant.
In summary, the catalysts obtained from the decavanadate-exchanged LDH precursors are more active than those prepared by the impregnation of NH4VO3 on ZnO or ZnAlO mixed oxide supports, in terms of both propane conversion and intrinsic activity.
3.2.2 Influence of the composition and the calcination time
Regarding the catalysts derived from the decavanadate-exchanged LDH precursors, the data in Table 5 show that the ZnAlVO–LDH3,y catalysts are more active than their corresponding ZnAlVO–LDH2,y ones, both in terms of propane conversion and intrinsic activity. This is obviously due to the higher V content in the former catalysts (Table 1). The propene selectivity is similar for all the ZnAlVO–LDHx,y catalysts and decreases to the benefit of COx from 60–70% at low temperature to 30–35% at high temperature. On the other hand, the ZnAlVO–LDHx,24 catalysts are more active than the ZnAlVO–LDHx,5 ones, the intrinsic activity of the former being ca. two times higher within all the temperature range studied. This behavior can be related to the higher Brønsted and Lewis acidity of the ZnAlVO–LDHx,24 samples. Moreover, these catalysts also give low amounts of cracking products, that is, 3.1% and 5.5% for x = 2 and 3, respectively, at 425 °C.
Regarding the catalysts prepared by impregnation of commercial ZnO, only ZnVO-I,5 calcined for 5 h presents propane conversion (0.7%) at 425 °C, with propene selectivity close to 54%. On the contrary, the ZnAlVO-I,y catalysts show propane conversion at temperatures much lower than the ZnVO-I,5 system, reaching conversions higher than 10% and 13% at 425 °C for y = 5 and 24, respectively. However, these catalysts show a noticeably lower activity than the LDH-derived systems (ZnAlVO–LDHx,y), both in terms of conversion and, mainly, intrinsic rate of propane transformation. This may be due, on the one hand, to the lower V content of the impregnated catalysts and their different phase composition compared with the LDH-derived ones. It is, on the other hand, consistent with their medium strength acidity, weaker than that found in the ZnAlVO–LDHx,y catalysts. The propene selectivity is similar for both ZnAlVO-I,5 and ZnAlVO-I,24 catalysts in the whole temperature range studied and decreases to the benefit of COx from 80–90% at 325 °C to 30–40% at 425 °C. It can be noticed that the catalytic performances of the ZnAlVO-I,y catalysts slightly increase with the calcination time.
The apparent activation energies (Eact) for propane conversion on the different catalysts have been determined (Table 5) from the Arrhenius plots displayed in Fig. 7. The most active catalysts, that is, the LDH-derived mixed oxides calcined for 24 h (ZnAlVO–LDHx,24) show, as expected, the lowest apparent activation energy, which follows the catalytic activity: the lower the activation energy, the higher the intrinsic rate of propane conversion. However, the variation in the activation energy spans in the same range (90 ± 10 kJ mol−1) for all the catalysts, and therefore it cannot solely explain their different activities. The pre-exponential factor, which depends on the catalyst structure and the number of the active sites, must also be taken into consideration to explain the differences in reactivity observed for the solids prepared from LDH precursors compared with the impregnated ones and for the solids calcined for different times. It is noteworthy that the values obtained for the apparent activation energy fall within the usual range measured for propane ODH over vanadium-based catalysts [33,38,39].
