

1 Introduction
The direct preparation of γ-lactones from linear substrates (vs carboxylic acids, ketoesters, and so on) has attracted much attention despite the main restrictive limitation of these linear molecules is that they are usually obtained through a multiple-step synthesis [1]. For this reason, the use of simple and easily available substrates is highly desirable. Among such alternative strategies, the carboesterification reaction of alkenes is a useful approach to γ-lactones [2] because of their high potential for application in natural product and drug synthesis [3].
Among the elements that have shown activity in this reaction, the case of manganese and copper must be emphasized. In this regard, manganese has not behaved as a true catalyst, because stoichiometric quantities of this element are needed to complete the reaction [4], whereas copper complexes have shown activity although with the problems of recovery and reuse [5]. In this context, a full mechanistic study has been conducted on different manganese-based oxides to understand the main physicochemical features of these solids that can lead to maximize both the activity and selectivity values during the carboesterification reaction of alkenes.
For achieving this, a variety of different manganese-based oxides have been prepared and some of them have been doped with copper [6], in an attempt to find if these elements were acting separately or jointly through a plausible synergistic mechanism.
Indeed, previous studies reported the use of copper(II) as co-oxidant in transformations of intermediate radicals to carbocations [7] and for improving the ability of manganese to exchange electrons in eventual oxidation/reduction processes [8].
In view of these precedents, here we developed a manganese/copper dual system that uses air as a terminal oxidant so that the carboesterification reaction of alkenes can take place. We have shown that the reaction mechanistic studies not only allow a better understanding of how the reaction occurs but also help to improve the catalyst design for achieving higher activities and selectivities.
2 Results and discussion
2.1 Screening of copper and manganese-based catalysts for the carboesterification reaction of styrene to obtain γ-lactones
Pioneering mechanistic studies on the synthesis of γ-lactones from olefins in the presence of the Mn(III) salt (Mn(OAc)3) [4] pointed to two possible reaction pathways to explain the formation of these cyclic esters: (1) a radical mechanism and (2) a single-electron transfer mechanism. A more recent study described the MnO2-promoted carboesterification of alkenes pointing to the intervention of a radical pathway as a plausible reaction mechanism in the presence of additives such as LiBr and NaOAc [9].
In this context, previous studies by Huang et al. showed that a copper(II) salt efficiently catalyzed the [3+2] cycloaddition of alkenes with acetic anhydrides to give γ-lactones [5] with good to excellent yields and a tentative mechanism based on the enolization of the starting anhydride was suggested [10]. A priori, both reaction routes with copper and manganese have in common the use of LiBr (20%) and bases as additives. Nonetheless, the route with copper differs in the participation of ionic intermediates unlike the manganese route.
Taking into account the aforementioned precedents, a series of amorphous (MnOx) and crystalline manganese-based oxides (Mn2O3, molecular sieve cryptomelane OMS-2 and the layered manganese-based oxide birnessite OL-Na) were synthesized via a variety of preparative methods (see Section 3). The resulting materials were properly characterized [6,11]. All these materials were applied as catalysts for the carboesterification reaction of alkenes with anhydrides as a simple approach to γ-lactones, using the styrene carboesterification reaction with acetic anhydride as a model reaction (Scheme 1).
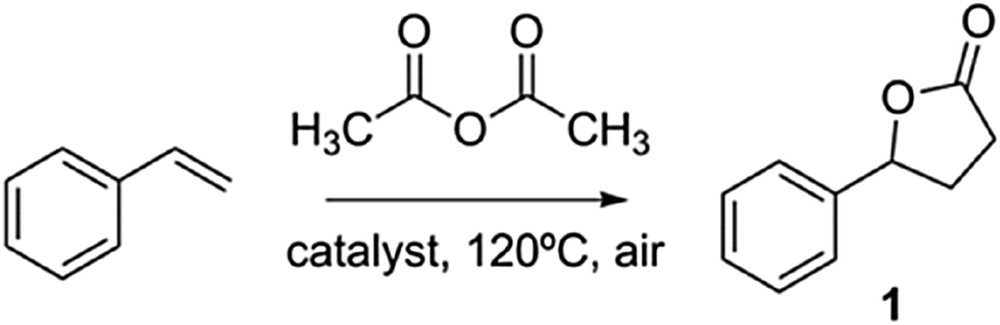
Reaction scheme for the [3+2] cycloaddition of styrene with acetic anhydride to afford γ-lactone 1.
Copper(II) was also incorporated as co-oxidant on these materials to assist in the transformation of hypothetically formed radical intermediates [7] and/or to improve the performance of manganese oxides in eventual redox processes (see Section 3) [8]. We envisaged that copper would influence positively the intermolecular or intramolecular addition to carbon–carbon multiple bonds through any of the hypothetical mechanisms that have been previously pointed out. Then, preliminary investigations focused on the styrene carboesterification reaction with acetic anhydride to give γ-butyrolactone (1) as a model reaction using this series of manganese solids in the presence of 20 mol % LiBr and 1 equiv of NaOAc, under air, at 120 °C. Under these reaction conditions, the aforementioned reaction proceeds to afford rather low yields of the cyclic ester 1 (entries 1–6, Table 1).
Results on the carboesterification reaction of styrene with acetic anhydride using different copper and/or manganese-based catalysts.
| Entrya | Catalyst | Additive | Mn (equiv) | Cu (mol %) | C (%)b | S (%)c | Y (%)d | MB (%)e |
| 1 | OMS-2 | LiBr | 0.6 | – | 71 | 20 | 14 | 44 |
| 2f | OMS-2 | LiBr | 0.6 | – | 50 | 34 | 17 | 69 |
| 3 | OMS-2 | LiBr | 1.2 | – | 90 | 35 | 32 | 44 |
| 4 | OL-Na | LiBr | 0.6 | – | 33 | 44 | 15 | 81 |
| 5 | Mn2O3 | LiBr | 0.6 | – | 45 | 42 | 19 | 74 |
| 6 | MnO2 | LiBr | 0.6 | – | 47 | 49 | 23 | 79 |
| 7 | Cu(1.66%)/OMS-2 | LiBr | 0.6 | 1.4 | 65 | 49 | 32 | 79 |
| 8 | Cu(0.35%)/OMS-2 | LiBr | 1.2 | 0.6 | 94 | 74 | 70 | 78 |
| 9 | Cu(0.8%)/OMS-2 | LiBr | 1.2 | 1.5 | 94 | 57 | 54 | 68 |
| 10 | Cu(1%)/OMS-2 | LiBr | 0.6 | 1 | 61 | 55 | 34 | 80 |
| 11 | Cu(1.66%)/OMS-2 | LiBr | 1.2 | 2.8 | 80 | 100 | 80 | 100 |
| 12 | Cu(1.66%)/OMS-2 | LiBr | 2 | 5 | 100 | 73 | 73 | 73 |
| 13 | Cu(0.5%)/Mn2O3 | LiBr | 0.6 | 0.4 | 30 | 79 | 24 | 95 |
| 14 | Cu(1%)/Mn2O3 | LiBr | 0.6 | 0.7 | 45 | 17 | 8 | 45 |
| 15 | Cu(0.5%)/MnOx | LiBr | 0.6 | 0.4 | 49 | 54 | 27 | 79 |
| 16 | Cu(1%)/MnOx | LiBr | 0.6 | 0.7 | 42 | 43 | 18 | 79 |
| 17 | Cu(1.6%)/Al2O3 | LiBr | – | 5 | 50 | – | – | 80 |
| 18 | Cu(1.8%)/ZrO2 | LiBr | – | 5 | 40 | – | – | 44 |
| 19 | Cu(1.8%)/MgO | LiBr | – | 5 | 92 | – | – | 50 |
| 20 | CuOx(1.6%), Mg, Al | LiBr | – | 5 | 90 | 10 | 9 | 41 |
| 21 | CuO | LiBr | – | 5 | 13 | 45 | 6 | 94 |
a Reaction conditions: styrene (0.25 mmol), LiBr (0.05 mmol), NaOAc (0.25 mmol), Ac2O (1 mL), n-dodecane (external standard).
