

1 Introduction
Nanometer-scale metallic particles (NSMPs) [1] display a wide variety of very exciting properties. Among them, the study of their chemical reactivity constitutes an interesting and fertile research field [2], their particular structural and electronic characteristics standing between the continuous energy bands in solids and the discrete energy levels in the isolated atoms [3]. In fact, many challenges arise in the adsorption of simple diatomic molecules like nitrogen monoxide (NO) or carbon monoxide (CO) on such NSMPs, which are intimately linked to major environmental and/or industrial aims, namely, the diminution of nitrogen oxide emissions from diesel car exhaust gases [4] or the Fischer–Tropsch process [5].
From an experimental point of view, we have already underlined the fact that through synchrotron radiation related techniques, the required abilities to characterize such NSMPs are present while the chemical reaction occurs [6–8]. From a theoretical point of view, we have also the possibility to describe, in a realistic way, the electronic structure of such entities to predict both their atomic and chemical rearrangements [9,10] and also to simulate (understand/predict) their activity/selectivity.
CO adsorption and activation on metal atoms, clusters, being supported or not, have been studied in the past. If one looks in the literature for works corresponding to the keywords CO, Metal, Cluster, DFT, almost 1000 articles are found (from Web of Science). Our aim is not to provide an exhaustive overview of this research topic, but a few relevant articles are summarized here, especially those during the “nanogold rush” initiated in the 1990s, that stimulated theoretical studies on metal clusters' reactivity. Since then we studied this topic, and in particular the reactivity of atomic, and gold nanostructures [11–19]. In this article, we make a concise overview of selected studies on this topic. We start by following an approach defined and discussed by Boudart in his article “Model catalysts: reductionism for understanding” [20]. Subsequently, CO molecules adsorbed on monometallic clusters are discussed. Then the different topics, namely, CO on atoms in the gas phase, on metal clusters in the gas phase, and supported on oxides, and also some examples on the effect of alloying are mentioned. Moreover, density functional theory (DFT) investigations on the adsorption and activation of small molecules such as CO are considered.
2 Adsorption of diatomic molecules on metallic surfaces
When the CO molecule adsorbs on a three-dimensional (3D) metal surface, it is well known that the electron population of CO increases steadily from Ni to Ti. The CO bond is considerably weakened, and its overlap population decreases to 0.43e on Ti(0001) from 1.02e on Ni(100) as compared with 1.21e in the free CO molecule. Consequently, a greater ease of CO cleavage is observed as one moves to the left in the transition metals [21].
When a diatomic molecule such as NO approaches a metal surface [3], it may encounter three kinds of potential wells corresponding, respectively, to a physisorbed state (far from the surface), a molecular chemisorbed state, and finally to a dissociative chemisorption state. As discussed by Desjonqueres and Spanjaard [3], dissociative chemisorption is the most stable situation if
| (1) |
On the basis of the different experimental results [24–29], it seems that one can relate quite simply to the adsorption mode (dissociative or molecular) and the behavior of the nanometer-scale monometallic clusters (sintering or dissociation) after this adsorption [30–32]. From the reviews of Broden et al. [22] and Brown and King [23], a model has already been proposed for the adsorption of NO molecules on NSMPs. It starts with a plot of the variation in the melting point versus the atomic number (Fig. 1). In this figure, a straight line separates two adsorption modes for NO: the molecular one and the dissociative one. Now, assuming this tendency, NSMPs that associated with molecular adsorption of the NO molecule have a tendency to grow. On the contrary, a fragmentation of the cluster is suspected for metals that are associated with the dissociation of the molecule.
