

1 Introduction
Lignin is the second most plentiful renewable natural polymer after cellulose, accounting for 15%–30% (wt) of the lignocellulosic biomass and almost 30% of all non-fossil–based carbon sources [1]. It has a highly branched three-dimensional optically inactive structure comprising three phenyl propane units (monolignols), for example, sinapyl alcohol (SA), coniferyl alcohol (CA), and p-coumaryl alcohol. The monolignols are cross-linked together by CC (5-5 and β-1 linkages) and COC (α-O-4 and β-O-4 linkages) bonds leading to a complex structure with a large variety of functional groups (e.g. hydroxyl OH, carbonyl CHO, and carboxyl COOH moieties) [2].
The ratio of monolignols in lignin composition entails classification of lignin into three major groups: (1) softwood lignin consisting mainly of CA and traces of SA units, (2) hardwood lignin with comparable ratios of SA and CA, and (3) grass lignin with major amounts of p-coumaryl alcohol [3]. In addition, softwood lignin has around 56 ether linkages, whereas hardwood lignin has around 72% [4]. This functionalized aromatic architecture recommends lignin as an energy supplier and aromatic precursor for fuel applications [5,6]. Consequently, its industrial exploitation is receiving tremendous attention nowadays.
The pulp and paper industry is an important provider of lignin as a major waste byproduct [7]. Unfortunately, only 2% of this waste is commercially exploited. The main reason is related to the high degree of physicochemical heterogeneity of lignin (e.g. built-in variety of aromatic units, interunit linkages, functional groups, and molecular size) [8].
However, lignin has several additional properties that can enhance its potential value. For example, lignin has adhesive properties (e.g. useful as rubber intensifier, polyolefin and rubber packing, and filler and comonomer for unsaturated polyester and vinyl ester materials). It can also be used as binder for glass wool building insulation, fiber nutritional source for pet and human food, dispensing agent, flocculent, and thickener or auxiliary agent in coatings and paints [3,9].
Another scenario for achieving proper exploitation of lignin may involve the development of derivatization methods of the lignin structures via chemical, enzymatic, and/or chemoenzymatic pathways. The enzymatic methods may even mimic the natural biosynthesis of lignin. Thus, peroxidase and laccase enzymes can provide oxidation of lignin building blocks leading to reactive species (radicals) with a multitude of coupling possibilities. Following this strategy, Dordick et al. [10] used horseradish peroxidase to incorporate phenols into lignin leading to the production of reactive polymers with properties of phenolic resins. Also, laccase alone or in combination with mediators was used to introduce phenolic-based “building block” functionalities onto lignocellulosic materials [11]. Similar strategies were developed for different comonomers such as guaiacol sulfonate, vanillylamine, or acrylamide grafted onto lignin [12,13]. However, achieving effective control over the heterogeneity of lignin structure is still an unsolved problem and constitutes an important drawback for industrial applications.
In this context, we propose the construction of polymers starting from the monolignols found in lignin, for example, SA and CA, leading to products similar to natural lignins. The oxidative polymerization (oxi-polymerization) of monolignol(s) was performed in the presence of hydrogen peroxide (H2O2) or tert-butyl hydroperoxide (t-BHP). The reaction was catalyzed by peroxidase enzyme leading to products with less heterogeneous polymeric structures. The polymeric products have been characterized using the gel permeation chromatography (GPC) technique. The versatility of the developed method has been proved by testing monolignol mixtures with different SA/CA molar ratios (oxi-copolymerization).
2 Experimental section
2.1 Chemicals and solutions
Enzyme screening for the investigated oxi-polymerization process was performed with three peroxidase enzymes from two sources, for example, PaDa-I (AaeUPO1 variant, unspecific peroxidase produced in Saccharomyces cerevisiae, 25.82 U mL−1 enzyme activity), R4, and 2-1B (versatile peroxidase originally from Pleurotus eryngii, expressed in S. cerevisiae, 12.95 U mL−1 enzyme activity) [14,15]. PaDa-I, R4, and 2-1B mutants were provided by Dr. Miguel Alcalde (Institute of Catalysis, CSIC, Madrid, Spain).
