

1 Introduction
Selective hydrogenation lies at the heart of several industrial processes. Concepts and frontiers developed in the last decade to improve catalyst chemoselectivity have been recently reviewed [1]. These include (1) the nanostructuration of less conventional metals to improve their ability to activate H2, (2) the use of oxides as active phases, (3) alloying, (4) the ensemble control in hybrid materials, and (5) site isolation approaches in single-site heterogeneous catalysts. Catalytic hydrogenation of organic compounds possessing multiple unsaturated bonds, such as α,β-unsaturated aldehydes, is particularly challenging, requiring active sites able to (1) discriminate closely related moieties, and in some cases, (2) achieve preferential activation of a more thermodynamically stable function [2]. The selective hydrogenation of cinnamaldehyde (CAL) to cinnamyl alcohol (COL) is of both fundamental and industrial interest. COL is one of the important perfumery chemicals, which is commercially manufactured from CAL by a well-known process called Meerwein–Ponndorf–Verley reduction, where aluminum triisopropoxide is used as a reagent. Although COL is obtained in high yields by this process, disposal of aluminum salts is a major drawback of this method [3]. Hence, it is necessary to develop a process based on catalytic hydrogenation, which will avoid the use of stoichiometric reagents.
The hydrogenation of CAL can produce COL, hydrocinnamaldehyde (HCAL), and/or hydrocinnamyl alcohol (HCOL; Scheme 1). The selective production of COL is difficult because the hydrogenation of the CC bond is thermodynamically favored over that of the CO one.
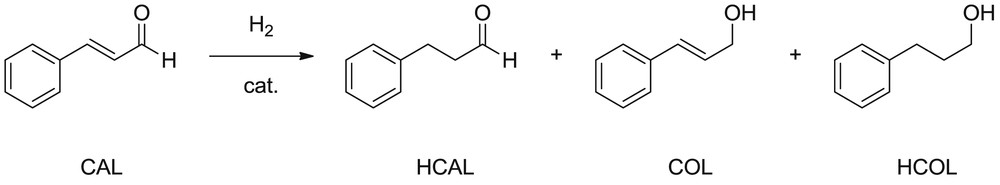
trans-CAL hydrogenation reaction.
Moreover, acetals and other unidentified high molecular weight compounds can also be produced in significant quantities. Although much research has already been focused on this issue, the selective hydrogenation of α,β-unsaturated aldehydes remains a challenge. High selectivity toward COL has been regularly reported on supported ruthenium catalysts [4–18]. In particular, ruthenium catalysts have been supported on a wide variety of carbon supports, including activated carbon (AC), fullerene C60, multiwalled carbon nanotubes (MWCNTs), single-walled carbon nanotubes (SWCNTs), graphite (G), organized mesoporous carbon (OMC), few-layer graphene (FLG), and carbon nanofibers (CNFs). Turn-over frequencies (TOF) ranging between 6 and 450 h−1 and selectivity values ranging between 0% and 90% have been reported for these systems (Table 1). Although a particle size effect has been shown for this reaction [8,14,19], the lower Ru dispersions usually lead to the higher COL selectivity. The results presented in Table 1 clearly point out a possible role of the metal support interaction on catalytic performances. Charge transfer between the support and the metallic phase has been analyzed as a possible lever to increase COL selectivity [6]. Thus, electron-rich active sites created on the catalyst surface normally allow to reach high selectivity toward COL. Such an increase in the surface electron density could not only enhance the repulsion between the active site and CC bond, but also favor the CO bond activation through favored back-bonding interactions between the catalyst and the π*CO of CAL orbitals, thus favoring the production of COL [20].
Catalytic systems based on carbon-supported ruthenium catalysts for CAL hydrogenation and their performances.