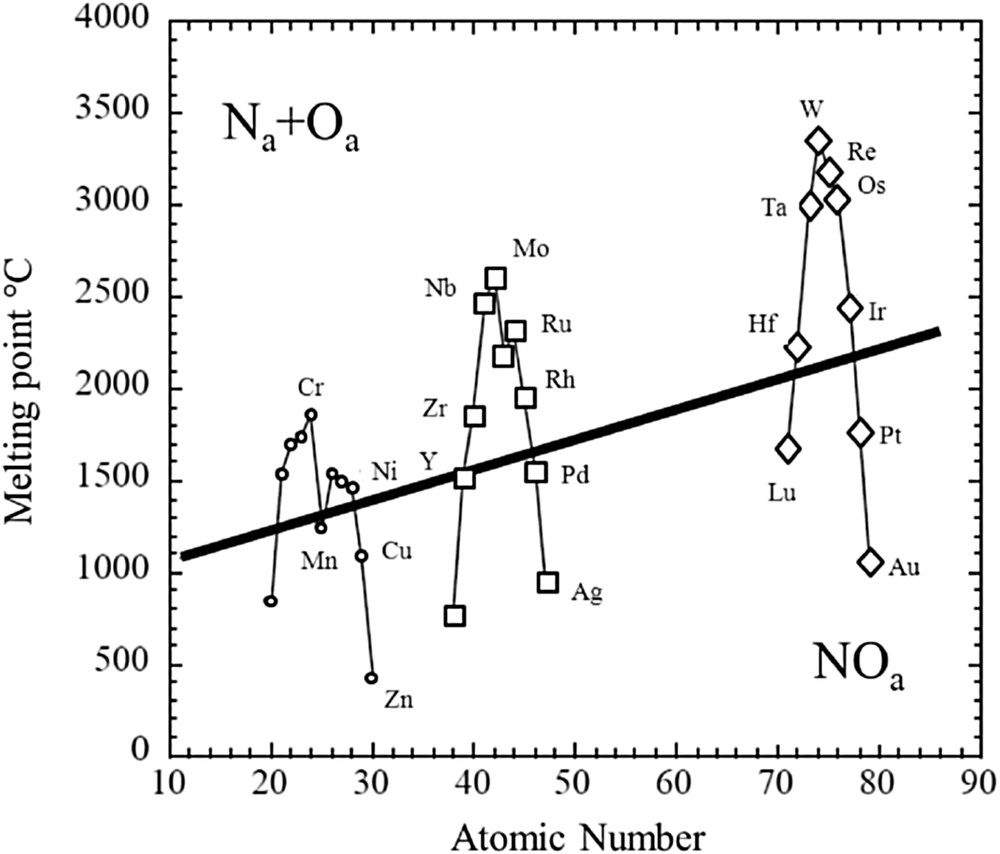
Correlation between the propensity for a metal to dissociate NO and its melting point (data taken from refs [22,23]).
This simple model is supported by a set of experimental data [24–29]. More precisely, Hashimoto et al. [33] have shown that NO adsorption on Ru clusters induces a significant structural change. Moreover, a similar result has been obtained by Schmidt and Krause [34] for Rh clusters. On the contrary, in Pt clusters, particle coalescence is observed under NO. Note that we are discussing here about NSMPs deposited on various supports.
Regarding DFT calculations [35,36], Endou et al. have considered the systems composed of a NO molecule interacting with tetrahedral precious metal clusters (M4, M = Rh, Pd, Ag, Ir, Pt, and Au). They noticed a substantial elongation of the NO bond as was found on the NO/Ir4 system nearly equal to that on the NO/Rh4 systems, which is reasonably in line with this simple model [37].
3 Role of the support and the cluster size on the CO adsorption process
In this section, the effect of the support through different experimental studies is emphasized, whereas the effect of particle size is pointed out through different theoretical works.
3.1 Role of the cluster size
Different theoretical studies have already been performed on the variation in the cohesive energy versus the size and the morphology of NSMPs [37–39]. In Fig. 2, such variations have been plotted for different metals. A significant decrease in the cohesive energy (around 30% between the bulk and a 13-atom cluster) is observed independent of the nature of the metal and the morphology of the cluster. In a first approximation, one can consider that for NSMPs the cohesive energy (or the melting temperature) is simply shifted as compared to that associated with the bulk. Also, the fact that the CO adsorption process occurs on metal atoms having a low coordination number is considered. Basically, this means that a shift (toward the bottom in Fig. 2) in the straight line has to be considered [40].