b Conversion (%) was obtained by gas chromatography (GC) on the basis of styrene converted.
c Selectivity (%) was obtained by GC on the basis of styrene converted.
d Yield (%) was obtained by GC on the basis of styrene converted.
e MB(%) = mass balance (%) calculated by GC (the integrated peak areas of starting reagents and products were corrected for their respective response factors, and the amount of unreacted starting material was not included in the mass balance).
f Reaction carried out under N2 atmosphere.
According to the results presented in Table 1, because most manganese-containing materials were active in the reaction (entries 1–6, Table 1), the presence of this element was taken into account along with copper in metal-doped manganese oxides for the interpretation of results (Table 1). Besides, when dealing with mixed valence oxides containing mainly trivalent and tetravalent manganese such as the case of cryptomelane OMS-2 and the laminar material OL-Na, all manganese centers were considered, in principle, as potential active sites [6].
In principle, the fact that similar yields of 1 were achieved with OMS-2 under the aerobic and inert atmosphere evidenced the lack of a regeneration process promoted by molecular oxygen (entries 1–2, Table 1) and reinforced the participation of OMS-2 as a stoichiometric oxidant as it will be shown subsequently. Effectively, we concluded that Mn neither behaved as a true nor as an effective catalyst in the carboesterification of styrene because even stoichiometric amounts of this element were insufficient to complete the conversion of the alkene and afford high yields of 1 (entries 1 and 3, Table 1). Moreover, looking closely at these results included in Table 1, it was observed that increasing amounts of Mn increased significantly the conversion of the vinyl aromatic alkene as well as the selectivity and yield of compound 1 but, in both cases, the mass balances were rather low (entries 1 and 3, Table 1).
Besides, it must be noted that with this series of manganese solids the most relevant secondary products detected by gas chromatography (GC) along with the main product, that is, γ-lactone 1 were (1,2-dibromoethylbenzene) (2) and 1-phenylethane-1,2-diyl diacetate (3), whereas 2-oxo-2-phenylacetate was detected at the level of traces (entries 1–6, Table 1) (Fig. 1).
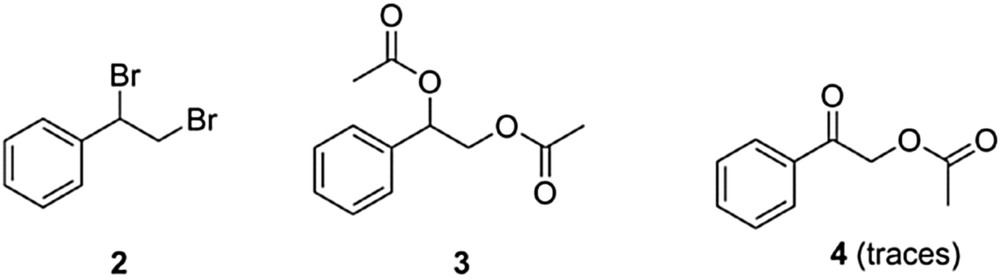
Subproducts detected by GC-MS in the carboesterification reaction of styrene with acetic anhydride in the presence of OMS-2.
At this point, we attempted to make the reaction catalytic by incorporating copper on the solids and reducing the amount of manganese accordingly. The incorporation of copper on the microporous material OMS-2 led to a slight improvement in the conversion and selectivity values, the mass balances notably improved, although the catalytic results were still rather far from optimum (entries 1 and 7, Table 1).
In parallel, a series of differently loaded copper catalysts were prepared using the same manganese oxide OMS-2 as support. All materials gave from low to moderate results as catalysts (entries 8–12, Table 1). Transmission electron microscopy (TEM) analysis of these copper-doped samples showed a rather heterogeneous distribution of CuO particles on the surface of OMS-2 regardless of the copper loading, evidencing the lack of an optimal and adequate dispersion and explaining the poor catalytic results.
From the aforementioned results, it was found that stoichiometric amounts of manganese from mixed valent oxide OMS-2 doped with copper Cu(1.66%)/OMS-2 afforded the best results of activity and selectivity for the synthesis of the lactone 1 (entry 11, Table 1), whereas other combinations with upper or lower copper and manganese loadings gave much poorer results (entries 8–12, Table 1). At this point, we could confirm that the lactonization reaction was taking place on the surface of Cu(1.66%)/OMS-2, and not by dissolved species because the elimination of the solid by hot filtration at a low conversion level stopped the reaction completely (Fig. 2).
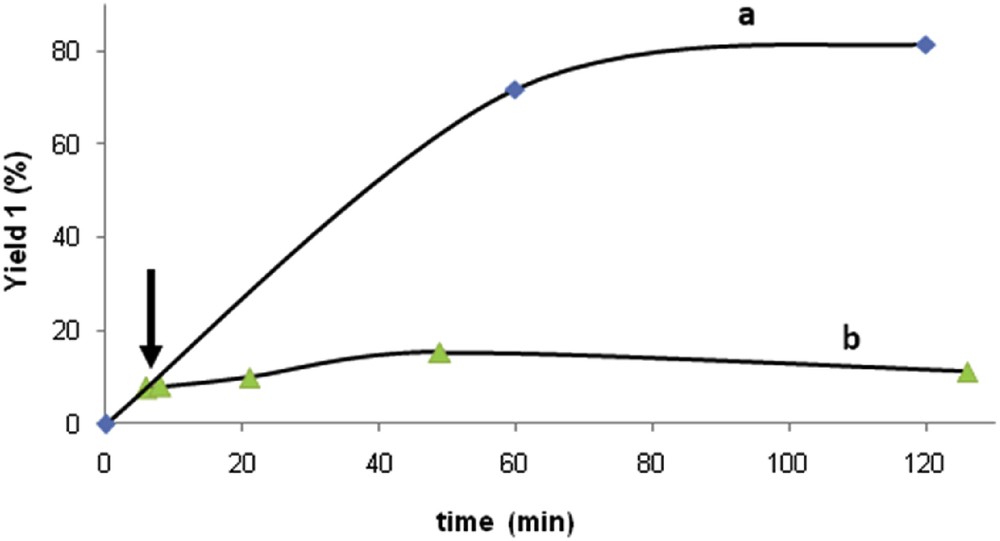
Graphics representing (a) the evolution of the yield of lactone 1 with time with Cu(1.66%)/OMS-2 and (b) the yield of lactone 1 with time when the solid Cu(1.66%)/OMS-2 is separated by hot filtration. The arrow shows the point when the reaction mixture is filtered.