| Catalyst | Ru NP size (nm) | T (°C) | (bar) | TOF (h−1) | Conversion | COL selectivity | Ref. |
| Ru@MWCNT | 1–2 | 100 | 20 | 450 | 88 | 30 | [11] |
| Ru@MWCNT | 1.6 | 70 | 10 | 49 | 40 | 32 | [18] |
| Ru/MWCNT | 2–4 | 80 | 30 | 15 | 70 | 25 | [7] |
| Ru/MWCNT | 2–3.5 | 70 | 10 | 45 | 40 | 39 | [17] |
| Ru/MWCNT | 1.6–2.8 | 70 | 10 | 41–52 | 40 | 32–43 | [18] |
| Ru/MWCNT | 1.7 | 100 | 20 | 50 | 66 | 35 | [21] |
| Ru/AC | 1.8 | 100 | 20 | 19 | 22 | 51 | [21] |
| Ru/AC | 10 | 60 | 1 | – | – | 61 | [8] |
| Ru/AC | <1 | 60 | 40 | 180 | 25 | 5 | [6] |
| Ru/C60 | Not given | 60 | 40 | 6 | 7 | 60 | [12] |
| Ru/OMC | <2 | 100 | 30 | 432 | 92 | 54 | [10] |
| Ru/SWCNT | 3.5 | 110 | 45 | – | 80 | 92 | [4] |
| Ru/CNF | 1–2 | 110 | 45 | 108 | 60 | 43 | [22] |
| Ru/G | Not given | 110 | 45 | 31 | 50 | 76 | [14] |
| Ru/G | <1 | 60 | 40 | 324 | 50 | 53 | [6] |
| Ru/FLG | 2.3 | 70 | 10 | 120 | 40 | 30 | [17] |
Nevertheless, electron-deficient gold nanoparticles (NPs) have shown remarkably high activity and selectivity for this reaction [23]. Furthermore, it is known that Lewis acids or metallic promoters can be used in this reaction to increase COL selectivity [24]. In that case, the electropositive metals or oxidized metal species on the surface act as electrophilic or Lewis sites for the adsorption and activation of the CO bond via the lone electron pair of the oxygen atom. For example, in bimetallic PtRu NPs, it has been proposed that electropositive Ru atoms act in this way. This second mechanism, referred to as the “electrophilic CO activation”, was the most frequently invoked to account for the promoting effect of electropositive species [25,26].
We recently reported the synthesis and complete physicochemical characterization of ruthenium fullerides and ruthenium fullerides decorated with ruthenium NPs [27]. The aim of the present study was to evaluate the reactivity of these materials in the selective hydrogenation of CAL. We have investigated the role of several parameters such as the choice of the solvent, the addition of a base, and the Ru/C60 ratio on the course of the reaction.
2 Materials and methods
All operations were carried out under argon atmosphere using standard Schlenk techniques or in an MBraun glovebox. Solvents were purified by standard methods or with an MBraun SPS-800 solvent purification system. [Ru(COD)(COT)] (COD = 1,5-cyclooctadiene, COT = 1,3,5-cyclooctatriene) was purchased from Nanomeps Toulouse, fullerene C60 (99.5%), CAL (>99.0%), and nonane (>99%) from Sigma–Aldrich, and H2 from Air Liquide. All these reactants were used as received. The ruthenium content was measured by inductively coupled plasma optical emission spectroscopy on a Thermo Scientific ICAP 6300 instrument.
2.1 Synthesis of Ru@C60 nanostructures
In a typical experiment, the [Ru(COD)(COT)] complex was introduced in a Fisher–Porter bottle, and a solution of fullerene C60 in CH2Cl2 was then introduced in the bottle. The resulting purple solution was stirred for 30 min at room temperature, after which the bottle was pressurized with 3 bar of H2. The solution, which turned black after few minutes of reaction, was kept under stirring overnight at room temperature. After this period of time, excess of H2 was purged and the volume of the solvent was reduced under vacuum. Pentane was then added to the colloidal suspension to precipitate the Ru@C60 nanostructures. After filtration under argon with a cannula, the black powder was washed twice with pentane and filtrated again before drying under vacuum. For each ratio studied, the quantities of reactants are detailed hereafter:
- Ru@C60 1/1: 30.0 mg (0.10 mmol) of [Ru(COD)(COT)]; 68.5 mg (0.10 mmol) of fullerene C60 and 300 mL of CH2Cl2. Yield: 68.1 mg. Ru: 10.6%
- Ru@C60 10/1: 400 mg (1.27 mmol) of [Ru(COD)(COT)]; 91.3 mg (0.126 mmol) of fullerene C60 and 400 mL of CH2Cl2. Yield: 188 mg. Ru: 48.7%
- Ru@C60 30/1: 150 mg (0.48 mmol) of [Ru(COD)(COT)]; 11.4 mg (0.016 mmol) of fullerene C60 and 50 mL of CH2Cl2. Yield: 48 mg. Ru: 54.7%
2.2 General procedure for the hydrogenation of CAL
Hydrogenation reactions were performed in a Top Industry high-pressure stainless steel autoclave. In a typical experiment, the autoclave was purged by three vacuum/argon cycles. The mixture of Ru@C60 catalysts (5 mg), nonane (1.5 mmol, 200 mg, as internal standard), trans-CAL (4.0 mmol, 528 mg), and 30 mL of the desired solvent was prepared in a glovebox and then transferred into the high-pressure autoclave under argon atmosphere. In several experiments, a few equivalents of a base related to CAL were added to the reaction mixture. The hydrogenation experiments were carried out under 20 bar H2 at 70 °C with a stirring rate of 1000 rpm. The products were analyzed on a Perkin–Elmer gas chromatograph equipped with Elite-5MS capillary column (30 m × 0.32 mm × 0.25 μm) with a flame ionization detector. The response factors of each component were determined with standard samples and were used to calculate the conversion and selectivity.