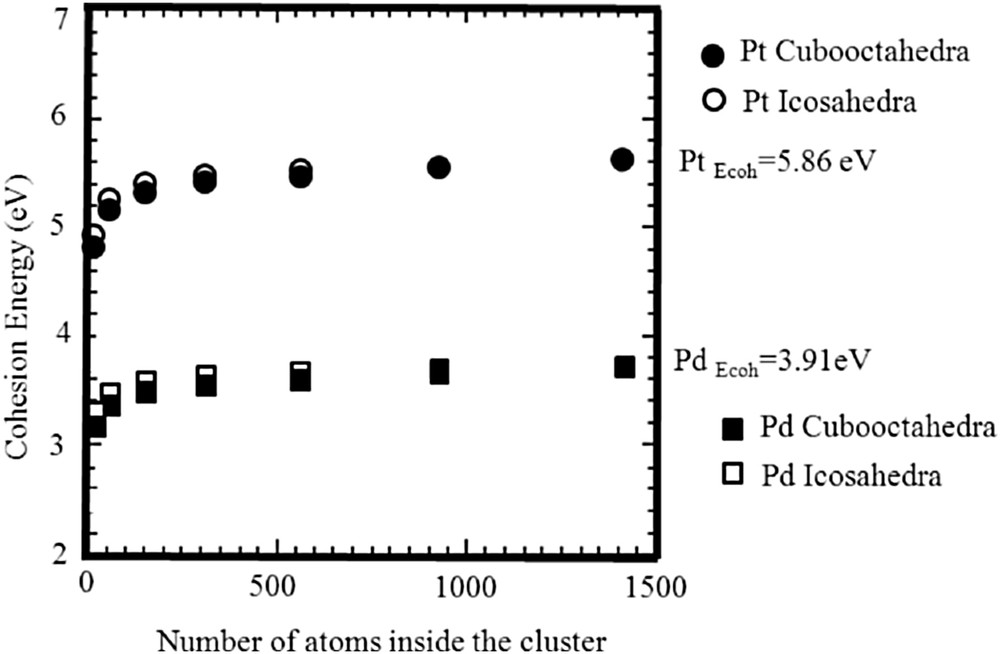
Variation in the cohesive energy versus the number of atoms inside the metallic cluster [39].
3.2 Role of the support
NO and CO behave differently as far as the support is concerned. This might be related to their bond-dissociation energy, that is, ΔEdis(CO) = 11.16 eV [41], ΔEdis(NO) = 6.50 eV3, and other reaction paths with different activation energies.
Regarding the CO adsorption performed on Pt-supported clusters, Benvenutti et al. [42] have considered different oxide supports namely TiO2, ZrO2, and Al2O3. Through a Fourier Transform Infrared (FTIR) study, they have observed a strong metal–support interaction. In fact, similar conclusions have been made by Rice et al. [43] and Tong et al. [44]. In Rh deposited on reduced cerium oxide thin films, the degree of dissociation of CO on Rh deposited on CeO2 is directly dependent on the degree of oxidation of the CeO2 [45]. As pointed out by Johanek et al. [46], the metal–substrate interaction plays an important role in the case of Pd too. Finally, among the different parameters, which play a key role, the effect of the substrate orientation has been emphasized by Nehasil et al. [47]. Note that, for the Au/TiO2 system, Grunwaldt and Baiker [48] have noticed that adsorption of CO on gold was weak, reversible, and not significantly influenced by the presence of TiO2. Now, we would like to consider different works that point out the influence of the adsorption of CO on the structural and/or electronic characteristics of the metallic cluster.
4 CO-induced structural changes in NSMPs
4.1 Copper
The adsorption of CO on Cu films grown on the oxygen-terminated ZnO (0001)O surface has been studied using temperature-programmed desorption and X-ray photoelectron spectroscopy by Ludviksson et al. [49]. Their results show that CO induces a dispersion of 3D Cu clusters into 2D islands.
4.2 Rhodium
Extended X-ray absorption fine spectroscopy and FTIR studies have revealed a reconstruction of reduced Rh particles supported on Al2O3 or TiO2 after admission of CO [50] resulting in monorhodium-carbonyl clusters. In addition, a significant size effect has been pointed out. Rh particles with an average first shell coordination number equal to 7 did not reconstruct under CO atmosphere, whereas very small metal particles (N = 4) formed Rh germinal carbonyl complexes [51].
