

1 Introduction
Low molecular weight gelators are one of the most interesting and promising examples of self-assembled soft matter [1]. It is believed that secondary forces, such as van der Waals, hydrogen bonding, hydrophobic, electrostatic, dipole–dipole, and π–π interactions, directed the monomers to self-assemble into the noncovalently bonded network that retains an organic solvent, exhibiting gel-like physical properties [1–3]. Although molecular self-assembly is not fully understood so far, researchers have found that the organogelation processes are affected by the molecular structures, solvents, temperature and the stimulus of ultrasound, and so on [4]. The rapid development on functional gelators with π-conjugated systems has been achieved due to the potential applications in optoelectronic devices, chemical sensors, and so on [5,6]. In particular, fluorescent π-organogels have attracted increasing interest and are extensively studied in the fields of sensors, organic light-emitting diode (OLED) and organic thin-film transistor (OTFT) [7]. It is well known that carbazole is a typical π-conjugated system [8] and a promising candidate for optoelectronic materials because of its intense luminescence and electron donating ability [9]. The first organogelator based on carbazole was synthesized by our group [8c]. Since then, many organogels based on carbazole derivatives with potential applications in sensors, photonics, switch, and soft optical materials were successively developed [8a,8c,10]. To generate new functional gels, we went on to extend the carbazole-based π-conjugated system and synthesized new fluorescent molecules in which N-phenylbenzamide was linked at 3-position or 3,6-position of carbazole via the spacer of cyanovinyl (CN-ODEC1, CN-DDEC1, CN-ODEC2, and CN-DDEC2; Scheme 1). It is interesting that the new synthesized carbazole derivatives can form stable gels in many aromatic solvents and in DMSO, and the molecular structures affected their gelation abilities. For instance, the DMSO gels formed from CN-ODEC1 and CN-DDEC1 could be stable for several months, and their critical gelation concentration values were of 0.8 w/v %. However, the unstable gels were formed from CN-ODEC2 and CN-DDEC2 in DMSO. In most aromatic solvents, the critical gelation concentrations of CN-ODEC1 and CN-DDEC1 were as high as 5.0 w/v %, whereas those for CN-ODEC2 and CN-DDEC2 were only 0.2 w/v %. Besides, it was found that H-bonding in gels of CN-ODEC1 and CN-DDEC1 was stronger than those in gels of CN-ODEC2 and CN-DDEC2. Notably, the photophysical properties of the organogelators depend on the molecular structures, and aggregation-induced emission (AIE) was observed during the self-assembling processes of CN-ODEC1 and CN-DDEC1, whereas this development for CN-ODEC2 and CN-DDEC2 was accompanied with a notable redshift in the emission band.
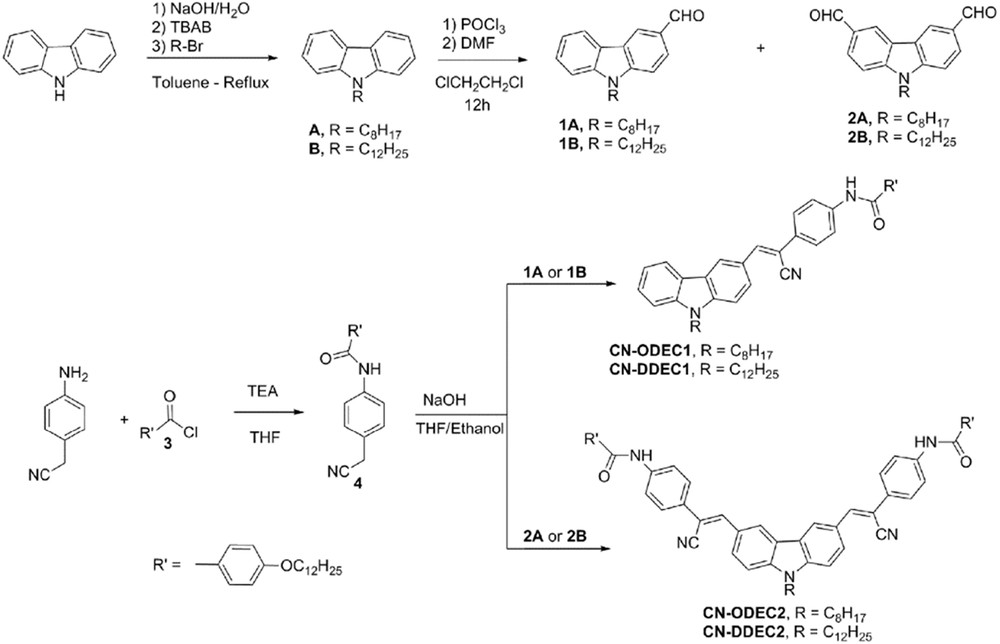
Synthetic route for CN-O/DDECn (n = 1, 2).