Similarly, other oxides and materials containing Mn and/or Cu were rather inefficient in forming the cyclic ester 1 regardless of its copper content (entries 13–21, Table 1).
Again, the formation of the secondary products 2 and 3 took place with Cu(1.66%)/OMS-2, but their formation was associated with the presence of manganese because the formation of these byproducts did not occur with CuO as a catalyst (entry 21, Table 1). In this context, it was assumed that copper was present as CuO on the OMS-2 surface as deduced from the diffuse reflectance spectrum of the Cu(1.66%)/OMS-2 sample [12].
Fig. 3 shows the evolution of the yield of lactone 1 and styrene with timing in the presence of catalytic amounts of OMS-2 and Cu(1%)/OMS-2 materials.
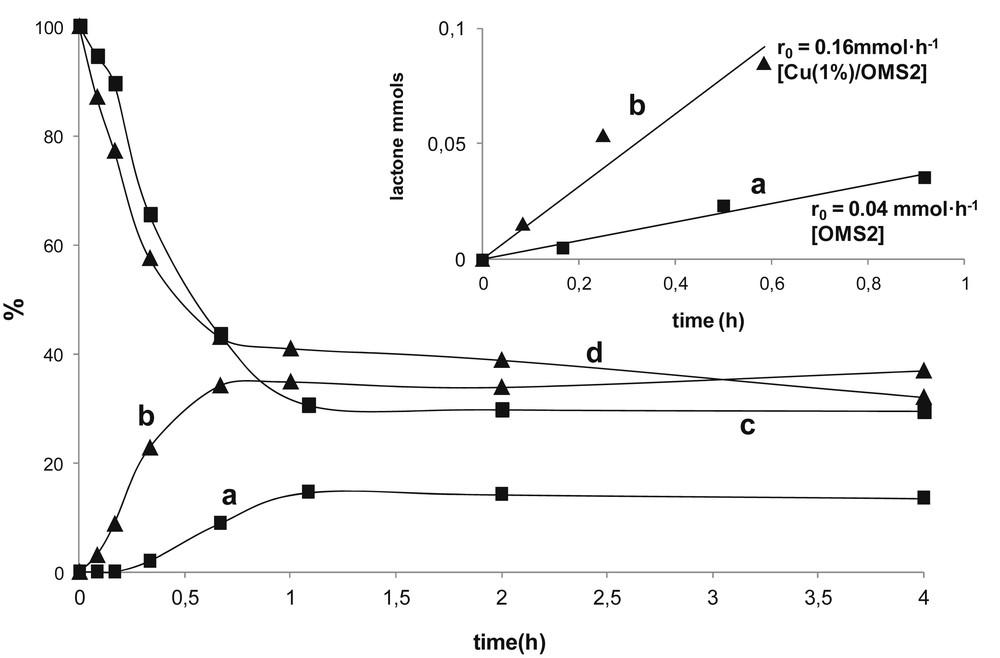
Evolution of the yield of γ-lactone 1 with time with (a) OMS-2 (entry 1, Table 1) and (b) Cu(1%)/OMS-2 (entry 10, Table 1). Evolution of the amount of starting reagent styrene with time in the presence of (c) OMS-2 and (d) Cu(1%)/OMS-2. The inset shows the initial reaction rate r0 obtained with (a) OMS-2 and (b) Cu(1%)/OMS-2 calculated from the respective tangent lines.
According to the results included in Fig. 3, both conversion and yield data of γ-lactone 1 were higher for Cu-doped OMS-2 (Cu(1%)/OMS-2) than that for OMS-2. But perhaps the most striking result was the higher initial reaction rate for forming 1 with Cu(1%)/OMS-2 (r0 = 0.16 mmol/h) than the rates obtained separately with OMS-2 (r0 = 0.04 mmol/h) and CuO (r0 = 0.008 mmol/h), a fact that would be pointing to the existence of a plausible cooperative effect between manganese and copper.
This fact together with the existence of a short induction period (approximately 20 min) with OMS-2, that clearly shortened with Cu(1%)/OMS-2 (Fig. 3), illustrates the beneficial effect of copper to accelerate and improve the selectivity and yield for the formation of 1 [13].