Stock solution of 1 M phosphate-buffered saline (PBS) was prepared by dissolving 8 g NaCl, 0.2 g KCl, 1.43 g Na2HPO4·2H2O, and 0.34 g KH2PO4 in 1 L distilled water. Dilution of the stock solution led to 10 mM PBS (pH = 7.4) used as aqueous buffer solution. CA and SA were of analytic purity and were purchased from Sigma–Aldrich together with the other reagents/solvents (30 wt % solution of H2O2, t-BHP, and THF).
Stock solution of monolignols (e.g. 10 mg mL−1 CA or SA) was prepared in methanol, that is, 10 mg of each monolignol was dissolved in 1 mL of methanol. These solutions were used further for the preparation of the reaction mixtures for the oxi-(co)polymerization process.
2.2 Oxi-(co)polymerization of monolignols
Oxi-(co)polymerization of monolignols (e.g. CA, SA, or CA/SA mixture) was performed using a peroxidase-based biocatalytic system. One hundred microliters of monolignol(s) stock solution was mixed with 300 μL PBS (10 mM, pH 7.4). The oxidation reagent (H2O2 or t-BHP) and peroxidase enzymes (PaDa-I, R4, and 2-1B) were then added to the solution to reach a final concentration of 0.6% and 2.582 U mL−1, respectively, and a final concentration of 2 mg mL−1 monolignol used for the oxi-(co)polymerization reaction. The reaction mixture was incubated for 2 h under controlled temperature (e.g. 25, 40, 50, and 60 °C) and gentle stirring (100 rpm).
2.3 Analysis of the reacted mixture
The reacted mixture was treated as indicated in Scheme 1. First, the mixture was separated into two phases (liquid and solid) by centrifugation. The liquid phase contained buffer salts, unreacted monolignol(s), and an excess of oxidation reagent, whereas the enzyme and polymeric products were deposited as a solid phase. For the liquid phase, the monolignol content was analyzed based on spectrophotometry (UV–vis; Specord 250, Analytic Jena), that is, the sample absorbance was read at 280 nm. The conversion (C, %) of monolignol has been calculated from the difference between the concentrations of monolignol before and after the reaction.
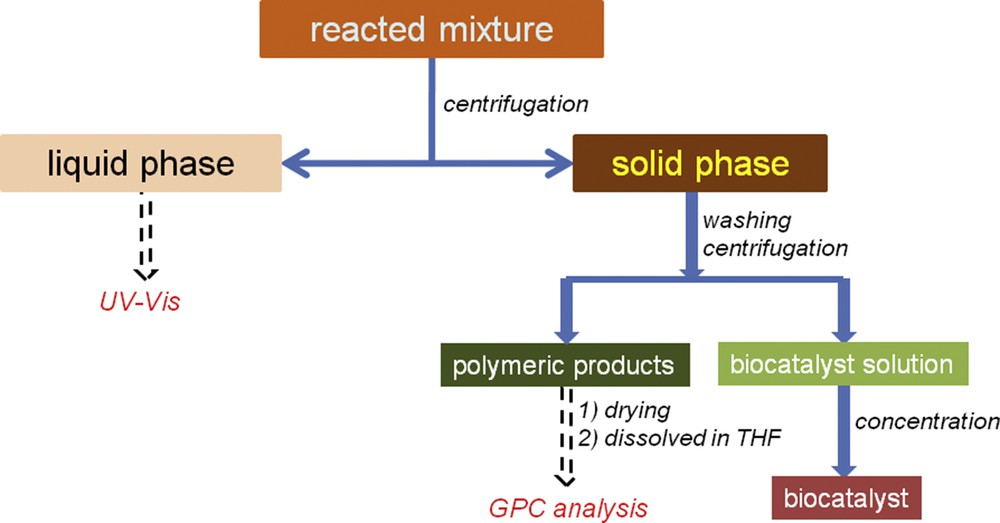
Monitoring of the reacted mixture.