2 Results and discussion
2.1 Synthesis
Compounds CN-ODEC1 and CN-DDEC1 were synthesized via Knoevenagel reactions of 2-(4-(4-dodecyloxybenzoylamino)phenyl)-1-acetonitrile (4) with formylcarbazoles 1A and 1B, respectively. Similarly, CN-ODEC2 and CN-DDEC2 were prepared from diformylcarbazoles 2A and 2B, respectively (Scheme 1).
It is worth to note that aldehydes 1A and 2A or 1B and 2B were synthesized [8c] in a one-step reaction from the 9-octylcarbazole (A) or 9-dodecylcarbazole (B), respectively, and the purification process via the silica gel column chromatography enabled as well in a one-step to separate the monoformylcarbazole and diformylcarbazole derivatives. The new compounds were characterized by 1H NMR, MALDI-TOF MS (matrix-assisted laser desorption ionization/time-of-flight mass spectrometry), FT-IR spectrometry, and elemental analysis.
2.2 Gelation abilities of CN-O/DDECn
The gelation abilities of CN-ODEC1, CN-DDEC1, CN-ODEC2, and CN-DDEC2 were evaluated in selected organic solvents (Table 1). It was found that they showed good gelation abilities in aromatic solvents (benzene, toluene, p-xylene, 1,2-dichlorobenzene, and bromobenzene) and DMSO. So, the carbazole derivatives were insoluble in the gelling solvents at room temperature, and clear yellow solutions appeared when heating to ca. 100 °C. Upon cooling to room temperature, immobile light-yellow or deep-yellow rigid gel phases were readily formed (Fig. S4 in the Supplementary Material). Even so, the gelation abilities were found to be different between monosubstituted (CN-ODEC1 and CN-DDEC1) and disubstituted (CN-ODEC2 and CN-DDEC2) carbazole derivatives. First, the onset gelation concentration (OGC) [4b] for monosubstituted derivatives in aromatic solvents was as high as 5.0 w/v % except for 1.0 w/v % in benzene, whereas that for disubstituted carbazole derivatives were as low as 0.2 w/v %. For DMSO gels, OGC for CN-ODEC1 and CN-DDEC1 was only 0.8 w/v %, and the formed gels could be stable at room temperature for several months. However, the gel phases obtained from CN-ODEC2 and CN-DDEC2 in DMSO, irrespective of the low or high concentration of the gelator, could be destroyed by slightly shaking, supposing that these gel phases are less stable. In addition, the disubstituted carbazole derivatives assembled into stable gels in 1,2-dichloroethane at an OGC of 0.6 w/v %. Moreover, the carbazole derivatives were virtually insoluble in some common organic solvents, such as n-hexane, cyclohexane, ethanol, methanol, and acetonitrile. CN-ODEC2 and CN-DDEC2 could not be dissolved in CCl4 and ethyl acetate, and precipitate appeared when cooling the hot solutions of CN-ODEC1 and CN-DDEC1 in the aforementioned solvents. Therefore, the gelation of carbazole derivatives was dependent on the molecular structures.
Gelation abilities of CN-O/DDECn in organic solvents.
| Solvents | CN-ODEC1 | CN-ODEC2 | CN-DDEC1 | CN-DDEC2 |
| Bromobenzene | G (5.0) | G (0.2) | G (5.0) | G (0.2) |
| 1,2-Dichlorobenzene | G (5.0) | G (0.2) | G (5.0) | G (0.2) |
| p-Xylene | G (5.0) | G (0.2) | G (5.0) | G (0.2) |
| Toluene | G (5.0) | G (0.2) | G (5.0) | G (0.2) |