2.2 Effects of Cu2+ on the structure of cryptomelane-type manganese oxide molecular sieve OMS-2: Cu(1.66%)/OMS-2
Although a priori, we do not know what level or in what way Cu2+ influences the performance of the manganese oxide OMS-2, we studied the oxygen desorption behavior by temperature-programmed desorption (TPD) analysis on both the undoped OMS-2 and the copper-doped Cu(1%)/OMS-2 [14]. In principle, TPD-MS data for Cu(1.66%)/OMS-2 and OMS-2 were similar. In fact, both materials exhibited a continuous desorption of water (ca. 200 °C) as well as abundant loss of O2 ranging from 300 to 500 °C and from 500 to 750 °C (Fig. 4):
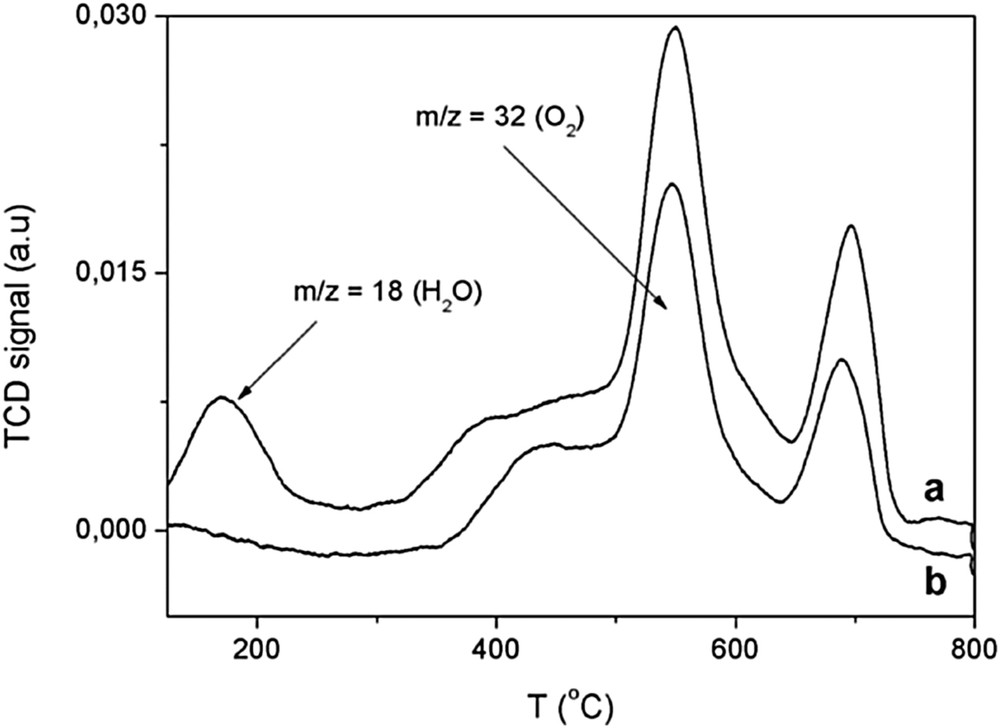
TPD plot for (a) OMS-2 under a He atmosphere (25–800 °C; 10 °C/min) and (b) TPD plot for Cu(1.66%)/OMS-2 under a He atmosphere (25–800 °C; 10 °C/min).
The sharp desorption peak at 550 °C was ascribed to liberation of lattice β-O2 species [15]. These β-O2 species are bound to Mn3+ being released at the middle temperature range during TPD. The shoulder next to this peak was associated with desorption of surface oxygen species (O2, O−) and/or labile oxygen species (β-O2) with different Mn-O bond strength [15].
Then, the second peak at higher temperature (700 °C) was associated with elimination of oxygen atoms bound to Mn4+ or γ-O2 species (Fig. 4) [15].
In summary, most oxygen species desorbed in OMS-2 were lattice oxygen species whose proportions remained almost invariable under the influence of copper. So it appears that copper does not exert any appreciable influence at the structural level, hence suggesting that this element is located on the surface. This assumption will be verified by other techniques later.
Because not only the reversible oxygen evolution but the reducibility of OMS-2 would account for the good performance in oxidation reactions, we have carried out in parallel H2-TPR (temperature-programmed reduction) studies of OMS-2 and Cu(1%)/OMS-2 for comparison. We have considered that the reduction of manganese oxides can be described by three successive main processes [16] [(Fig. 5).
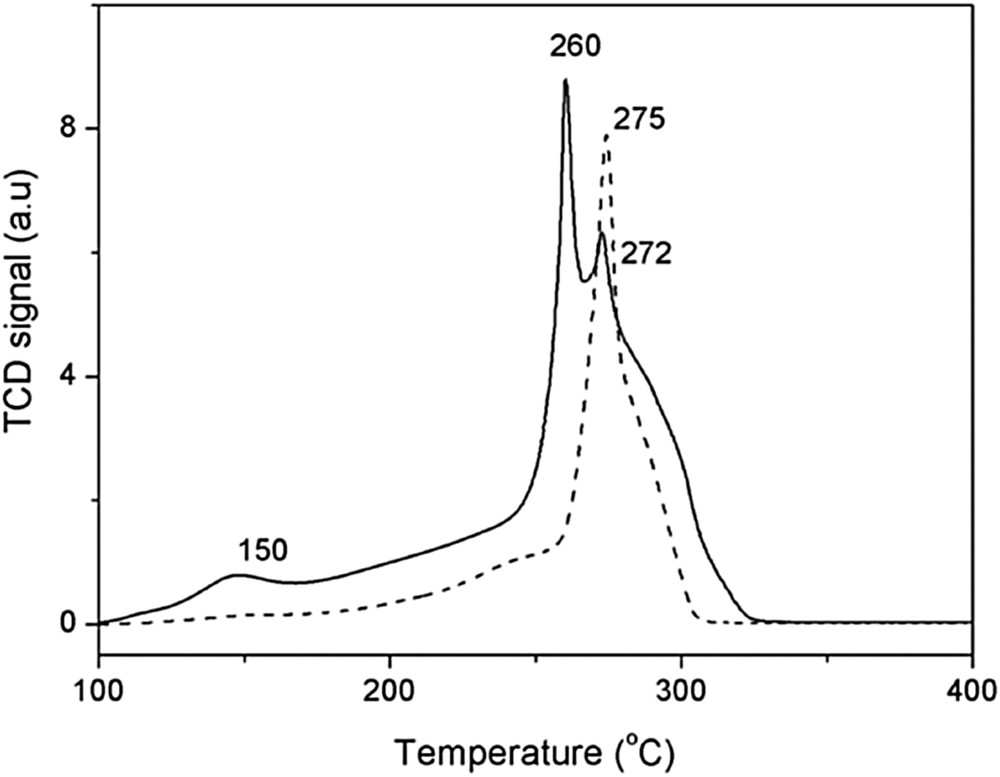
Graphics showing the TPR profile of OMS-2 (dashed line) and Cu(1.66%)/OMS-2 (solid line).
The TPR profile of OMS-2 showed a unique peak around 275 °C that could be decomposed into different overlapping components and a shoulder around 245 °C. All these components were assigned to the reduction of structural Mn+3 and Mn+4 cations involving different phases (i.e., MnO2, Mn2O3, Mn3O4 and MnO) [17]. The incorporation of copper had a strong influence on the TPR profile, showing a shifting of the bands mentioned previously at lower temperatures and a new peak at 150 °C. These facts evidenced a higher oxygen mobility or reducibility of the solid under the influence of Cu.
In this context, it has been suggested that the presence of Cu in the material influences the reactivity of the oxygen atoms due to the formation of Cu–O–Mn bridges. These Cu–O–Mn species are much easier reducible and should probably give higher reactivity [17]. Indeed formation of those mixed oxide species would contribute to delocalize oxygen, that is, facilitating the reduction of manganese and explaining the acceleration of the reaction promoted by copper.
So as a conclusion the major effect of CuO on the cryptomelane is increasing the reducibility of manganese oxide, hence explaining the acceleration of the styrene carboesterification reaction with acetic anhydride.