The solid phase was washed several times, alternating between the distilled water and buffer solutions to eliminate the peroxidase enzyme from the polymeric product. After centrifugation, the supernatant was collected and the precipitate (polymeric product) was dried under vacuum conditions. For GPC analysis, the dried polymeric product was dissolved in THF. GPC analysis was carried out using an Agilent Technologies instrument (Model 1260) equipped with two columns (Zorbax PSM 60-S, 6.5 × 250 mm, 5 μm and Polargel-M, 300 × 7.5 mm) and a multidetection unit (260 GPC/SEC (size-exclusion chromatography) system containing RID (Refractive Index Detector), LS (Light Scattering), and VS (Viscosity) detectors). Optimum GPC conditions were set up (1 mL min−1 THF as mobile phase, 100 μL injection volume of sample, and 35 °C temperature at the detectors and columns). Polystyrene standards in the range of 162–10,000 g mol−1 were used for system calibration. Agilent GPC/SEC Software (Version 1.1, Agilent Technologies) allowed the determination of average molecular weight (MW), number-averaged molecular weight (Mn), and polydispersity index (PD).
Collected supernatants containing the peroxidase enzyme were mixed together and concentrated using Amicon ultra-0.5 mL centrifugation filters (30K device NMWL (Nominal Molecular Weight Limit), >95% typical retention, Sigma–Aldrich) under spin conditions of 14,000g (rpm), 10 min spin time, room temperature, and 500 μL starting volume. The recovered enzyme was used in a further reaction cycle for testing the stability of the biocatalyst under reaction conditions.
3 Results and discussion
3.1 Oxi-polymerization of monolignols
Oxi-polymerization of monolignols (e.g., SA or CA) has been performed via a radical mechanism using peroxidase enzyme in the presence of an oxidation reagent (H2O2 or t-BHP) [16]. The general principle of the oxi-polymerization process for SA and CA is summarized in Scheme 2.
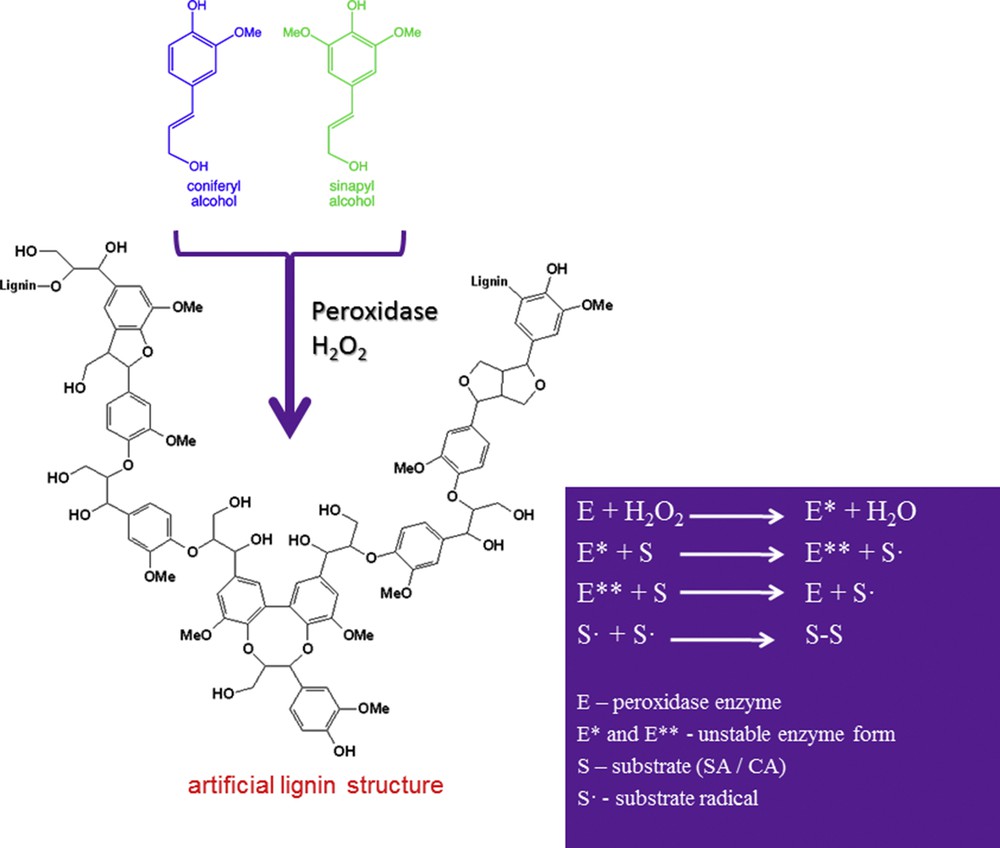
Oxi-polymerization of monolignols.