| DMSO | G (0.8) | G (4.0) | G (0.8) | G (4.0) |
| 1,2-Dichloroethane | P | G (0.6) | P | G (0.6) |
| Benzene | G (1.0) | G (0.2) | G (1.0) | G (0.2) |
| Benzaldehyde | P | S | P | S |
| 1,4-Dioxane | P | P | P | P |
| n-Hexane | Is | Is | Is | Is |
| Chloroform | IG | IG | IG | IG |
| Cyclohexane | Is | Is | Is | Is |
| CCl4 | P | Is | P | Is |
| THF | S | S | S | S |
| Ethyl acetate | P | Is | P | Is |
2.3 Self-assembly property of CN-O/DDECn
The scanning electron microscopy (SEM) images were obtained to gain insight into the morphologies of organogels of CN-O/DDECn in different solvents. From Fig. 1, one can see that well-defined tapes with width of about 1.0 μm and lengths varying from 4.0 μm to greater than 10.0 μm (Fig. 1a and c) were generated from monosubstituted carbazole derivatives CN-ODEC1 and CN-DDEC1 in bromobenzene, and amorphous three-dimensional structures consisting of entangled fibrous assemblies were formed from CN-ODEC2 and CN-DDEC2 (Fig. 1b and d) in bromobenzene. In addition, the SEM images of CN-ODEC1 and CN-ODEC2 xerogels obtained from toluene and DMSO showed similar microstructures (Fig. 2). However, it is important to note that the tapes in xerogel CN-ODEC1 obtained from toluene (Fig. 2a) were short in length, whereas the xerogel obtained from DMSO exhibited network of tapes, which are densely intertwined and built up larger ones in some cases (Fig. 2c). This observation was consistent with the result that in monosubstituted derivatives (CN-ODEC1 and CN-DDEC1) the DMSO gel was more stable than the ones formed in aromatic solvents. Moreover, optical microscopy images (Fig. S1 in the Supplementary Material) showed that fibrous microstructures were obtained from monosubstituted carbazole derivatives and amorphous aggregates were generated from disubstituted carbazole derivatives. On the basis of these observations, one can assess that the morphologies of the xerogels mainly depended on the structures of the gelator instead of organic solvents. X-ray diffraction (XRD) measurements were performed to reveal the packing modes in the self-assemblies. Unfortunately, no obvious diffraction peaks appeared in low-angle regions for the xerogels, illustrating poor long-distance ordering of the four carbazole derivatives in bromobenzene (Fig. S2a). However, the presence of diffraction peaks in the wide-angle regions for the xerogels obtained from monosubstituted carbazole derivatives instead of disubstituted ones suggested their different packing modes in organogels, which will be further discussed subsequently (Chart 1).

SEM images of the dried gels based on CN-ODEC1 (a), CN-ODEC2 (b), CN-DDEC1 (c), and CN-DDEC2 (d), obtained in bromobenzene (5.0 w/v % for CN-ODEC1 and CN-DDEC1; 0.2 w/v % for CN-ODEC2 and CN-DDEC2).

SEM images of the dried gels based on CN-ODEC1 (a, in toluene; c, in DMSO) and CN-DDEC2 (b, in toluene; d, in DMSO) (5.0 w/v % for CN-ODEC1 and CN-DDEC1; 0.2 w/v % for CN-ODEC2 and CN-DDEC2).
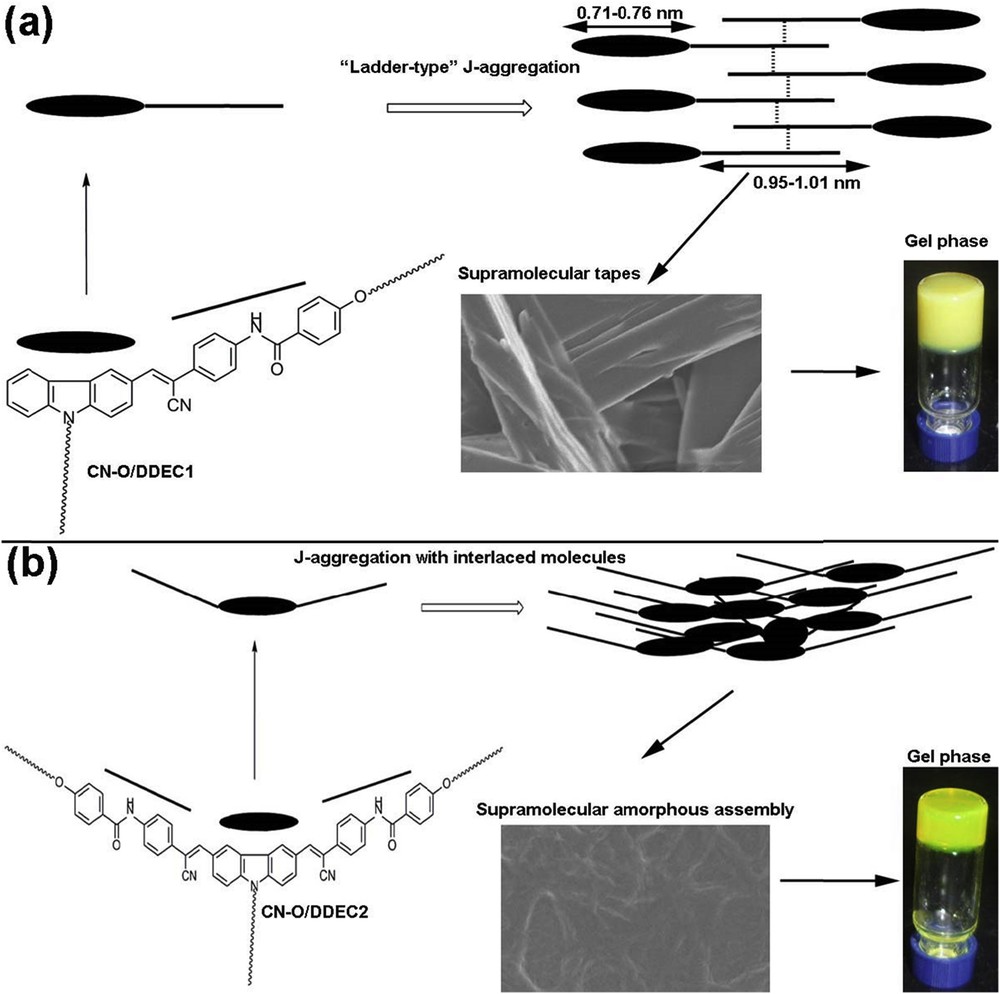
Schematic representation of the possible aggregation mechanisms for (a) CN-O/DDEC1 under lateral view, (b) CN-O/DDEC2 under lateral view.