2.3 Effect of Cu2+ on the reactivity of cryptomelane-type manganese oxide molecular sieve OMS-2: Cu(1.66%)/OMS-2
By looking closely at the reactivity and material balance data included in Table 1, we concluded that, in general, polymerization of styrene was the most important factor responsible for the decrease of selectivity and yields of 1, preventing in most cases the achievement of proper mass balances (Table 1). Indeed, in most experiments the detection by GC-MS of styrene-derived oligomers of high molecular weight at long retention times confirmed this experimental observation (Table 1) [18]. Moreover, the reaction mixture turned into a red color with time.
A priori, the poor material balances were associated with the presence of Mn, because the mass balances were almost completed when using CuO as a catalyst (entry 21, Table 1). Moreover, the aforementioned reddish color was reproduced when both, styrene and acetic anhydride, were put in contact in the absence of a catalyst and any other reagent. Under these reaction conditions (without catalyst) styrene transformed but the formation of lactone 1 did not occur indicating that the alkene polymerization was possibly taking place.
Cationic polymerization of styrene was discarded under our reaction conditions [19], but the possibility of a chain reaction process launched by the redox couple styrene/anhydride is possible in accordance to their half reduction potentials [20]. This reaction would lead to the formation of a styryl radical cation intermediate. However, we could not detect this radical cation either spectroscopically or via trap experiments. Nevertheless, the formation of this styryl radical cation intermediate was indirectly confirmed through the formation of the secondary products 2 and 3 following the reaction in Scheme 2 [7].
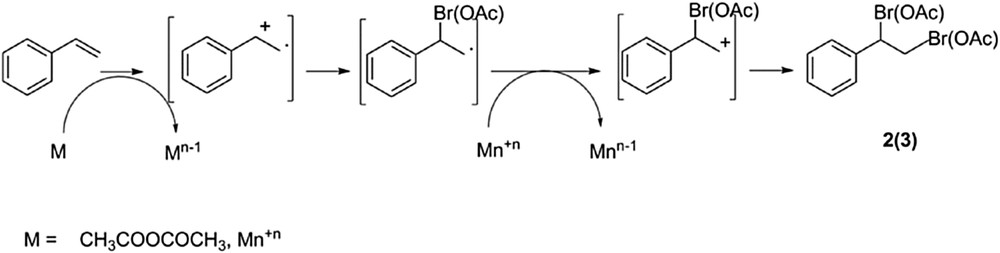
Plausible reaction scheme for the formation of compounds 2 and 3 through a styryl radical cation.
Moreover, given that formation of compounds 2 and 3 was not observed with CuO as a catalyst (entry 21, Table 1), we assumed the exclusive participation of manganese (and not copper) in the formation of these secondary products (Scheme 2).
At this point, to minimize the undesired polymerization reaction, styrene was slowly added in portions in an attempt to keep a low concentration of the vinylic monomer and consequently to slow down the formation of polystyrene (see entry 4, Table 2). However, this strategy failed in improving the outcome of the reaction probably because of a faster reaction rate for polymerization over lactonization (compare entries 3–4, Table 2).
Results of the aerobic carboesterification reaction of styrene and 1-dodecene with acetic anhydride under different experimental conditions.
| Entrya | Catalyst | Substrate | Additive | Mn (equiv) | Cu (%) | C (%)b | S (%)c | Y (%)d | MB (%)e |
| 1 | Cu(1.66%)/OMS-2 | Styrene | LiBr | 1.16 | 2.78 | 80 | 100 | 80 | 100 |
| 2 | Cu(1.66%)/OMS-2 | Styrene | LiBr | 0.7 | 1.68 | 61 | 54 | 33 | 75 |
| 3 | Cu(1%)/OMS-2 | Styrene | LiBr | 0.7 | 1 | 59 | 59 | 35 | 78 |
| 4f | Cu(1%)/OMS-2 (portions) | Styrene | LiBr | 0.7 | 1 | 78 | 30 | 23 | 57 |
| 5g | Cu(1%)/OMS-2 | Styrene | LiBr | 0.56 | 0.8 | 31 | 0 | 0 | 63 |
| 6h | Cu(1%)/OMS-2 | Styrene | LiBr | 0.5 | 0.7 | 2 | 0 | 0 | 96 |
| 7 | Cu(1%)/OMS-2 | 1-Dodecene | LiBr | 0.5 | 0.7 | 29 | 60 | 18 | 91 |
| 8g | Cu(1%)/OMS-2 | 1-Dodecene | LiBr | 0.62 | 0.89 | 15 | 28 | 4 | 89 |
| 9i | Cu(1%)/OMS-2 | Styrene | LiBr | 0.6 | 1 | 66 | 33 | 22 | 66 |
| 10 | Cu(1%)/OMS-2 | Styrene | – | 0.64 | 0.9 | 70 | 6 | 4 | 34 |
| 11g | Cu(1%)/OMS-2 | Styrene | – | 0.5 | 0.8 | 8 | 0 | 0 | 73 |
| 12 | OMS-2 | Styrene | – | 0.46 | – | 60 | 13 | 8 | 49 |
| 13g | OMS-2 | Styrene | – | 0.66 | 0.94 | 28 | – | – | 72 |
a Reaction conditions: styrene (0.25 mmol), LiBr (0.05 mmol), NaOAc (0.25 mmol), Ac2O (1 mL) and n-dodecane as external standard.
b Conversion (%) was obtained by GC on the basis of styrene converted.
c Selectivity (%) to γ-lactone was obtained by GC on the basis of styrene converted.
d Yield (%) was obtained by GC on the basis of styrene converted.
e MB (%) = mass balance (%) calculated by GC (the integrated peak areas of starting reagents and products were corrected for their respective response factors, and the amount of unreacted starting material was not included in the mass balance).
f Acetic anhydride was added in small portions.
g 1 mmol (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was added.
h Acetic anhydride was replaced by ethyl acetate.
i Reaction carried out without base NaOAc.
Because according to the literature references formation of 1 can occur through a free radical mechanism, mediated by Mn or Cu, the radical scavenger TEMPO was incorporated into the reactor. Under the standard conditions used here the formation of γ-lactone 1 as well as secondary products 2 and 3 was completely inhibited (compare entries 3 and 5, Table 2), hence confirming that the formation of these products came from a radical pathway. Then, as a probe of concept and considering that the introduction of acetate groups onto aromatic rings or double bonds mediated by radicals can account for the formation of carboxymethyl CH2COOH intermediates, we tried to reproduce the same radical synthon using ethyl acetate ester (instead of acetic anhydride) [4a]. However, the fact that ethyl acetate did not add to styrene argued against the intervention of such short-lived radical intermediates, questioning a possible free radical lactonization process (see entry 6, Table 2) [4a]. Moreover, because the mass balance reached almost 100% (entry 6, Table 2) with ethyl acetate, this led us to conclude that acetic anhydride was absolutely essential for obtaining the five-membered γ-lactone 1, being also one of the main factors responsible for the polymerization process as previously pointed out (vs through a single-electron transfer process).