2.3 Transmission electron microscopy analyses
Some transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HREM) analyses were performed at the “Centre de MicroCaracterisation Raimond Castaing, UMS 3623, Toulouse” by using a JEOL JEM 1011 CX-T electron microscope operating at 100 kV with a point resolution of 4.5 Å and a JEOL JEM 1400 electron microscope operating at 120 kV. The high-resolution analyses were conducted using a JEOL JEM 2100F equipped with a field emission gun operating at 200 kV with a point resolution of 2.3 Å and a JEOL JEM-ARM200F cold field emission gun operating at 200 kV with a point resolution of 1.9 Å. The approximation of the particle mean size was made through a manual analysis of enlarged micrographs by measuring at least 200 particles on a given grid.
2.4 Density functional theory calculations
To model the catalytic sites and to study the different adsorption modes of CAL on the Ru fullerides, density functional theory (DFT) calculations were carried out using the Vienna ab initio simulation package VASP [28–31], which uses the full-potential projector augmented wave framework [32,33]. Spin-polarized calculations were performed with approximating exchange-correlation effects using the Perdew, Burke, and Ernzerhof (PBE) exchange-correlation functional [34]. A kinetic-energy cutoff of 400 eV was found to be sufficient to achieve a total-energy convergence within several millielectron volts, considering a k-point sampling in Gamma-point only calculations for isolated molecules and complexes, in conjunction with a Gaussian smearing with a width of 0.05 eV. During geometry optimization runs, all the atoms were fully relaxed until forces on individual atoms were smaller than 0.01 eV/Å. Calculation cells for isolated molecules and complexes were (25 × 26 × 27) Å3 to avoid spurious interactions between periodic images. Figures of the different geometries were produced with the three-dimensional visualization program VESTA [35]. The calculations including MeOH solvent effects were performed with the implicit solvent model described in Mathew et al. [36] and compared with an explicit treatment of the interaction between the adsorbed CAL and three MeOH molecules. As in a previous study [37], we have used a C60Ru13C60 complex to investigate the most stable adsorption modes of CAL over the Ru NPs. In addition, the hydrogenation of the Ru NP surface was also considered with a coverage value of 1H per surface Ru atom.
3 Results and discussion
3.1 Catalyst synthesis and characterization
A series of Ru@C60 catalysts with a Ru/C60 molar ratio ranging between 1 and 30 were prepared by decomposing [Ru(COD)(COT)] under H2 (3 bar) in the presence of fullerene C60 at room temperature in CH2Cl2 [27]. The decomposition reaction of [Ru(COD)(COT)] in the presence of C60 results in the selective formation of spherical particles that can be surface decorated with metallic Ru NPs. The mean size diameters of Ru NPs and spheres of Ru@C60 according to the Ru/C60 ratio are given in Table 2.
Mean size diameters of Ru NPs and spheres of Ru@C60 according to the Ru/C60 ratio.
| Ru/C60a | %Rub (%) | Ru NP size (nm)c | Nanosphere size (nm)c |
| 1/1 | 10.6 | Not observed | 36.2 ± 1.2 |
| 10/1 | 48.7 | 1.26 ± 0.03 | 32.4 ± 0.3 |
| 30/1 | 54.7 | 1.34 ± 0.01 | 56.4 ± 4.7|103.2 ± 1.0 |
a Produced by decomposing [Ru(COD)(COT)] under H2 (3 bar) in the presence of C60 at room temperature in CH2Cl2.
b Determined by inductively coupled plasma optical emission.
c Manual measurement from enlarged micrographs of at least 200 objects.