The UV–vis absorption and FT-IR spectral measurements were performed to investigate the driving forces for the gelation of CN-O/DDECn. As shown in Fig. 3a and b, CN-ODEC1 and CN-DDEC1 gave one strong absorption band at about 382 nm in dilute solutions in bromobenzene, which redshifted to 390 and 394 nm for CN-ODEC1 and CN-DDEC1, respectively, with a shoulder at about 420 nm in gel states, suggesting the formation of J-aggregates in gel states. The broadening of the absorption bands in gel states was possibly due to π–π interactions. In the cases of CN-ODEC2 and CN-DDEC2, two peaks at about 336 and 408 nm can be observed in dilute solutions. As the gel phases were prepared, the absorption redshifted and the intensities decreased. We could detect three absorption peaks at about 382, 432, and 465 nm in gel phases. The absorption spectral changes during sol–gel transitions illustrated that π–π interactions played key roles on the gelation and J-aggregates were formed, in which the gelators intertwined one another with no ordered arrangement in the gel states (Chart 1). Fig. S3a and b showed the FT-IR spectra of the xerogels obtained from bromobenzene. A strong vibration band at 3332 cm−1, which was attributed to the N-H stretching mode of the amide bonds, emerged in the spectra of xerogels of CN-ODEC1 and CN-DDEC1. The amide I bands of CN-ODEC1 and CN-DDEC1 appeared at 1642 cm−1. It revealed that strong intermolecular H-bonding via amide groups occurred in the xerogels of monosubstituted carbazole derivatives [11,12]. On the other hand, the bands at 3410 and 3425 cm−1 due to N-H stretching vibration appeared in xerogels of CN-ODEC2 and CN-DDEC2, respectively, and their vibration absorption bands ascribed to amide I appeared at about 1653 cm−1. It indicated that the strength of H-bonding in the gels of CN-O/DDECn was in an order of CN-ODEC1 ∼ CN-DDEC1 > CN-ODEC2 ∼ CN-DDEC2. The strong H-bonding that could be detected in the xerogels formed from monosubstituted carbazole derivatives might be explained by the presumable head-to-tail J-type arrangement [13], in which the molecules were arranged in a “ladder-type”, and the weak H-bonding between disubstituted carbazole derivatives in xerogels was possibly due to the hindrance of U-shaped conformation [8c], which made the gelators to interlace one another creating a “heavy entangled” amorphous structures (Chart 1).