In accordance with this reasoning, the carboesterification reaction of a long chain aliphatic alkene and far less prone to polymerize, such as 1-dodecene, was studied giving the corresponding five-membered γ-lactone 4 with a proper mass balance (entry 7, Table 2). Interestingly, no secondary product analogs like those observed with styrene (2 and 3) were detected with 1-dodecene indicating that these two carboesterification reactions (from styrene and 1-dodecene) may undergo different reaction pathways.
Again, the radical scavenger TEMPO was incorporated in the reactor during the lactonization reaction of 1-dodecene and, in contrast to styrene, both conversion and selectivity dropped to half (entries 5 and 8, Table 2), which clearly demonstrates that a radical as well as a non-radical reaction pathway might be operative with linear aliphatic alkenes.
Moreover, when the carboesterification reaction of styrene with acetic anhydride was carried out in the presence of CuO and the radical trap TEMPO, the catalytic parameters remained invariable, hence confirming that the reaction mechanism with copper does not go through radicals [5]. Further control experiments were performed to elucidate the influence of the base and the LiBr salt. In this regard, we obtained that in the absence of CH3COONa or the additive LiBr, the performance of the copper-doped material Cu(1%)/OMS-2 was severely inhibited giving low yields of the lactone 1 (entry 9–10, Table 2). The incorporation of TEMPO in the experiment without LiBr completely inhibited the synthesis of lactone 1, hence confirming that in the absence of LiBr a radical pathway was simultaneously taking place.
These experimental facts prove that two operating mechanisms are presumably coexisting during the carboesterification reaction of alkenes: (1) an enolization route involving ionic intermediates that operate in the presence of the additive LiBr and (2) an electron transfer process that in parallel accounts for the formation of the γ-lactone as well as the undesired polymerization reaction and can take place without LiBr (Scheme 3).
Tentative mechanisms for the Cu(1.66%)/OMS-2 catalyzed carboesterification of alkenes.
At this point, because we had previously hypothesized that Mn (and not Cu) was involved in the styryl radical cation formation we considered important to detect the plausible formation of reduced manganese species during the [3+2] cycloaddition of styrene with the copper-doped material Cu(1.66%)/OMS-2. For achieving this, the lactonization reaction of styrene with acetic anhydride was monitored by EPR spectroscopy and the detection of relatively stable high spin EPR Mn2+ species at different reaction times confirmed this assumption.
To carry out this study the reaction was stopped at different reaction times and the solid was separated by centrifugation. The recovered material was frozen at 77 K into a quartz tube and then the EPR spectrum was recorded. Because initially the EPR spectra of fresh Cu(1.66%)/OMS-2 was EPR silent, this confirmed that the Mn2+ content was negligible (Fig. 6).
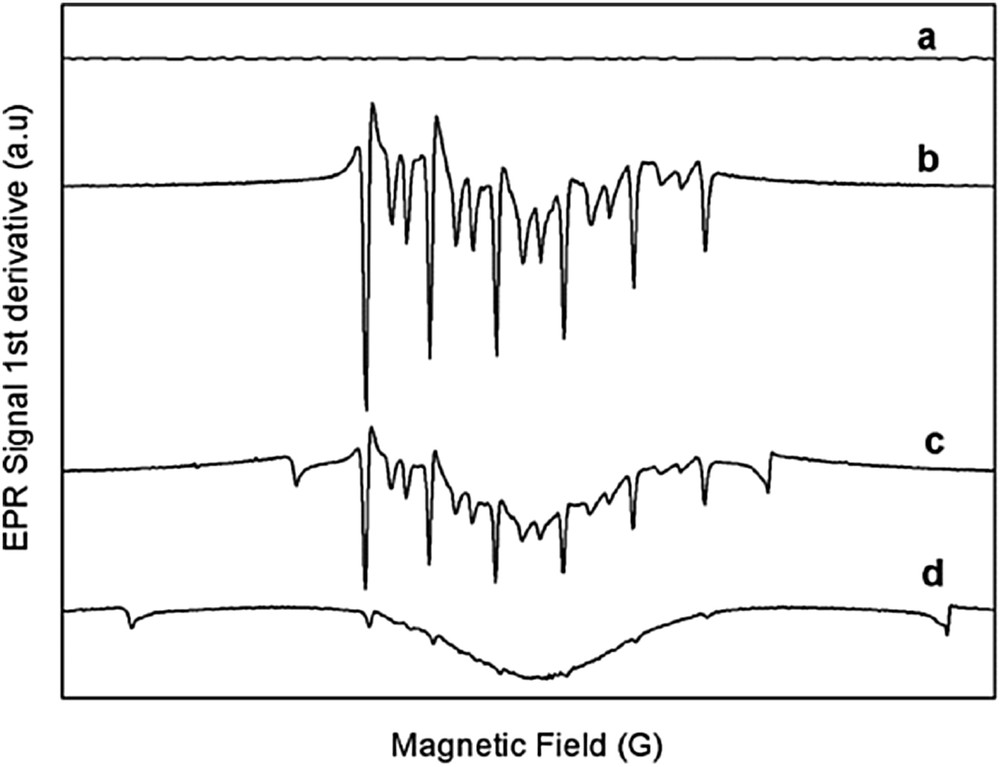
Electron paramagnetic spectra of transient manganese species detected in solid Cu(1%)/OMS-2 during the lactonization reaction for forming 1 from styrene and acetic anhydride at (a) 0 min; (b) 20 min; (c) 45 min and (d) 240 min.
However, the high spin Mn2+ signal soon appeared during the lactonization process with Cu(1.66%)/OMS-2, hence confirming the continuous and gradual reduction of manganese in the copper-doped cryptomelane (Fig. 6).
Effectively, according to the result in Fig. 6, a pattern of six signals could be observed at short reaction times, which was attributed unequivocally to Mn2+ (g = 2.008, I = 5/2). The hyperfine splitting constants of 90.5, 92, 94.5, 98 and 100 indicate that the cation was predominantly placed at octahedral positions (Fig. 4) [21]. At longer reaction times the signal widened, a phenomenon that was previously observed in other manganese materials and which has been assigned to Δm = ±1 forbidden transitions [22]. Interestingly, in this case, no g values associated with Cu(II) were found.
In parallel, the X-ray photoelectron spectroscopy (XPS) surface elemental analysis of OMS-2 and Cu(1.66%)/OMS-2 before and after the reaction was also carried out (see Fig. 7).