In addition, observations by M. Baumer et al. [52] suggest that the aggregates are liable to structural rearrangements when exposed to CO. More precisely, if this effect is temperature dependent (measurements have been collected at 390 and 300 K), the experimental data can be explained by a spreading of Rh over the surface resulting in larger island diameters. Moreover, Nehasil et al. [53] have pointed out an effect of the particle size on the value of Ea, the activation energy of desorption. More precisely, they noticed that Ea decreases with decreasing particle size. Regarding the effect of temperature, Berko et al. [54] have noticed that the adsorption of CO at 300 K induces the disruption of Rh crystallites as indicated by a drastic decrease in the size of Rh particles. On the contrary, at higher temperature, it leads to the reforming of the Rh cluster.
4.3 Palladium
Different studies have been devoted to the adsorption of CO on Pd particles supported on different light oxides such a-Al2O3 [55,56], MgO [57], SiO2 [58–60], and TiO2 [61]. Sheu et al. [58] have noticed that the fraction of CO in the bridging mode increases and that of the linear mode decreases with increasing size of the Pd particles. Also, one of the striking points in the study of Goyhenex et al. [57] is the possibility of spillover of CO between the particles and physisorption of CO on the support.
Voogt et al. [62] have investigated by ellipsometry the CO adsorption on SiO2/Si(100)-supported palladium particles. They have concluded that, for this system, there are no particle size effects with the adsorption of CO. Because other studies have pointed out such dependence, they have concluded that the particle size effects are dependent on the support or that the observed particle size effect is actually a support effect.
4.4 Iridium
As pointed out by Berko et al. [54], the supported iridium nanoparticles of 1–3 nm size exhibit a very high reactivity toward CO. As a result of the CO adsorption at 300 K, crystallites disrupt into smaller particles and finally into atomically dispersed Ir.
4.5 Platinum
Mojet et al. [63] have shown that at room temperature the platinum particles supported on SiO2 are stable under CO atmosphere. On the contrary, the smaller platinum particles in zeolite LTL reconstruct with the formation of very small PtCO aggregates upon admission of CO at room temperature. Gan et al. [64] have noticed that on Pt-covered surfaces (TiO2) the chemistry of CO adsorption and desorption strongly depends on the size of Pt nanoclusters. When decreasing the cluster size, CO is found to desorb at higher temperatures. Maniguet et al. [65] have performed an extended X-ray absorption fine spectroscopic study of carbon monoxide oxidation on supported Pt fuel cell electrocatalysts. They have noticed that the adsorption of CO does not result in a change in the particle structure as indicated by the structural parameter associated with the first PtPt shell.
4.6 Gold
For this metal, Wallace and Whetten [66] have measured the carbon monoxide adsorption activity of small gold-cluster anions (AuN−, N = 4–19) by pulsed helium flow-reactor methods at room temperature. The reaction product distribution reveals a highly size-dependent adsorption activity and saturation.
5 Associated model
Following the approach already performed for nitrogen monoxide, we used here the early work of Anderson [67] to propose a similar model for carbon monoxide (Fig. 3). Thus, we can consider three families of metals with respect to their position versus the straight line corresponding to the CO adsorption at room temperature. This straight line separates two adsorption modes for CO: the molecular one and the dissociative one. The first family is composed of metals above the curve, the second one close to this line, and the third below the line. Now, for each family, we would like to consider the influence of the cluster size.