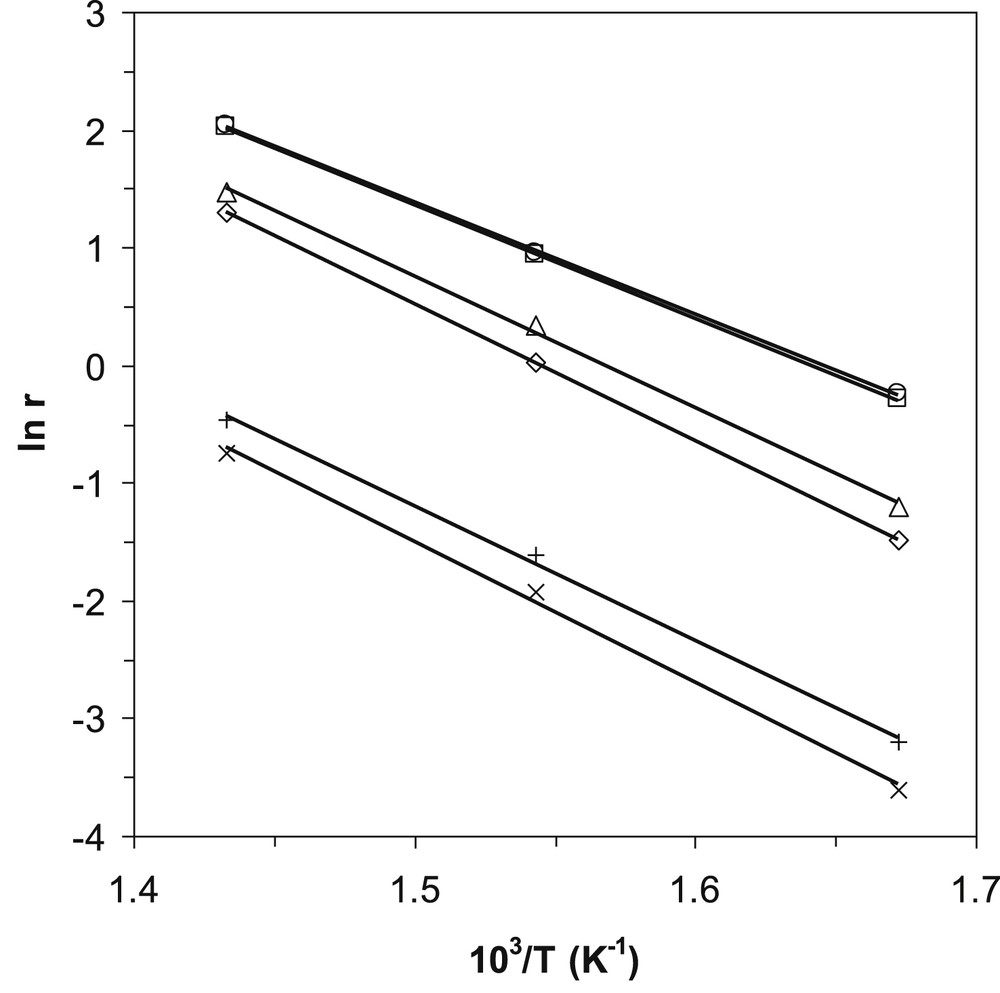
Arrhenius plots for the propane conversion over the different catalysts: ZnAlVO–LDH2,5 (⋄), ZnAlVO–LDH2,24 (□), ZnAlVO–LDH3,5 (▵), ZnAlVO–LDH3,24 (○), ZnAlVO-I,5 (×), and ZnAlVO-I,24 (+), in the temperature range from 325 to 425 °C.
The effect of the propane conversion on the propene selectivity is shown in Fig. 8, the conversion change for each sample being obtained by modifying the reaction temperature. Comparing the data obtained at different temperatures is justified in the case of vanadium-based catalysts because of the similarity of the activation energies for the propene formation and its consecutive oxidation to carbon oxides [40]. According to the data in Fig. 8, all the catalysts show similar selectivities to propene, which, as expected, decrease with increasing conversion. The extrapolation to zero conversion reveals that propene is not the only primary product, clearly suggesting that the direct oxidation of propane represents an important pathway toward the total oxidation products (COx) on these catalysts, likely because of their relatively high vanadium content.
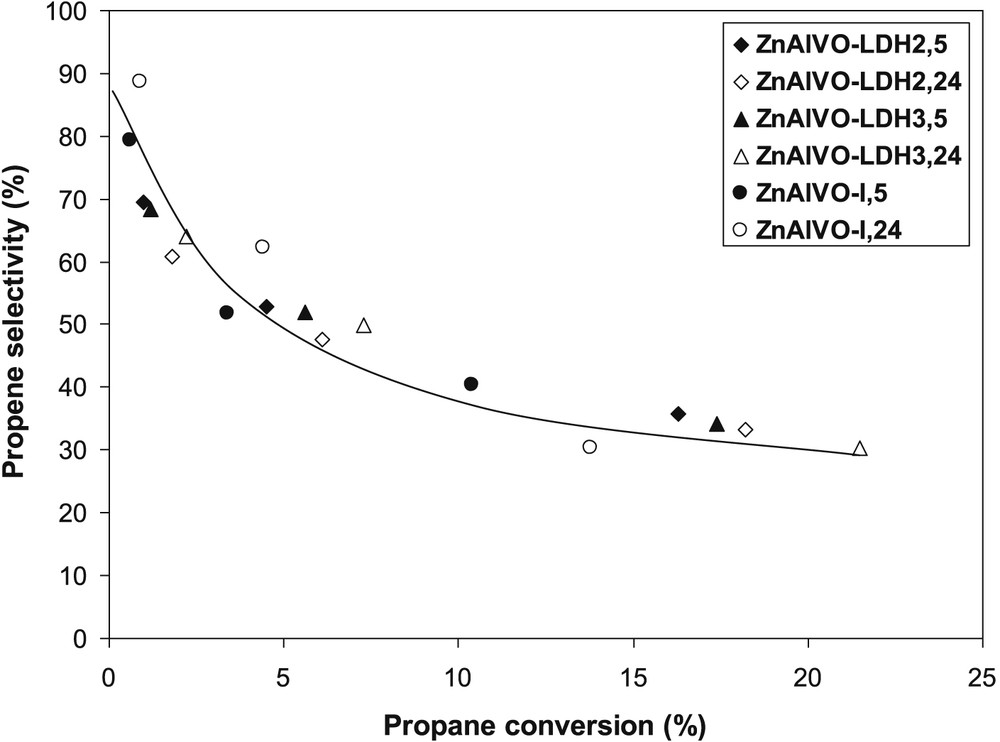
Effect of the conversion on the propene selectivity in the ODH of propane over the ZnAlVO–LDHx,y and ZnAlVO-I,y catalysts.