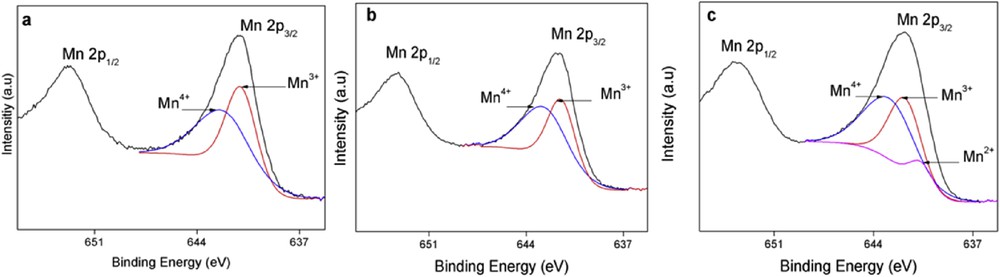
XPS measurements for fresh prepared (a) OMS-2, (b) fresh prepared Cu(1.66%)/OMS-2 and (c) Cu(1.66%)/OMS-2 after reaction.
According to these measurements, the coexistence of different oxidation states for manganese was confirmed for both solids (Fig. 7). Indeed Mn 2p3/2 peaks belonging to Mn3+ and Mn4+ species were detected at 641.9 and 642.8 eV (binding energy [BE]), respectively, for cryptomelane-type manganese oxide OMS-2 (52.6% Mn4+ and 47.4% Mn3+). The incorporation of copper did not change substantially the Mn3+/Mn4+ ratio, albeit the abundance slightly increased for Mn4+ (55.4%) and decreased for Mn3+ (44.6%). The signal assigned to copper was very weak and it did not allow its deconvolution.
Interestingly, the XPS copper signal disappeared after reaction, whereas the Mn 2p3/2 peak could be deconvoluted in three main components: Mn4+, Mn3+ and Mn2+ (640.6 eV BE) [23] giving an abundance of 50.6%, 38.7% and 10.7%, respectively, for these three manganese ions. These results point to a clear reduction of the high valent manganese components (Mn4+ and Mn3+) to lower manganese oxidation states in the copper-doped cryptomelane material Cu(1.66%)/OMS-2 and suggest a plausible inability of the latter to reoxidize and restore the original surface composition of the catalyst.
In summary, the manganese-mediated intermolecular coupling between an acetate group and an alkene with copper-doped cryptomelane-type manganese oxide tends to result in variable yields of the cyclic ester and accounts for two possible reaction mechanisms: (a) an enolization reaction pathway involving ionic intermediates that it operates in the presence of LiBr and (b) an electron transfer process that in parallel accounts for the formation of the γ-lactone as well as the undesired polymerization reaction and takes place without LiBr.
A clear synergistic effect between copper and manganese has been devised in copper-doped cryptomelane-type manganese oxide that takes place at the surface level because copper is not incorporated into the structure. In fact it appears that Cu2+ dispersed on the surface of cryptomelane may induce a more releasable lattice oxygen, thus enhancing the redox properties of the manganese ions.
3 Experimental section
3.1 Analyses and instrumentation
3.1.1 Inductively coupled plasma–atomic emission spectroscopy analysis
The chemical compositions were measured by inductively coupled plasma–atomic emission spectroscopy (ICP-AES) analysis. The chemical analyses were carried out in a Varian 715-ES ICP Optical Emission spectrometer, after solid dissolution in HNO3/HCl/HF aqueous solution.
3.1.2 Area measurements
Textural properties were obtained from the CO2 adsorption isotherms measured at 273 K fitting the results with Dubinin–Astakhov equation [25] using a Micromeritics ASAP 2010 apparatus.
3.1.3 X-ray diffraction
The crystal structure of the as-prepared samples was verified by X-ray powder diffraction. X-ray diffraction (XRD) pattern analysis was obtained using a Panalytical CUBIX diffractometer with monochromatic Cu Kα radiation (λ = 0.15417 nm) at 45 kV and 40 mA. The angle (2θ) was measured in a scan range of 2.00–90.03° in steps of 0.04018° with a counting time of 34.92 s.
3.1.4 EPR spectroscopy
The EPR spectra were recorded using a Bruker EMX-12 spectrometer operating at the X band, with a modulation frequency of 100 kHz and amplitude of 1.0 Gauss. All spectra were measured at 77 K. Samples were measured at different reaction times. For achieving this, the reaction was stopped and the catalyst was separated by centrifugation. The catalyst was introduced in a quartz tube being frozen with liquid nitrogen (77 K).
3.1.5 Temperature-programmed desorption
TPD profiles were obtained using AutoChem II 2920. Fifty milligrams of a catalyst (granulometry, 0.4–0.8 mm) was placed in a quartz tube, heated to 105 °C and purged with helium gas (He) for 30 min. The desorption was carried until 800 °C at a heating rate of 10 °C/min purged with helium. O2 and H2O desorption were followed with a thermal conductivity detector (TCD).
3.1.6 Temperature-programmed reduction
H2-TPR profiles were obtained using Autochem 2910 with a TCD. Fifty milligrams of a catalyst (granulometry, 0.4–0.8 mm) was placed in a quartz tube, heated to 105 °C and purged with argon gas (Ar) for 30 min, and then reduced in a stream of a mixture of 10% H2/Ar (50.01 mL/min) at a heating rate of 5 °C/min to 600 °C.
3.1.7 X-ray photoelectron spectroscopy
The chemical state of Mn was determined by XPS. Photoelectron spectra were recorded using a SPECS spectrometer equipped with a 150 Magnetic Circular Dichroism (MCD)-9 Phoibos detector and using a monochromatic Mg Kα (1253.6 eV) X-ray source for doped OMS-2 materials and a non-monochromatic Al Kα (1486.6 eV) X-ray source for the undoped material. Spectra were recorded using an analyzer pass energy of 30 eV, an X-ray power of 50 W and under an operating pressure of 10−9 mbar.
During data processing, BE values were referenced to C1s peak (284.5 eV). Spectra treatment has been performed using the CASA software.
3.2 Catalysts preparation
All reagents were of analytical grade and purchased from Aldrich. Microporous and laminar materials (OMS-2 and OL-Na, respectively) were synthesized according to the reported procedures [11a,b]. Microporous and laminar copper-doped materials (Cu/OMS-2 and Cu/OL-Na, respectively) were also synthesized according to the reported procedures [24].
3.2.1 Synthesis of MnOx, MnOx (0.5% Cu), MnOx (1.0% Cu) and MnOx (3.0% Cu) catalysts
Mn(NO3)·H2O (0.025 mol) and the required amount of Cu(NO3)2·2.5H2O (according to the metal loading) were dissolved in 50 mL of Milli-Q water. A 0.5 M Na2CO3 solution was added dropwise to the previous aqueous solution until the pH became ca. 8. A precipitate was formed that was aged at 298 K for 1 h. Then the solid was separated by filtration, being washed several times with distilled water. The solid was dried under air at 393 K overnight and it was calcined in static air (with a heating rate of 5 °C/min) at 623 K for 4 h.