For a Ru/C60 ratio of 1, a ruthenium fulleride is obtained, in which each Ru atom is coordinated to C60, with a η2(6)–η6 coordination mode [27]. HREM performed on this sample did not reveal the presence of any Ru NP (Fig. 1a). However, scanning transmission electron microscopy (STEM) images (Fig. 1b) allow visualizing extremely small objects, isolated atoms, and few-atom clusters. They are seen as light gray dots distributed uniformly through the fulleride spheres (Fig. 1b). This fulleride phase is the kinetic product of the reaction. Interestingly, single atom catalysis has been proposed as an effective strategy to maximize the product selectivity in hydrogenation reactions [1]. Then, for the Ru/C60 ratio >1, Ru NPs are produced (Fig. 1c and d), presumably from the atom/cluster germs on the surface of the fulleride spheres.

a) High-resolution electron microscopy micrograph of Ru@C60 1/1 (scale bar 5 nm); (b) STEM of Ru@C60 1/1 (scale bar 5 nm); (c) TEM of Ru@C60 10/1 (scale bar 50 nm); and (d) TEM of Ru@C60 30/1 (scale bar 50 nm).
The presence of Ru NPs only on the surface of the ruthenium fulleride has been confirmed by electron tomography [27]. Interestingly, the Ru mean NP size does not change with the Ru loading (Table 2), and 1.34 ± 0.01 nm Ru NPs can be produced by this method for a Ru loading as high as 54% w/w. X-ray powder diffraction (XRD) was not informative relative to Ru NPs' size, because of their very small size, as confirmed by Wide-angle X-ray scattering (WAXS) analyses. Thus, after corrections and Fourier transforms, the related pair-distribution functions (PDF) are very close and consistent with metallic Ru NPs with low structural disorder and sizes in the 1.5–2.5 nm range [27]. This is in agreement with the TEM measurements.
Fullerene are well-known electron acceptor materials [38], and significant charge transfer from ruthenium to fullerene has been evidenced by Raman spectrometry and X-ray photoelectron spectroscopy (XPS) for all the prepared materials. It has been shown that in Raman spectroscopy, the energy of the Ag(2) mode (1469.3 cm−1 for pure C60) is sensitive to charge transfer in transition metal fullerides [39]. We have observed here a spectral shift as large as −8.7 cm−1 for the Ru@C60 1/1 sample and −11.8 cm−1 for the Ru@C60 10/1 sample [27]. The charge transfer was also evidenced by XPS, by comparing the binding energy of Ru 3p3/2 in samples Ru@C60 1/1 and 20/1 with that of metallic ruthenium (461.2 eV): the measured binding energies were 462.2 and 461.5 eV for the Ru@C60 1/1 and 30/1 samples, respectively [27].