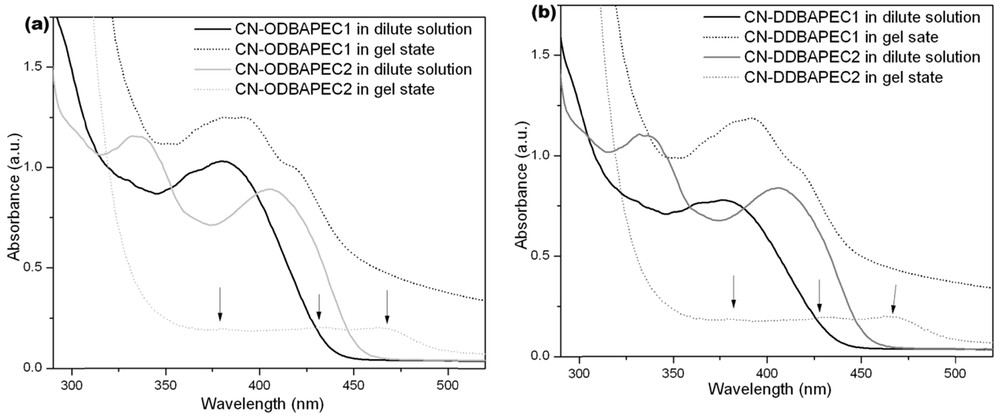
UV–vis absorption spectra in bromobenzene of (a) CN-ODEC1 (black solid line, dilute solution at 0.05 w/v %; black dotted line, gel at 5.0 w/v %) and CN-ODEC2 (light gray solid line, dilute solution at 0.05 w/v %; dotted light gray line, gel at 0.2 w/v %); (b) CN-DDEC1 (black solid line, dilute solution at 0.05 w/v %; black dotted line, gel at 5.0 w/v %) and CN-DDEC2 (gray solid line, dilute solution at 0.05 w/v %; dotted gray line, gel at 0.2 w/v %). Masquer
UV–vis absorption spectra in bromobenzene of (a) CN-ODEC1 (black solid line, dilute solution at 0.05 w/v %; black dotted line, gel at 5.0 w/v %) and CN-ODEC2 (light gray solid line, dilute solution at 0.05 w/v %; dotted light gray ... Lire la suite
2.4 AIE in gel states
When irradiated with 365 nm light (insets of Fig. 4 and Fig. S4c and d), we found that the obtained gels of CN-ODEC1 and CN-DDEC1 emitted greenish yellow light and the ones obtained from CN-ODEC2 and CN-DDEC2 exhibited yellowish emission. The fluorescence emission spectral changes during sol–gel transitions of the gelators in bromobenzene were recorded. From Fig. 4a, one can see that the emission band of CN-ODEC1 in solution (5.0 w/v %) appeared at 485 nm. Upon the formation of stable gel, the emission intensity of CN-ODEC1 increased significantly, meaning the gelator showed AIE behavior. Meanwhile, the gel of CN-ODEC1 gave two emission bands at 475 and 495 nm. Similarly, CN-DDEC1 also exhibited AIE property [14a,b]. On the other hand, during the sol-to-gel processes, CN-ODEC2 and CN-DDEC2 not only exhibited AIE phenomenon, but also showed notable redshift in emission bands [14b,c]. From Fig. 4b and d, the emission bands of CN-ODEC2 and CN-DDEC2 in solutions are located at 482 nm, and redshifted to 522 nm in gel states. Such a significant redshift in the emission during gelation processes illustrated that strong π–π interaction was involved in gels of CN-ODEC2 and CN-DDEC2. Therefore, we deemed that the intermolecular interactions between the neighboring monosubstituted gelator molecules occurred via the extended π-backbones and the alkyl chains (Chart 1), and the gelators were arranged in a “ladder-type” disposal [13,15]. On the other hand, the disubstituted gelator molecules were interlaced with one another [15,16], which is in agreement with the absence of Bragg peaks in wide-angle X-ray scattering (WAXS) region as well as the weakness of H-bonding for these derivatives. A schematic representation of the possible molecular packing modes in gel states is illustrated in Chart 1.
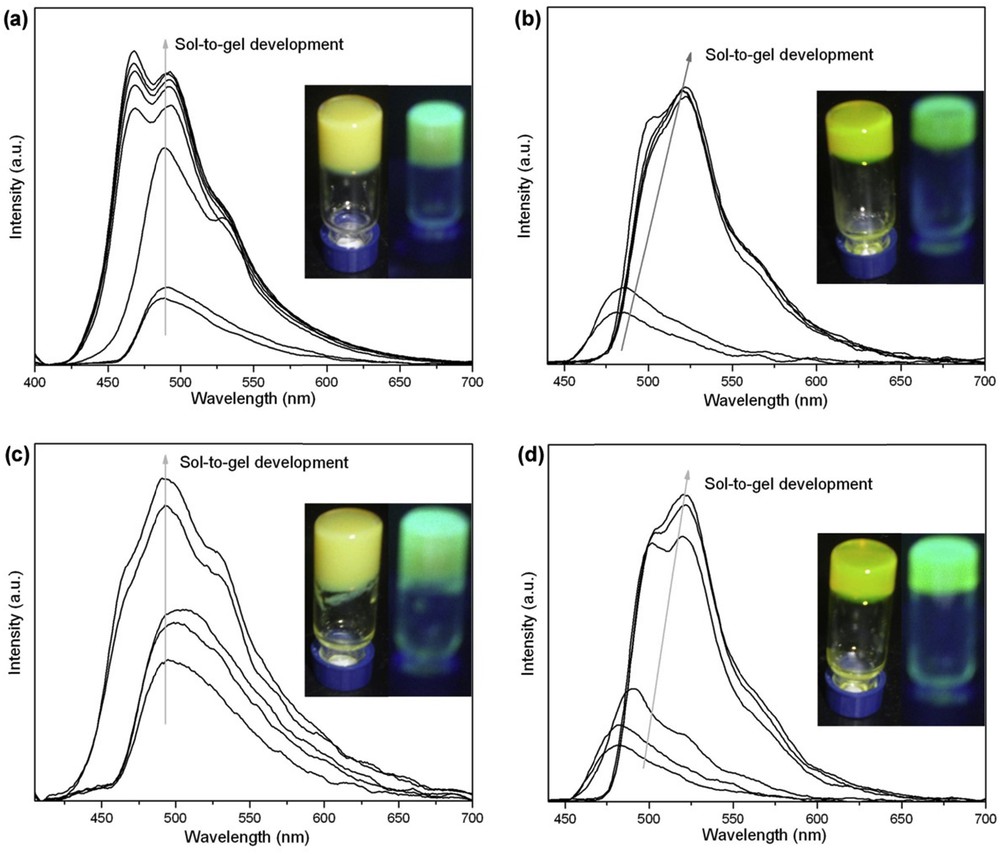
Fluorescence emission spectra in bromobenzene of (a) CN-ODEC1 (5.0 w/v %, λexc = 392 nm), (b) CN-ODEC2 (0.2 w/v %, λexc = 432 nm), (c) CN-DDEC1 (5.0 w/v %, λexc = 392 nm), and (d) CN-DDEC2 (0.2 w/v %, λexc = 432 nm). The insets show the corresponding gel phases under day light (left) and 365 nm UV light (right).