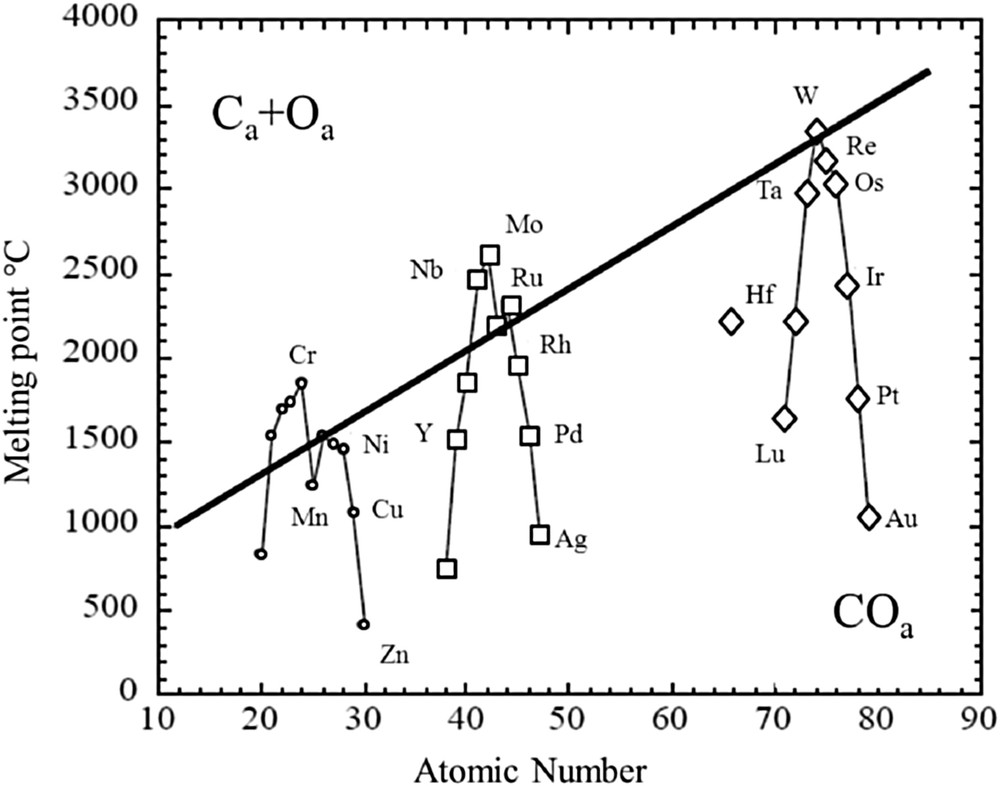
Correlation between the propensity for a metal to dissociate CO and its atomic number. Reprinted from ref [68].
5.1 The first family of transition metals
For this family of transition metals, the adsorption of CO leads to a dissociation of the metallic cluster. Now, when the size of the cluster decreases, the cohesive energy also decreases, and thus there is a possibility to observe for very small clusters, namely, for those which contain a few number of atoms, a sintering process.
5.2 The second family of transition metals
For the second family of transition metals, that is, those close to the line (Fig. 3), a significant size effect has to be considered. Rhodium belongs to this family and clearly numerous works already published [51–54] underline such dependence. Moreover, because these transition metal elements are close to the line, different parameters such as the morphology of the metallic cluster, the presence of a second metal, or another element like chlorine, which can originate from catalyst preparation procedures [69], may change the behavior of the metallic cluster drastically.
5.3 The third family of transition metals
Regarding this last family in which we find copper, palladium, platinum, and iridium, the size of the cluster cannot be considered as a significant parameter because all are below the curve whatever the diameter of the particle. For copper [48] and palladium [61], the behavior of these metals versus the CO adsorption is in line with this trend. In fact, Voogt et al. [62] show clearly for this metal the observed particle size effect is a support effect.
On the contrary, for iridium and platinum, a size effect has been observed. For iridium [54], small crystallites disrupt into smaller particles and finally into atomically dispersed Ir. Nevertheless, for large particles, the authors have observed at least a corrosion process. Also, for platinum, the size dependence related to the CO adsorption has been pointed out on different supports, that is, silica and zeolite [63]. Moreover, the role of zeolite in the CO adsorption process is not clear [66]. Regarding the work performed on TiO2, a support effect is clearly suspected. Gan et al. [64] have observed CO dissociation on the Pt-supported surface, possibly because of the reduction by Ti3+.
6 Mechanism of the sintering process
When we consider a nondissociative CO adsorption, metallic-carbonyl M-(CO)x clusters are formed. A possible explanation of our model is linked to the fact that such entities being mobile on the support and unstable at high temperature lead to a sintering process of metallic particles. To sum up, the final structural characteristics of the metallic part of the sample when CO adsorption is considered are those associated with “bulk” particles.