Screening of the peroxidase enzymes allowed the selection of the most suitable biocatalyst for the oxi-polymerization of monolignols (Fig. 1). PaDa-I, R4, and 2-1B peroxidase enzymes were tested using H2O2 as oxidation reagent. The conversion of SA/CA monolignol was carried out in the liquid phase. The enzymes exhibited a similar catalytic behavior for SA substrate, that is, conversion of 84%–90% SA for all of the three enzymes at 50 °C (Fig. 1a). A maximum conversion of 90% was reached for SA monolignol and R4 peroxidase enzyme. For CA monolignol (Fig. 1b), the conversions were recorded in the range of 49%–53% at 50 °C. Maximum conversion of 53% was achieved for PaDa-I peroxidase at 50 °C. This indicates that the peroxidase mutants preferred to catalyze oxi-polymerization of SA than CA.
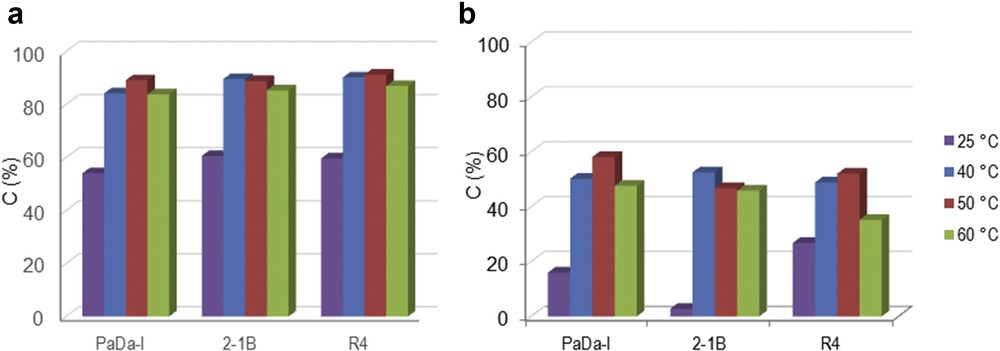
Oxi-polymerization of (a) SA and (b) CA using different peroxidase enzymes and H2O2 as oxidation reagent at different temperatures. Experimental conditions: 2 mg mL−1 monolignol, 0.6% H2O2 and 2.582 U mL−1 enzyme, 100 rpm, 2 h.
Temperature variation in the range of 25–60 °C influenced the catalytic behavior of these enzymes (Fig. 1). Thus, an increase in temperature (from 25 to 50 °C) led to a notable improvement in conversion. This behavior was noted for both monolignols (SA and CA). However, the oxi-polymerization of monolignol was negatively affected when the temperature was further increased to 60 °C. Again, this observation was made in both cases (Fig. 1a and b). The main reasons for reduced conversion at higher temperature should be enzyme denaturation and/or precarious stability of H2O2 [14,17,18]. On the basis of the temperature variation studies, 50 °C appeared to be the optimal temperature for oxi-polymerization of SA/CA monolignol.
Polymeric products resulting in the experiments described previously were characterized by GPC analysis (Table 1). For both monolignols SA and CA, R4 peroxidase led to polymeric structures with high molecular weight (MW of 3188 and 859 Da for SA and CA, respectively), whereas PaDa-I enzyme yielded polymers with a modest mass (MW of 226 and 586 Da for SA and CA, respectively). It should be noted that the combination of SA and R4 peroxidase produced the largest polymeric structure with a relatively homogeneous distribution (MW = 3188 Da, Mn = 1115 Da, and PD = 2.8).