3 Conclusions
In summary, four new carbazole derivatives, in which N-phenylbenzamide was linked at 3-position or 3,6-positions of carbazole via the spacer of cyanovinyl (CN-ODEC1, CN-DDEC1, CN-ODEC2, and CN-DDEC2), were synthesized. It is found that they could self-assemble into organogels in aromatic solvents and DMSO. The xerogels obtained from monosubstituted derivatives showed well-organized tapes, and those formed from disubstituted derivatives exhibited three-dimensional amorphous structures. The UV–vis absorption, FT-IR, and fluorescence emission spectra illustrated that the strong H-bonding and moderate π–π interactions were the main driving forces for the gelation of CN-ODEC1 and CN-DDEC1, and head-to-tail “ladder-type” J-aggregates were formed in gel state. On the other hand, strong π–π interactions happened during the gelation of CN-ODEC2 and CN-DDEC2, and “staircase-type” J-aggregates were obtained. Notably, significant AIE was observed during the gelation processes of the synthesized gelators. Such new π-gels with intense emission are of decisive significance in potential photonics applications and related fields.
4 Experimental section
4.1 General information
The solvents were dried using conventional methods. All others materials are used as received. 1H NMR spectra were recorded on Mercury plus 500 MHz using CDCl3 or DMSO-d6 as solvents. Mass spectra were performed on Agilent 1100 MS series and AXIMA CFR MALDI-TOF MS (COMPACT). UV–vis absorption spectra were determined using a Shimadzu UV-1601PC spectrophotometer. Fluorescence emission spectra were carried out using a Shimadzu RF-5301 luminescence spectrometer. FT-IR spectra were measured using a Nicolet-360 FTIR spectrometer by incorporating samples in KBr pellet. XRD patterns were carried out on a Japan Rigaku D/max-γA instrument. XRD was equipped with graphite monochromatized Cu Kα radiation (λ) 1.5418 Å, using a scanning rate of 0.02 s−1 in the 2θ range from 0.7 to 10 and higher. SEM observations were carried out using a Japan Hitachi model X-650 San electron microscope. The samples for these measurements were prepared by casting the organogel on silicon wafers and drying at room temperature, and then coating with gold.
4.2 Synthesis
The alkoxybenzol chloride 3 and 2-(4-(4-dodecyloxybenzoylamino)phenyl)-1-acetonitrile (4) were synthesized according to a general procedure and used without any further distillation [17]. Unless otherwise noted, all starting materials were obtained from commercial suppliers and used without further purification.
9-Octylcarbazole (A). Carbazole (6 g, 0.036 mol), tetrabutylammonium bromide (TBAB, 0.2 g), and 1.5 equiv of 1-bromooctane were dissolved in toluene (20 mL), then NaOH aqueous (50%, 10 mL) was added, and the obtained mixture was stirred and refluxed for 5 h. Then the toluene was removed under the reduced pressure. The resulted liquid phase was dissolved in dichloromethane and subjected to column chromatography eluting with ethyl acetate/petroleum ether (1:4) to obtain a dark liquid. Yield: 6.5 g, 65%; Rf: 0.7. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.11 (d, 2H, Ar–H), 7.45 (d, 2H, Ar–H), 7.42 (d, 2H, Ar–H), 7.23 (m, 2H, Ar–H), 4.29 (t, 2H, NCH2C), 1.86 (m, 2H, CCH2C), 1.23–1.41 (m, 10H, C(CH2)5C), 0.87 (t, 3H, CH3). FT-IR (KBr, cm−1): 3051, 2917, 2845, 1626, and 1592.