In the literature, different works regarding different metals are in accordance with this simple model. Regarding rhodium, Gaussmann and Kruse [70] have already pointed out such mechanism using (atom-probe) pulsed field desorption mass spectrometry. Also, for rhodium, Baumer et al. [52] have proceeded to an exposition of the sample to CO during deposition, which leads to the formation of a carbonyl-like species. In the case of platinum, the formation of very small PtCO aggregates upon CO admission at room temperature is observed [63]. Evaporation of Pd in the presence of a CO atmosphere leads to the formation of a palladium carbonyl-like compound, which is stable only at low temperature. Finally, such entities have been observed also in the case of iridium [71].
7 Theoretical studies on the interaction of metals with small diatomic molecules
CO adsorption, considered as the first step in the CO oxidation reaction, on the metal surface has been studied theoretically since 1980s [68] and 1990s [72]. For metal systems in the gas phase, Gajdos et al. [73] assessed the state-of-the-art DFT Perdew–Burke–Ernzerhof (PBE) and revised PBE functionals on the trends in CO adsorption on close-packed metal surfaces: Co, Ni, Cu from the 3d row, Ru, Rh, Pd, Ag from the 4d row, and Ir, Pt, Au from the 5d row. The trends in adsorption energy, geometry, vibrational properties, and other parameters derived from the electronic structure of the substrate were cautiously evaluated. It was shown that although the geometrical and vibrational properties of the adsorbate–substrate complex are calculated with high accuracy, the adsorption energies calculated with the gradient-corrected Perdew–Wang exchange-correlation energies are overestimated. In addition, the calculations tend to favor adsorption sites with higher coordination, resulting in the prediction of the wrong adsorption sites for the Rh, Pt, and Cu surfaces (hollow instead of top). The revised PBE functional leads to lower (i.e., more realistic) adsorption energies for transition metals, but to the wrong results for noble metals—for Ag and Au, endothermic adsorption was predicted. The site preference remained the same [73]. This study was refined in 2008 [74].
Concerning the noble metals and gold, in particular [75], Lopez et al. showed that the catalytic properties of supported nanosized gold particles were related to the availability of many low-coordinated gold atoms on the small particles. Au1Au13 gold clusters were investigated by of DFT calculations to study the interaction with CO. The overlap between CO orbitals and cluster frontier orbitals was shown to be important. As a result, the authors found no simple relationship between CO chemisorption energy and the frontier orbital energies of Aun clusters [76]. Nevertheless, DFT calculations showed how gold nanoparticles are more active catalysts for CO oxidation than other metal nanoparticles. The high catalytic activity of nanosized gold clusters at low temperature was related to the ability of low-coordinate metal atoms to activate reactants [77].
Hybrid density functional calculations have been carried out for AuO2, AuCO, Au13, Au13O2, Au13CO, Au13H2, and Au55 clusters to determine the catalytic behavior of Au clusters with different sizes and structures for CO oxidation [78]. From these calculations, it was found that O2 and CO could adsorb onto several Au model systems. Especially, icosahedral Au13 cluster has a relatively weak interaction with O2, whereas both icosahedral and cubooctahedral Au13 clusters have interactions (∼20 kcal/mol) with CO. These findings suggest that the surfaces of the Au clusters are the active sites for the catalytic reactions on the supported and unsupported Au catalysts.
Besides Au, paladium was also investigated. DFT calculations were performed to investigate CO adsorption on neutral, cationic, and anionic Pdn (n = 1–7) clusters [79]. From the results, it was observed that the binding of CO molecule to neutral and cationic palladium clusters takes place via onefold, twofold, and threefold coordination. Then again, only terminal adsorption of CO molecule is possible in anionic clusters excluding bridging adsorption in Pd7 cluster. On Pd, Pd2, and Pd4 small clusters, the theoretical results obtained were compared with experimental vibration frequencies of CO adsorbed on supported Pd nanoparticles [80]. The results indicated that the CO adsorption over Pd2 and Pd4 clusters gives CO vibration frequency values close to the experimental values. Also, the theoretical results for the threefold hollow sites were compared with other nanoclusters, with n > 50 atoms, where the Pd4 cluster also gives values close to the experimental values. The size dependence of adsorption properties of metal nanoparticles for CO as a probe on Pdn clusters was studied and was further investigated for large particles (n = 13–116 atoms) [81], the values slowly decrease with cluster size from the asymptotic value for an (ideal) infinite surface. For clusters of 13–25 atoms, starting well above the asymptotic value, the adsorption energies drop quite steeply with increasing cluster size.