3.2 CAL hydrogenation
CAL hydrogenation was studied at 20 bar of H2 and 70 °C using different solvents and bases. The Ru@C60 10/1 catalyst was used to optimize the reaction conditions. In isopropanol and in the absence of any base the Ru@C60 catalyst formed very large amounts (≈90%) of acetals from the condensation reaction between CAL or HCAL and the solvent. CAL and HCAL acetals were formed as acid-catalyzed products. This could be explained by the fact that the Ru NPs are highly electron deficient, and thus an acidic surface should operate. We independently checked that the Ru@C60 catalyst was active for the condensation reaction of CAL and iPrOH under argon (78% conversion after 20 h at 70 °C). This reactivity is surprising, because a significant amount of acetals is usually obtained when using highly acidic supports such as zeolites [16,40–42]. A selectivity toward acetals of 80%, similar to the one obtained with the Ru@C60 catalyst, was obtained on a Ru/Y catalyst [42]. This result points out that thanks to the high acidity of the Ru NPs, the CO bond can be activated efficiently. However, such electron-deficient NPs should be less active for H2 dissociative chemisorption, so that condensation reaction predominates. At 20% conversion, 61% of COL was obtained with this catalyst without any base (selectivity calculated on the hydrogenated products only), which is a relatively high selectivity for a Ru catalyst (Tables 1 and 2), particularly if we consider the metal particle electronic density. To modulate the surface acidity and to prevent acetal formation, we investigated the addition of bases to this system. In general, the addition of a base in the reaction mixture significantly improves the selectivity toward COL [24]. Two types of bases are generally used, amines and alkali hydroxides. The mechanism of CO activation with addition of amines in hydrogenation or transfer hydrogenation is still uncertain [43]. The improved selectivity to COL can be attributed to a steric blocking of the sites that give rise to CC hydrogenation and/or to an electronic effect. One may assume that these bases interact with the metal surface via their lone electron pair, which may disfavor the hydrogenation of the CC bond, as discussed previously. A nonclassical metal–ligand bifunctional mechanism, where a hydride on Ru and a proton of the amine ligand are simultaneously transferred to the CO function via a six-membered pericyclic transition state, has been proposed in homogeneous catalysis [44]. A steric effect in long-chain amines prevents α,β-unsaturated aldehydes lying flat on the surface and, therefore, avoids direct contact of the CC bonds with the catalytic surface [45]. For the alkali hydroxides, the improvement of the selectivity toward CO bond hydrogenation was attributed to the polarization of the CO bond via an interaction between the metal cation that acts as a Lewis acidic site and the lone electron pair of oxygen atom in the CO group [24,46].
Several bases were tested as additives and the results of the catalysis are summarized in Table 3. The addition of 1.5 equiv of a strong base, such as KOH, suppresses completely the acetal formation and leads to a decrease in the selectivity toward COL, which drops from 60% to 40%. With trimethylamine, a significant increase in the TOF was measured accompanied by a decrease in selectivity to COL from 60% to 40%. The addition of 1.5 equiv of pyridine (related to CAL, 0.5 mL), a weaker base than trimethylamine, was beneficial in terms of selectivity. It allows increasing the selectivity toward COL up to 77% and suppressing completely the formation of acetals.
Effect of the base on CAL hydrogenation using Ru@C60 10/1.a
| Solvent | TOFb (h−1) | Selectivity %c | ||
| HCAL | HCOL | COL | ||
| No based | – | 14 | 25 | 61 |
| KOH | 11.6 | 56 | 1 | 43 |
| Et3N | 54.7 | 45 | 13 | 42 |
| Pyridine | 17.7 | 20 | 3 | 77 |
| Pyridine (excess)e | 23.8 | 27 | 2 | 71 |
a Reaction conditions: 4.0 mmol CAL (528 mg), 1.5 mmol of nonane (200 mg), isopropanol 30 mL, 70 °C, 1.5 equiv base, 20 bar H2, 1000 rpm.
b TOF was calculated at 2 h of reaction.
c At 20% conversion of CAL.
d Acetals are the main products of the reaction, and the given selectivity do not take them into account.
e 3 mL (9 equiv).
The addition of an excess of pyridine does not allow further improvement in the selectivity but increases the activity. Although we will see later on that the nature of the solvent has a pronounced effect on both activity and selectivity, these results can be tentatively rationalized by a modulation of the electronic density on the ruthenium surface by coordination of the amine ligands. First, the σ-donor amine ligand coordination reduces the electron deficiency, so that acetal formation is completely suppressed. Second, according to the basicity of the amine, an effect is noticed both on catalyst activity and selectivity. For the stronger bases, the increase in the electron density on the Ru NPs should improve the H2 dissociation rate and thus the hydrogenation rate, as observed for N(Et)3. In this case, however, the Ru is not acidic enough to activate the CO bond and selectivity decreases. The use of a weaker base such as pyridine enables a better balance between activity and selectivity toward COL.