Peroxidase screening for the oxi-polymerization of monolignols (SA and CA). Experimental conditions: 2 mg mL−1 monolignol, 0.6% H2O2 and 2.582 U mL−1 enzyme, 100 rpm, 50 °C, 2 h.
| Sample | SA | CA | ||||
| PaDa-I | 2-1B | R4 | PaDa-I | 2-1B | R4 | |
| C (%) | 84 | 90 | 90 | 50 | 53 | 49 |
| MW | 226 | 721 | 3188 | 586 | 677 | 859 |
| Mn | 214 | 582 | 1115 | 575 | 670 | 733 |
| PD | 1.0 | 1.2 | 1.8 | 1.0 | 1.0 | 1.2 |
It is already known that H2O2 might inhibit the catalytic activity of peroxidase enzyme. For this reason, t-BHP was also tested as a potential oxidation reagent. Fig. 2 shows the conversion of the monolignols (a) SA and (b) CA recorded in the liquid phase using t-BHP. The ascendant trend of the conversion was noted for both monolignols when the temperature was varied in the range of 25–60 °C. Maximum conversion of 82% SA was achieved for 2-1B peroxidase at 60 °C. Therefore, t-BHP allowed reaching a high conversion of monolignols at 60 °C, which was opposite to the behavior observed with H2O2. This can be attributed to the higher stability of t-BHP relative to H2O2 [18,19]. In this case, enhanced thermostability of t-BHP compensated any reduction in efficiency due to enzyme denaturation at 60 °C.
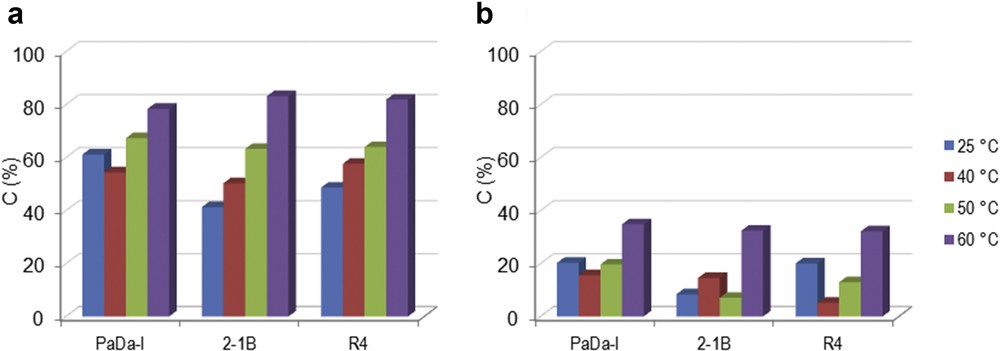
Oxi-polymerization of (a) SA and (b) CA using different peroxidase enzymes and t-BHP as oxidation reagent at different temperatures. Experimental conditions: 2 mg mL−1 monolignol, 0.6% t-BHP and 2.582 U mL−1 enzyme, 100 rpm, 2 h.
Table 2 shows the characteristics of the lignin polymers produced via the oxi-polymerization of SA/CA with both oxidation reagents (H2O2 or t-BHP) in the presence of R4 peroxidase at different temperatures (25, 40, 50, and 60 °C). Higher temperature was correlated with the production of extended polymeric structures for both monolignols (SA and CA). For SA, H2O2 favored the formation of larger polymers than did t-BHP. For CA, however, the opposite trend was observed. Moreover, t-BHP led to a more chaotic mass distribution with an irregular MW variation with temperature for both monolignols. On the basis of these results, H2O2 seems to be the optimum oxidation reagent for this system.
Temperature and oxidation reagent effects on the oxi-polymerization of monolignols (SA and CA). Experimental conditions: 2 mg mL−1 monolignol, 0.6% oxidation reagent, 2.582 U mL−1 R4 enzyme, 100 rpm, 2 h.