It should be noted that the presence of subsurface C in Pd has been reported [82,83] with a barrier of 0.74 eV from a surface position to a subsurface one [84]. Some interesting questions arise with this property. In particular, the dissociative adsorption is a source of atomic C that could migrate irreversibly to the subsurface and could be proposed in novel metallic particle synthesis.
On Pd38 nanoparticulate, CO adsorption [85] for different coverages and its effect together with alloying on atom ordering in the nanoparticles [86–88] have been studied.
It is also important to underline that the coverage of the adsorbate (e.g., CO) on the substrate (e.g., metallic nanoparticles) is important in the activation process (see e.g., references cited above). For instance, at low coverage CO can be dissociated on one metal, but at high coverage it will not be the case.
Other metals such as Pt and Cu have also been investigated, especially the particular cluster containing 13 atoms [89]. At the DFT level, the adsorption properties of CO, NO, and OH were investigated on the Cu13, Pt7Cu6, and Pt13 clusters in the cationic, neutral, and anionic states [90]. The adsorption energies of CO and NO are found to be substantially stronger on Pt13 than on Cu13. Hence, CO and NO bind preferentially on Pt-sites on Pt7Cu6, and as a consequence, it has the potential to increase Pt composition in the surface of large PtCu nanoclusters. The CO and NO adsorption energies on the binary cluster are enhanced by few percent as compared with the unary clusters, which shows that the PtCu interaction can contribute to increase the adsorption energy even for the cases in which the charge transfer among the chemical species is negligible.
Subsequently, investigations were performed on binary metal systems as seen in the later study. DFT calculations were performed to study Pt/Au clusters of different size, structure, and composition and their interactions with a CO molecule [91]. Among the Pt/Au isomers studied, the planar structure is the most stable structure in many Pt compositions, although 3D structures become more stable with increasing Pt composition. Furthermore, structures with the Pt atoms surrounded by Au atoms are more stable among homotops. However, these conclusions will be altered if ligands are attached to the Pt/Au bimetallic clusters, as evidenced from the results of CO adsorption. When both Au and Pt sites are exposed, CO adsorption at the Pt site is stronger. If only a Au site is available for CO adsorption, the strongest adsorption occurs at ∼25% Pt composition, which may correlate with the experimentally observed reactivity of the core−shell structured Pt/Au nanoparticles. As far as the reaction is concerned, it was shown that the mechanism CO oxidation catalyzed by several PtmAun (m + n = 4) clusters [92] preferred in all situations to proceed via the single-center pathway to the two-center pathway according to the two-step mechanism. In PtAu bimetallic clusters, Pt sites are the catalytically active centers, whereas Au sites are “formally spectators” for CO oxidation. The calculated barriers for the reactions mediated by bimetallic clusters Pt3Au, Pt2Au2, and PtAu3 are comparable with that catalyzed by monometallic Pt4 cluster, implying that the catalytic activity of Pt centers in the bimetallic clusters seems not to be dependent on its surroundings.
The adsorption of CO on clusters of 13 noble metal atoms, with composition ranging from 100% Pt to 100% Au was studied [93]. Different adsorption sites were tested and the CO adsorption energy, CO bond stretching frequency, geometry of the CO cluster system, and HOMO (Highest Occupied Molecular Orbital)–LUMO (Lowest Unoccupied Moleculas Orbital) gaps were investigated. The CO adsorption energy on Pt is >1 eV more favorable than that on Au. It was found that when CO adsorbs on Pt, increasing Au content decreases the adsorption energy. In contrast, when CO adsorbs on Au, increasing Pt content increases the adsorption energy. In general, higher adsorption energies lead to lower CO stretching frequencies, that is, activation of the CO bond.