3.2.2 Synthesis of Mn2O3, Mn2O3 (0.5% Cu), Mn2O3 (1.0% Cu) and Mn2O3 (3.0% Cu) catalysts
Mn(NO3)·xH2O (0.025 mol) and the required amount of Cu(NO3)2·2.5H2O (according to the metal loading) were dissolved in 50 mL of Milli-Q water. Then, 0.5 M Na2CO3 solution was added dropwise to the previous aqueous solution until the pH became ca. 8 and a precipitate was formed. The suspension was aged at 298 K for 1 h. The precipitate was filtered, washed several times with distilled water and dried under air at 393 K overnight. The resulting solid was calcined under air at 873 K (heating rate = 5 °C/min) for 4 h.
3.2.2.1 Synthesis of CuOx(1.66%)/Al2O3; CuOx(1.8%)/ZrO2 and CuO(1.8%)/MgO catalysts
The required quantity of Cu(NO3)·3H2O was dissolved in 20 mL of Milli-Q water. Then, 1 g of the support (Al2O3, ZrO2 or MgO) previously calcined at 723 K under N2 (heating rate = 5 °C/min for 5.5 h) was added and the mixture was stirred at room temperature for 1 h. Solvent was removed under reduced pressure and the resulting solid was dried under vacuum at 393 K overnight. Before each use, the catalyst was calcined at 723 K under air for 6 h (heating rate = 2 °C/min).
3.2.2.2 Synthesis of CuOx(1.66%), Mg, Al catalysts
The required amount of Mg(NO3)2·6H2O (18.24 g), Al(NO3)3·9H2O (9.29 g) and Cu(NO3) ·3H2O (0.25 g) was carefully dissolved in 100 mL Milli-Q water. Then, 100 mL of an aqueous solution containing NaOH (5.95 g) and Na2CO3 (7.07 g) was slowly added (1 mL/min) under vigorous stirring, and a precipitate was formed, being aged at 333 K during 18 h. The slurry was filtered and the solid was washed several times with Milli-Q water until the pH of the filtrate became ca. 7. The resulting solid was dried by heating at 333 K for 12 h.
The solid was calcined under air at 873 K for 8 h (heating rate = 2 °C/min).
3.3 Catalyst characterization
3.3.1 Characterization data of OMS-2 and Cu(1%)/OMS-2 catalysts
Chemical and textural analyses of OMS-2 and Cu(1%)/OMS-2 catalysts
| Catalysts | ICP-AESa | Area (m2/g)b | Average crystallite size (nm)c | |
| % Mn | % Cu | CO2 (273 K) | ||
| OMS-2 | 67.5 | – | 96.8 | 11.6 |
| Cu(1%)/OMS-2 | 64.3 | 1 | 96.7 | 10.6 |
a Chemical composition obtained by ICP-AES analysis.
b Textural properties were obtained from the CO2 adsorption isotherms measured at 273 K fitting the results with the Dubinin–Astakhov equation [25].
c Measured by TEM (the average crystallite size was calculated from the Scherrer equation for the reflection at 2θ = 28.842° in the XRD pattern).
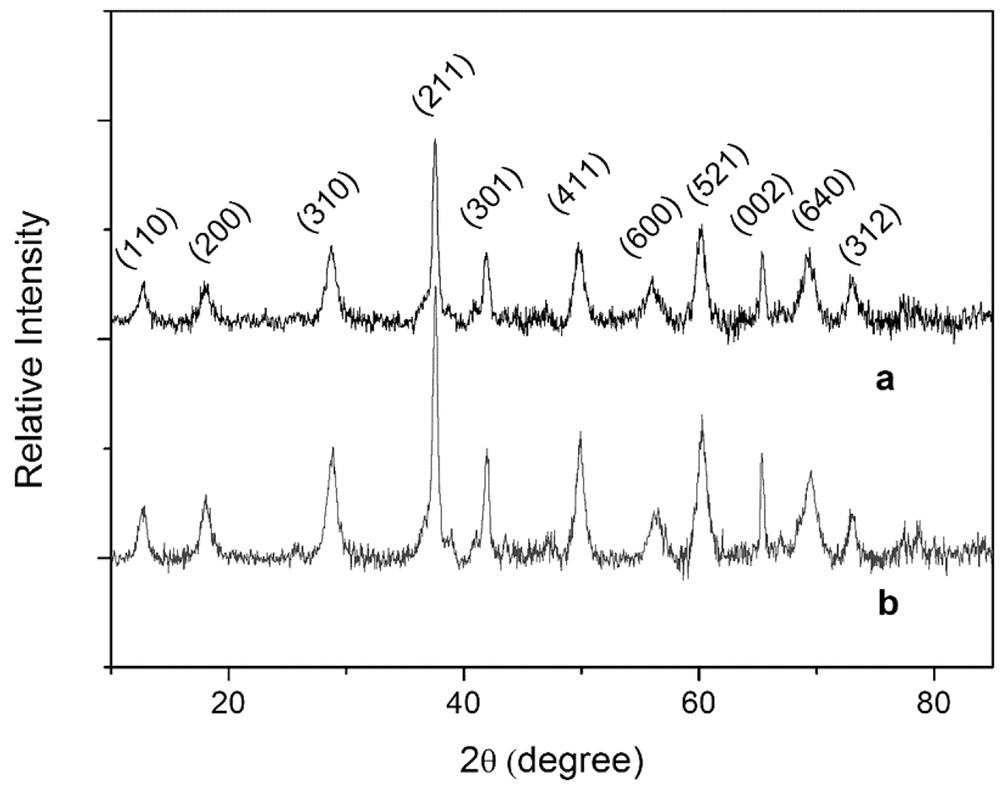
XRD patterns of Cu/OMS-2 (a) and OMS-2 (b) catalysts.
3.4 Catalytic activity experiments
The reactions were carried out by adding 0.25 mmol of the olefin, 0.05 mmol LiBr, 0.25 mmol NaOAc and 1.0 mL Ac2O in a glass reactor, using n-dodecane as external standard and 0.6, 1.2, or 1.9–2 equiv of catalyst (15 mg, 30 mg or 47.5 mg of catalyst, respectively). Then the temperature was increased to 120 °C under air atmosphere. Reaction samples were extracted at regular reaction times with a microsyringe, being analyzed by GC using an HP-5 capillary column (5% phenylmethylsiloxane, 30 m × 320 μm × 0.25 μm).
Products were identified by GC-MS using an Agilent 6890N8000 equipment equipped with a mass spectrometry detector (Agilent 5973N quadrupole detector).
3.5 Hot filtration experiment
The experiment was carried out by separating the catalyst by filtration from the reaction mixture and monitoring the formation of the lactone 1 from the filtrate by GC.
Acknowledgements
Financial support by the Ministerio de Economia y Competitividad, Programa Severo Ochoa (SEV2016-0683) is gratefully acknowledged.