The influence of the Ru/C60 ratio was also studied (Table 4); the amount of catalyst was adjusted to maintain a constant Ru concentration. On small clusters or atoms (Ru/C60 1/1), the TOF is slightly lower than that on the 10/1 sample, and the selectivity to COL decreases from 77% to 65%. These results constitute the first experimental evidence of a size effect in this reaction at the subnanometer scale. DFT calculations on the catalytic performances of Ptn clusters (n = 6, 10, 14, 18) for CAL hydrogenation have shown a relationship with the size of the cluster and the catalytic performances, with an optimum in activity and COL selectivity for n = 14 [47]. Increasing the Ru/C60 ratio to 30/1 has a positive effect on the TOF, which increases from 17.7 h−1 for Ru/C60 ratio 10/1 to 32 h−1 for Ru/C60 ratio 30/1, and a negative effect on selectivity that decreases from 77% to 65%. In that case, the reason cannot be the particle size because these catalysts display very similar Ru NP sizes (see Table 2). These two catalysts present a peculiar structure, in which the Ru NPs are present in a shell surrounding the ruthenium fulleride core. One difference between these two catalysts is the proportion of Ru presents as NPs compared with Ru in the fulleride (atoms and clusters). The sample 30/1 presents a higher proportion of Ru NPs and thus allows a higher activity. The thickness of the shell of Ru NPs increases when passing from sample 10/1 to 30/1. This thickness has been measured by three-dimensional TEM to be 7 nm for the sample 30/1 [27]. It is possible that the proximity between the Ru NPs in sample 30/1 but also the concentration of fullerene around these NPs that might be different (lower concentration of C60 for the 30/1 sample) have a negative effect on the selectivity.
Effect of the Ru/C60 ratio on CAL hydrogenation using Ru@C60.a
| Ratio | TOFb (h−1) | Selectivity %c | ||
| HCAL | HCOL | COL | ||
| 1/1 | 12.6 | 23 | 12 | 65 |
| 10/1 | 17.7 | 20 | 3 | 77 |
| 30/1 | 32 | 33 | 2 | 65 |
a Reaction conditions: 4.0 mmol CAL (528 mg), 1.5 mmol nonane (200 mg), 30 mL of isopropanol, pyridine (4.5 equiv), 70 °C, 20 bar H2, 1000 rpm.
b TOF was calculated at 2 h of reaction.
c At 20% conversion of CAL.
It has been shown that the nature of the solvent used has a significant effect on the rate and selectivity of catalytic hydrogenation reactions [48]. For CAL hydrogenation, only fragmentary information is available on the influence of the nature of the solvent [16,42,49–56]. For ruthenium catalysts, the activity and selectivity were influenced by the selected solvent in a different way according to the support. For zeolite supports, a pronounced solvent effect was reported both on the activity and the selectivity [16,42]. In that case, the catalysts were the most active in alcohols and relatively inactive in apolar solvents. The highest selectivity toward COL was obtained in apolar solvents. For carbon supports, the activity increases in polar and protic solvents, such as isopropanol, and the selectivity toward HCAL is higher in aprotic solvents such as hexene [42,54]. Generally speaking, solvents have different roles in addition to the usual ones (heat management, solubilization). They can (1) affect hydrogen solubility, (2) compete with the reactants for adsorption on the metal surface, (3) catalyze side reactions, (4) provoke catalyst agglomeration, and (5) interact with the reactant [57]. Concerning the latter effect, favorable thermodynamic interactions between the solvent and the reactant are expected to reduce the adsorption of the reactant on the catalyst, whereas unfavorable interaction should aid this adsorption. The most important solvent effects in the hydrogenation of α,β-unsaturated aldehydes are (1) solvent polarity, (2) hydrogen solubility, (3) interactions between the catalyst and the solvent, and (4) solvation of reactants in the bulk liquid phase [2].
Methanol, isopropanol, acetone, and dioxane were compared in the presence of pyridine. Hydrogen solubility in these solvents is expected to follow the order: acetone > iPrOH > dioxane ≈ MeOH [58]. The relative permittivity (ɛr) follows the order MeOH (ɛr = 32.7) > acetone (ɛr = 20.7) > iPrOH (ɛr = 17.9) > dioxane (ɛr = 2.25). A significant solvent effect was noticed both on catalyst activity and selectivity. In an apolar solvent such as dioxane a modest TOF is obtained (14.7 h−1), which cannot be related only to the hydrogen solubility if we consider the results obtained in methanol (128.4 h−1 for a similar H2 solubility). In addition, in this apolar solvent a drastic change in selectivity is observed because HCAL is obtained with 73% of selectivity, pointing to a role of the solvent on the activation of the CO bond of the aldehyde. In this solvent, where acetals cannot be formed, the removal of pyridine induces a decrease in the TOF and a low COL selectivity (<20%). In a polar and aprotic solvent such as acetone, the TOF increases (21.1 h−1). This result can be correlated to the H2 solubility. The selectivity shifts toward the preferential formation of COL (S = 67%). In a polar and protic solvent such as isopropanol, the TOF is lower than in acetone, which is in phase with the lower solubility of H2 in this alcohol. The selectivity toward COL is further increased to 77%.