9-Dodecylcarbazole (B). Compound B was synthesized following the similar procedure as that of compound A. Yield: 7.5 g, 62%; Rf: 0.73. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.10 (d, 2H, Ar–H), 7.46 (d, 2H, Ar–H), 7.41 (d, 2H, Ar–H), 7.23 (m, 2H, Ar–H), 4.29 (t, 2H, NCH2C), 1.86 (m, 2H, CCH2C), 1.23–1.41 (m, 18H, C(CH2)8C), 0.87 (t, 3H, CH3). FT-IR (KBr, cm−1): 3051, 2917, 2845, 1626, and 1592.
9-Octyl-3-formylcarbazole (1A) and 9-octyl-3,6-diformylcarbazole (2A). A solution of N,N-dimethylformamide (19.6 g, 0.268 mol) in 1,2-dichloroethane (10 mL) was added dropwise to phosphoryl chloride (27 mL, 0.179 mol) at 0 °C. Then the reaction mixture was stirred for 30 min at room temperature. Then, 9-octylcarbazole (A, 5 g, 0.018 mol) in 1,2-dichloroethane was added dropwise under 0 °C. After being stirred overnight at 90 °C, the mixture was poured into water (250 mL), extracted with chloroform, and the organic layer was washed with water and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (DCM as an eluent).
9-Octyl-3-formylcarbazole (1A). Yield: 2.46 g, 45%; Rf: 0.69; mp = 47–50 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 10.10 (s, 1H, CHO), 8.61 (s, 1H, Ar–H), 8.16 (d, 1H, Ar–H), 8.01 (d, 1H, Ar–H), 7.53 (t, 1H, Ar–H), 7.48 (t, 2H, Ar–H), 7.32 (t, 1H, Ar–H), 4.33 (t, 2H, NCH2C), 1.88 (t, 2H, CCH2C), 1.24–1.35 (m, 10H, C(CH2)5C), 0.85 (t, 3H, CH3). FT-IR (KBr, cm−1): 3046, 2920, 2847, 2802, and 2709 (C–H of CHO), 1690 (CO), 1625, 1592, 1566, 1497. MALDI-TOF MS m/z: 309.8 [M+H]+.
9-Octyl-3,6-diformylcarbazole (2A). Yield: 1.34 g, 22%; Rf: 0.35; mp = 126–129 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 10.14 (s, 2H, CHO), 8.68 (s, 2H, Ar–H), 8.10 (d, 2H, Ar–H), 7.56 (d, 2H, Ar–H), 4.39 (t, 2H, NCH2C), 1.92 (m, 2H, CCH2C), 1.24–1.55 (m, 10H, C(CH2)5C), 0.86 (t, 3H, CH3). FT-IR (KBr, cm−1): 3047, 2920, 2847, 2803, and 2722 (C–H of CHO), 1685 (CO), 1626, 1592, 1566, 1497. MALDI-TOF MS m/z: 337.1 [M+H]+.
9-Dodecyl-3-formylcarbazole (1B) and 9-dodecyl-3,6-diformylcarbazole (2B). Compounds 1B and 2B were synthesized following the similar procedure as those of compound 1A and 2A.
9-Dodecyl-3-formylcarbazole (1B). Yield: 2.73 g, 50%; Rf: 0.7; mp = 68–71 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 10.10 (s, 1H, CHO), 8.62 (s, 1H, Ar–H), 8.16 (d, 1H, Ar–H), 8.00 (d, 1H, Ar–H), 7.53 (t, 1H, Ar–H), 7.47 (t, 2H, Ar–H), 7.33 (t, 1H, Ar–H), 4.34 (t, 2H, NCH2C), 1.89 (t, 2H, CCH2C), 1.23–1.30 (m, 18H, C(CH2)9C), 0.87 (t, 3H, CH3). FT-IR (KBr, cm−1): 3049, 2918, 2847, 2802, and 2711 (C–H of CHO), 1686 (CO), 1625, 1593, 1568, 1465, 1383, 1349, 1288, 1250, 1229, 1205, 1126. MALDI-TOF MS m/z: 363.6 [M+H]+.
9-Dodecyl-3,6-diformylcarbazole (2B). Yield: 1.46 g, 25%; Rf: 0.37; mp = 122–125 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 10.14 (s, 2H, CHO), 8.68 (s, 2H, Ar–H), 8.09 (d, 2H, Ar–H), 7.56 (d, 2H, Ar–H), 4.39 (t, 2H, NCH2C), 1.92 (m, 2H, CCH2C), 1.23–1.34 (m, 18H, C(CH2)9C), 0.87 (t, 3H, CH3). FT-IR (KBr, cm−1): 3050, 2918, 2847, 2803, and 2722 (C–H of CHO), 1691 (CO), 1625, 1593, 1568, 1465, 1383, 1349, 1288, 1250, 1229, 1202, 1125. MALDI-TOF MS m/z: 392.2 [M+H]+.