| Sample | SA | CA | ||||||
| 25 °C | 40 °C | 50 °C | 60 °C | 25 °C | 40 °C | 50 °C | 60 °C | |
| H2O2 | ||||||||
| C (%) | 60 | 90 | 90 | 87 | 27 | 49 | 52 | 35 |
| MW | 450 | 618 | 3188 | 3910 | 653 | 802 | 859 | 853 |
| Mn | 444 | 412 | 1115 | 3330 | 435 | 625 | 733 | 757 |
| PD | 1.0 | 1.5 | 1.8 | 1.2 | 1.5 | 1.3 | 1.2 | 1.1 |
| t-BHP | ||||||||
| C (%) | 49 | 58 | 64 | 80 | 20 | 5 | 13 | 32 |
| MW | 441 | 488 | 255 | 391 | 611 | 1002 | 1025 | 1553 |
| Mn | 307 | 240 | 246 | 333 | 468 | 621 | 899 | 1257 |
| PD | 1.4 | 2.0 | 1.0 | 1.2 | 1.306 | 1.6 | 1.1 | 1.2 |
Thus, the best performance of the oxi-polymerization system of monolignols was achieved for SA monolignol oxidized with H2O2 in the presence of R4 peroxidase at a temperature of 50 °C (90% SA conversion in the liquid phase and polymeric products with MW = 3188 Da, Mn = 1115 Da, and PD = 2.8). The biocatalyst (R4 peroxidase) was recovered using centrifugation filters. For the next reaction cycle, the enzyme exhibited an approximately 30% loss of catalytic activity.
3.2 Oxi-copolymerization of SA/CA mixtures
Mixtures of SA and CA were also subjected to oxi-polymerization using the peroxidase-catalyzed system. All of the three peroxidase enzymes (PaDa-I, R4, and 2-1B) were tested with both H2O2 and t-BHP. Fig. 3 shows the experimental results for SA/CA = 50:50 (molar ratio). For H2O2 (Fig. 3a), an upward trend of conversion was noted for all of the enzymes in the temperature range of 25–50 °C. Again, further increase in the temperature to 60 °C was associated with a decrease in conversion. Using t-BHP, the variation in the conversion was not monotonic (Fig. 3b). The conversion of the monolignol mixture was higher for H2O2 compared to t-BHP, which is a similar behavior to what happened with oxi-polymerization of individual SA and CA (see Section 3.1.). However, it should be noted that oxi-copolymerization occurred with slower rates compared to oxi-polymerization.
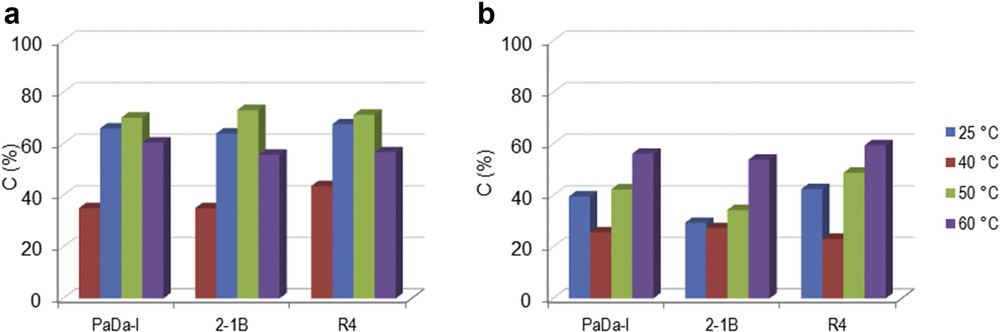
Oxi-copolymerization of lignin monomer mixture of SA/CA = 50:50 (molar ratio) catalyzed by different peroxidase enzymes using (a) H2O2 or (b) t-BHP as oxidation reagent at different temperatures. Experimental conditions: 2 mg mL−1 monolignols mixture, 0.6% oxidation reagent and 2.582 U mL−1 enzyme, 100 rpm, 2 h.
Table 3 shows the results of the GPC analysis for polymeric products in the case of R4 enzyme. The MW of the products displayed a positive trend with temperature up to 50 °C followed by a decrease at 60 °C for both oxidation reagents (H2O2 and t-BHP). The homogeneity of the polymeric products was not influenced by temperature or peroxide type. In addition, the oxi-copolymerization system allowed the generation of more homogeneous polymers than the oxi-polymerization system (PD in the range 1.0–1.6).