Using methanol as a solvent instead of isopropanol does not affect the selectivity toward COL, which remains high (76%). However, in the case of this alcohol, a pronounced enhancement of the activity is observed, which cannot be related to hydrogen solubility. Such an influence of the nature of the alcoholic solvent on the activity of catalysts for CAL hydrogenation (MeOH > EtOH > iPrOH) has already been reported in the case of Pt/FLG [56] and Pt/SiO2 [53] catalysts. For these systems, however, significant amounts of acetals were produced in methanol, which is not the case in our work because of the presence of pyridine. Considering the TOF obtained in acetone, isopropanol, and methanol, we can discard a possible contribution of catalytic transfer hydrogenation to explain the differences in reactivity. Considering the permittivity values, we can conclude that the higher TOF are obtained in the solvent of higher permittivity. Because methanol should be prompter to form hydrogen bonds than isopropanol, owing to the higher acidity of its proton, the possible implication of the solvent to lower an activation barrier through hydrogen bonds can be proposed. As far as selectivity is concerned, we have performed DFT calculations on the adsorption of CAL on a complex model of Ru fulleride, consisting of 2 C60 and a bare or hydrogenated Ru13 cluster, as depicted in Fig. 2.
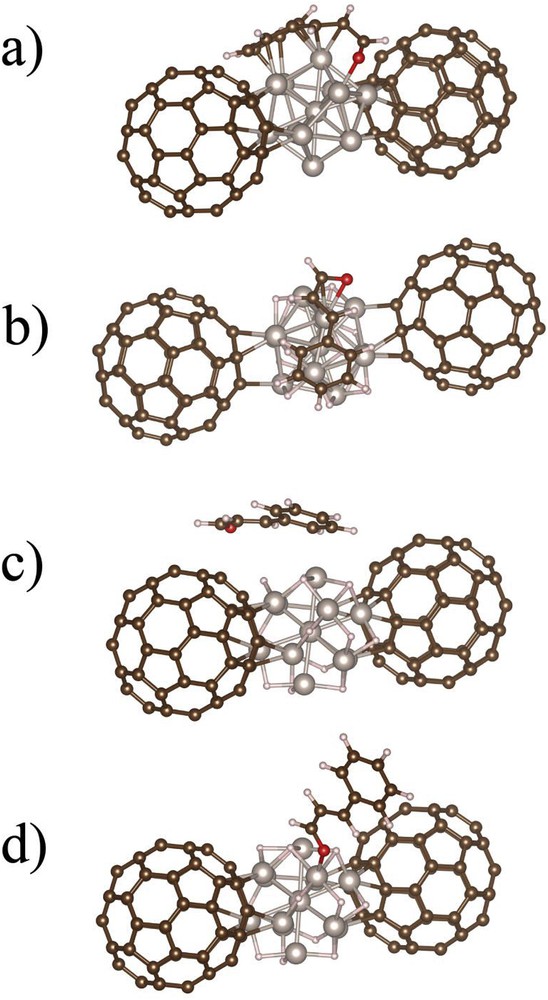
a) Side view of the most stable adsorption mode of CAL on naked Ru fulleride model (CC-mode) in vacuum. (b) Side view of the most stable adsorption mode of CAL on the hydrogenated Ru fulleride model (CC-mode) in vacuum. (c) Side view of the cycle-mode of CAL on the hydrogenated Ru fulleride model in vacuum. (d) Side view of the O-mode of CAL on the hydrogenated Ru fulleride model in vacuum.