It is well known that the acid–base properties of the catalyst have a strong influence on its behavior in alkane ODH [11,18,39,41,42]. The catalytic behavior of the different mixed oxides can be explained taking into account the surface acidity determined by FTIR of pyridine adsorption. Thus, propane activity followed the Lewis and Brønsted acid strengths of the different families of mixed oxides. Notably, as the impregnated samples (ZnAlVO-I,y and ZnVO-I,5), which exhibit larger amounts of highly reducible species, are less active than the decavanadate-exchanged LDH-derived systems exhibiting higher acid strength, it results that the acidity mainly governs the catalytic activity of these catalysts. This suggests that the acid sites are involved in the propane activation by the first CH bond breaking [43,44]. On the other hand, the presence of strong acid sites favors the total oxidation reaction of propane [44], in line with the direct propane oxidation pathway evidenced from the conversion-selectivity plot.
The higher acidity could also explain the higher intrinsic activity observed for ZnAlVO–LDH3,24 than for ZnAlVO–LDH2,24, probably due to the presence of a larger amount of coordinatively unsaturated vanadium cations on the surface of the former, acting as Lewis acid sites.
Regarding the selectivities, olefins have a more basic character than alkanes and, therefore, propene interacts more strongly with the surface of the catalysts as the acid strength increases. The longer retention time enhances the subsequent oxidation of propene, as revealed by the high amounts of carbon oxides observed. However, no significant differences in the propene selectivity are observed between the different catalysts, as revealed by the conversion-selectivity plot (Fig. 8), in line with their similar basic site density determined by CO2-TPD (Table 3).
ZnAlVO and, particularly, ex-LDH ZnAlVO catalysts have shown comparable propylene yield to the LDH-derived MgAlVO systems [18], but their higher acidity favored the formation of CO and CO2, which are the main products at higher temperatures. Moreover, ZnAlVO mixed oxides obtained by the calcination of V-containing ZnAl–LDH have shown better performance than similar systems, such as zinc-modified alumina-supported vanadium oxides with similar V/Zn ratios [45]. However, with ca. 6% propylene yield (at 425 °C), the ZnAlVO–LDH catalysts have lower catalytic performance in the propane ODH than the classical VMgO catalysts that can give propylene yields of 20% (in the temperature range 500–600 °C) [10].
4 Conclusions
The catalytic properties of different ZnAlVO mixed oxides prepared from calcined decavanadate-exchanged ZnAl–LDH precursors have been studied for the first time in the ODH of propane. They have been compared with supported catalysts obtained by the impregnation of a vanadium salt on ZnO and ZnAlO mixed oxide. The mixed oxides derived from LDH precursors show 10 times higher catalytic activity than those obtained by impregnation, being also higher than that observed for other ZnAlVO catalysts reported in the literature. The catalytic activity increases with the calcination time, this effect being more significant for the LDH-derived oxides. The most active catalyst is that prepared by the calcination for 24 h of the decavanadate-exchanged LDH precursor, that is, ZnAlVO–LDH3,24, which has the highest vanadium content and acidity. Indeed, it has been established that the acidity rather than the nature of the crystalline phases, the reducibility, or the specific surface area of the catalysts governs their catalytic activity in the propane ODH. All the catalysts show similar selectivities to propene, which decrease with increasing conversion (through reaction temperature) to the benefit of carbon oxides. The similar selectivities to propene observed have been correlated to the similar surface basic sites densities of the catalysts. The carbon oxides are formed by both direct propane combustion and sequential oxidation of propene.
Acknowledgments
Financial support from MICINN (grant MAT2009-08526) and ERDF is greatly acknowledged.