Oxi-copolimerization of the lignin monomer mixture (molar ratio SA/CA = 50:50). Experimental conditions: 2 mg mL−1 monolignol mixture, 0.6% oxidation reagent, 2.582 U mL−1 R4 enzyme, 100 rpm, 2 h.
| Oxidizing reagent | H2O2 | t-BHP | ||||
| 25 °C | 50 °C | 60 °C | 25 °C | 50 °C | 60 °C | |
| C (%) | 44 | 71 | 57 | 42 | 49 | 59 |
| MW | 609 | 937 | 567 | 444 | 1690 | 1270 |
| Mn | 588 | 805 | 355 | 421 | 1333 | 831 |
| PD | 1.0 | 1.2 | 1.6 | 1.0 | 1.4 | 1.5 |
The composition of the monolignol mixture was varied by choosing different molar ratios of SA and CA. The results are presented in Table 4. Higher CA content was associated with an improvement in monolignol conversion. This behavior was noted for H2O2 and 20%–50% CA as well as t-BHP and 20%–40% CA. The same trend has been observed for the MW of the polymeric products. When CA dominated the monolignol mixture (>50% CA), the conversion and the MW of the products decreased significantly. A slight variation in the PD was recorded in all cases. Consequently, CA as comonomer (<50% CA content) can have a positive effect on the system, improving the conversion, and allowing the production of large polymeric products. However, an opposite behavior was noticed for CA dominating the monolignol mixture (>50% CA content).
Effects of the composition of the lignin monomer mixture on the oxi-copolymerization process. Experimental conditions: 2 mg mL−1 monolignol mixture, 0.6% oxidation reagent and 2.582 U mL−1 R4 enzyme, 100 rpm, 50 °C, 2 h.
| Mixture of lignin monomers | H2O2 | t-BHP | |||||||||
| SA (%) | 80 | 60 | 50 | 40 | 20 | 80 | 60 | 50 | 40 | 20 | |
| CA (%) | 20 | 40 | 50 | 60 | 80 | 20 | 40 | 50 | 60 | 80 | |
| C (%) | 61 | 66 | 71 | 57 | 42 | 61 | 64 | 49 | 41 | 23 | |
| MW | 383 | 398 | 937 | 859 | 874 | 2049 | 2033 | 1690 | 1852 | 2221 | |
| Mn | 361 | 382 | 805 | 733 | 723 | 1812 | 1720 | 1333 | 1320 | 1955 | |
| PD | 1.0 | 1.1 | 1.2 | 1.2 | 1.2 | 1.1 | 1.2 | 1.4 | 1.4 | 1.1 |
4 Conclusions
A biocatalytic system for the oxi-polymerization of monolignols such as SA and CA has been developed. The system mimics natural lignin synthesis by using monomeric compounds often found in the lignin structure (SA and CA monolignols). R4 peroxidase enzyme efficiently catalyzed the oxidation of SA using H2O2 as oxidation reagent leading to a conversion of up to 90% SA and polymeric products with MW = 3188 Da, Mn = 1115 Da, and PD = 2.8. Also, the whole biocatalytic system displayed a promising thermostability/activity for temperatures as high as 50 °C. A higher thermostability/activity was noted for t-BHP compared to H2O2. However, the characteristics of the polymeric products demonstrated that H2O2 would be a better choice for the oxi-polymerization of SA and CA monolignols. For the oxi-copolymerization of mixtures of SA and CA, an antagonistic effect has been noticed. Indeed, including CA as comonomer at appropriate concentrations (<50% CA content) led to an improvement in the conversion of the monolignol mixture. For high content of CA in the monolignol mixture (>50% CA content), lower transformation of the monolignols was achieved. In addition, similar trend of the MW for polymeric products was noticed.
As a general remark, the use of constituent lignin monomers offers the opportunity to produce polymeric mixtures with an increased homogeneity compared to natural lignin. Moreover, the polymeric products can be designed with controlled structure/properties including the molecular weight. Thus, the developed system is a valuable alternative for the production of (bio)polymers with predictable/requested properties. In addition, the system can find applicability for clean up of phenolic residues from saccharide syrup obtained after pyrolysis of lignocellulosic materials.
Acknowledgments
This work was supported financially by the PN III TE program, contract no. 103/2015 from MEN-UEFISCDI. The authors thank Dr. Miguel Alcalde (Institute of Catalysis, CSIC, Madrid, Spain) for supplying the peroxidase enzymes and Dr. Curt Reimann for his support in preparing this manuscript.