In Table 6, we have summarized the different adsorption energies obtained after geometric optimizations with three different starting geometries: (1) cycle-mode when CAL is presenting its aromatic cycle to the apex of the Ru13 cluster, (2) CC-mode when the CC double bond is concerned, or (3) the O-mode when the oxygen points to one of the Ru apexes. In vacuum, for bare Ru fullerides, the most stable geometry is yielded when the CC is presented but a strong distortion of CAL is also observed, with a strong interaction of the CAL cycle with a facet of the Ru NP and an ORu interaction (Fig. 2a). Having this picture in mind, one could assume that HCAL, COL, and HCOL are equivalent products, but this is clearly in contradiction with the results of the CAL hydrogenation in dioxane as shown in Table 5. As soon as hydrides are present on the Ru NP surface, the most stable state is still the CC mode (Fig. 2b), but now with a slightly longer RuO bond (2.18 Å) than in the O-mode (2.08 Å) and more importantly no interaction between the aromatic cycle and the Ru cluster. This situation should favor CC bond hydrogenation. In addition, the energy difference between the CC and the O-mode of CAL decreases.
DFT adsorption energies for the three most stable anchorage modes in vacuum (in kcal/mol).
| Solvent | Naked NP-Ru | Hydrogenated NP-Ru | |||
| Vacuum | MeOH (implicit) | Vacuum | MeOH (implicit) | MeOH (explicit) | |
| Cycle-mode | −76 | – | −16 | – | – |
| CC-mode | −94 | −87 | −40 | −29 | −45 |
| O-mode | −28 | −23 | −27 | −17 | −19 |
Effect of the solvent on CAL hydrogenation using Ru@C60 10/1.a
| Solvent | TOFb (h−1) | Selectivity %c | ||
| HCAL | HCOL | COL | ||
| Dioxane | 14.7 | 73 | 6 | 21 |
| Acetone | 21.1 | 22 | 11 | 67 |
| Isopropanol | 17.8 | 20 | 3 | 77 |
| MeOH | 128.4 | 17.2 | 6.9 | 76d |
a Reaction conditions: 4.0 mmol CAL (528 mg), 1.5 mmol nonane (200 mg), 30 mL of solvent, pyridine (0.5 mL), 70 °C, 20 bar H2, 1000 rpm.
b TOF was calculated at 2 h of reaction.
c At 20% conversion of CAL.
d At 77% conversion of CAL.
Globally, the MeOH solvent effect, with an implicit solvent model that considers only a change in the dielectric constant in the surrounding of the molecular complex, is rather weak both on the geometries and on the adsorption energies. Indeed, we have observed a slight reduction in the absolute values, on the order of 10 kcal/mol for all the tested geometries, when the systems were placed in MeOH.
This appears to be not sufficient to make the O-mode more stable when compared to the CC mode as one can expect from the high selectivity toward COL obtained in MeOH (Table 5). Moreover, adding three molecules of MeOH in the vicinity of the CAL molecule to allow for extra H-bonds that may stabilize more the O-mode over the CC mode is still not sufficient because the energy difference remains in favor of the CC mode by more than 15 kcal/mol.
4 Conclusions
We have prepared original catalysts consisting of ruthenium NPs deposited on ruthenium fulleride nanospheres by a simple one-pot procedure. These catalysts are characterized by a high metal loading, ranging between 10% and 50% w/w, a small ruthenium NP size (<1.5 nm), and a significant charge transfer from the metal to the fullerene. These electron-deficient ruthenium NPs do not allow the hydrogenation of CAL but catalyze the condensation of CAL with an alcoholic solvent to produce acetals. Such reactivity is commonly observed when using highly acidic supports such as zeolites. To prevent acetal formation and promote hydrogenation it is necessary to modulate the catalyst acidity by adding a soft base such as pyridine. In that case, and when using methanol as a solvent, these catalysts performed very well for the selective hydrogenation of CAL to COL, and the value obtained (TOF = 128 h−1, SCOL = 77%) are among the best one ever reported for a Ru/carbon catalytic system (see Table 1). The use of an aprotic and apolar solvent such as dioxane induces a remarkable selectivity shift toward HCAL formation and a significant decrease in the TOF. Preliminary DFT calculations suggest that this shift is not correlated to a specific precoordination of CAL on the ruthenium particles. The question of treating solvent dynamical effects may be addressed to solve this issue, but this is definitively beyond the scope of the present study.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique (CNRS), which we gratefully acknowledge. The authors acknowledge financial support from the program of China Scholarships Council (CSC) to F.L. I.C.G. acknowledges the Calcul en Midi-Pyrénées initiative-CALMIP (Project p0812) and GENCI-CINES and GENCI-IDRIS (x2016096649) for allocations of computer time.