Many efforts were undertaken to understand the effect of the support for the metal nanoclusters. One of the first effects of the support studied is the effect on the presence of the MgO surface on the oxidation of CO on supported gold aggregates [94]. DFT calculations showed that the supporting oxide also takes an active role in the bonding and activation of adsorbates bound to the gold. The oxide stabilizes a peroxolike reaction intermediate, CO⋅O2, and causes steric repulsion to CO. The most reactive site at Au/MgO appears where the gold shelters the MgO, thereby creating a cavity where several low-coordinated Au atoms and Mg2+ cations from the substrate can interact simultaneously with an adsorbate.
On TiO2 [95], the prevailing view of CO oxidation on gold (Au/TiO2) catalysts is that the reaction occurs on metal sites at the Au/TiO2 interface. Dual catalytic sites were observed at the perimeter of 3-nm TiO2-supportedAu particles during CO oxidation. The activation barriers for the formation and bond scission of the COO2 complex confirm this model, as well as the measured apparent activation energy of 0.16 eV. The observation of sequential delivery and reaction of CO first from TiO2 sites and then from Au sites indicates that catalytic activity occurs at the perimeter of Au nanoparticles [96].
Atomically dispersed supported metal catalysts offer unique opportunities for designing highly selective catalysts and maximizing the utility of precious metals that have potential applications in a wide variety of industrial chemical reactions [97]. Although substantial advances in understanding the origin of the activity of such highly dispersed metal catalysts have been made for a few chemical reactions, the reaction mechanisms and the nature of the active site small metal clusters versus single atoms are still highly debated. Using a combination of DFT and microkinetic modeling, a positively charged single Pt atom on TiO2(110) was shown to exhibit a very high low-temperature activity for the water–gas shift reaction (TOF >0.1 s−1 at 473 K). A comparison of these results with his very recent work [97] on TiO2-supported Pt cluster models provides clear evidence that different active sites are responsible for the experimentally observed activity at low and high temperatures. Finally, they explain why contradictory experimental conclusions have been reported.
It is suggested that there may be several effects contributing to the special catalytic properties of supported nanosized gold particles, and that it is useful to order them in a hierarchy [75].
8 Conclusions
The preceding model associated with the adsorption process of NO and based on a purely energetic point of view has been considered for the CO adsorption process. Such model considers that a simple relationship exists between the adsorption mode (dissociative or molecular) and the behavior of the nanometer-scale monometallic clusters (sintering or fragmentation). In this article, we review interesting and systematic results regarding the adsorption of CO on NSMPs but this adsorption process still remains a challenge for both experimentalists and theoreticians. Moreover, several questions arise as follows:
- (1) Is it possible to predict the behavior of the nanometer-scale bimetallic particles? In fact, the presence of a second metal alters significantly the physical and chemical properties of metallic surfaces as compared with the corresponding single metal systems. CO hydrogenation over ZrO2- and Mo/ZrO2-supported Rh catalysts, using TEM and FTIR, and chemisorption of H2, O2, and CO have been studied by Guglielminotti et al. [98]. These authors have noticed that the two metals, Mo and Rh, form a very complex interacting system. It seems that Mo inhibits the sintering of highly dispersed Rh obtained by Rh4(CO)12 decomposition.
- (2) Is it possible to predict the behavior of the NSMPs in the case of adsorption of a different molecule? In fact, numerous works are devoted to the adsorption of a CO + O2 (see e.g. ref [99]) or a mixture of NO + O2, for example. The behavior of NSMPs in the presence of such a mixture is not obvious. Recently, we have clearly pointed out that when the catalyst is submitted to a mixture of either NO + O2 or C3H6 + O2, the particles are not fully oxidized. On the contrary with the total mixture, namely NO +O2 + C3H6 + O2, the particles both grow and are oxidized [25]. Work is in progress to confirm or modify this simple model.
Acknowledgments
D.B. is indebted to Dr D. Spanjaard from the Laboratoire de Physique des Solides (Paris XI University, France) and Dr M. C. Desjonquères from the SPCSI (CEA Saclay, France) for invaluable discussions on the topic of nanometer-scale metallic clusters.