9-Octyl-3[2-(4-dodecyloxybenzoylaminophenyl)-2-cyano-ethenyl]carbazole (CN-ODEC1). 2-(4-(4-Dodecyloxybenzoylamino)phenyl)-1-acetonitrile 4 (0.68 g, 1.63 mmol), 1A (0.5 g, 1.63 mmol), and NaOH (25 mg) were dissolved and gently refluxed in a mixture of ethanol/THF (v/v = 1/5, 25 mL) for 12 h. After cooling to room temperature, the mixture was poured into water (250 mL) and the solid was collected by filtration. After being purified on silica gel column chromatography (DCM), a yellow solid was obtained (0.77 g, 67%); Rf: 0.6; mp = 135.0–138.0 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.64 (s, 1H, Ar–H), 8.14 (t, 2H, Ar–H), 7.86 (d, 2H, Ar–H), 7.81 (s, 1H, N–H), 7.70–7.74 (d+s, 5H, Ar–H and vinyl-H), 7.45–7.51 (d, 4H, Ar–H), 6.99 (d, 2H, Ar–H), 4.33 (t, 2H, CH2N), 4.03 (t, 2H, CH2O), 1.82–1.90 (d, 4H, CH2), 1.27 (m, 28H, CH2), 0.87–0.88 (t, 6H, CH3). FT-IR (KBr, cm−1): 3326, 2918, 2849, 2211, 1643, 1608, 1517, 1504, 1470, 1438, 1413, 1331, 1312, 1276, 1260, 1243, 1181. MALDI-TOF MS m/z: 710.8 [M+H]+.
9-Octyl-3,6-di[2-(4-dodecyloxybenzoylaminophenyl)-2-cyano-ethenyl]carbazole (CN-ODEC2). The synthetic method of CN-ODEC2 was similar to that of CN-ODEC1 using 2-(4-(4-dodecyloxybenzoylamino)phenyl)-1-acetonitrile 4 (0.68 g, 1.63 mmol), 2A (0.12 g, 0.357 mmol), and NaOH (25 mg) as reagents. After being purified on silica gel column chromatography (DCM/THF, v/v = 25/1), a yellow solid was obtained (0.16 g, 40%) Rf: 0.49; mp > 200.0 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.61 (s, 2H, Ar–H), 8.52 (s, 1H, Ar–H), 8.31 (s, 1H, Ar–H), 8.22 (d, 2H, Ar–H), 8.00 (s, 1H, N–H), 7.90 (d, 7H, Ar–H + N–H), 7.76 (d, 8H, Ar–H), 7.56 and 7.49 (s, 2H, vinyl-H), 7.01 (t, 2H, Ar–H), 4.34 (t, 2H, CH2N), 4.06 (t, 4H, CH2O), 1.86 (t, 6H, CH2), 1.30 (m, 46H, CH2), 0.91 (t, 9H, CH3). FT-IR (KBr, cm−1): 3432, 2919, 2850, 2211, 1655, 1605, 1516, 1504, 1467, 1325, 1308, 1242, 1172. MALDI-TOF MS m/z: 1142.9 [M+H]+.
9-Dodecyl-3[2-(4-dodecyloxybenzoylaminophenyl)-2-cyano-ethenyl]carbazole (CN-DDEC1). The synthetic method of CN-DDEC1 was similar to that of CN-ODEC1 using 2-(4-(4-dodecyloxybenzoylamino)phenyl)-1-acetonitrile 4 (0.68 g, 1.63 mmol), 1B (0.5 g, 1.63 mmol), and NaOH (25 mg) as reagents. Yield: 0.90 g, 68% Rf: 0.61; mp = 118.0–121.0 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.64 (s, 1H, Ar–H), 8.15 (t, 2H, Ar–H), 7.86 (d, 2H, Ar–H), 7.82 (s, 1H, N–H), 7.70–7.74 (d+s, 5H, Ar–H and vinyl-H), 7.45–7.51 (d, 4H, Ar–H), 6.99 (d, 2H, Ar–H), 4.33 (t, 2H, CH2N), 4.03 (t, 2H, CH2O), 1.82–1.89 (d, 4H, CH2), 1.27 (m, 36H, CH2), 0.88 (t, 6H, CH3). FT-IR (KBr, cm−1): 3349, 2921, 2847, 2214, 1655, 1605, 1514, 1467, 1408, 1413, 1324, 1245, 1174. MALDI-TOF MS m/z: 766.7 [M+H]+.
9-Dodecyl-3,6-di[2-(4-dodecyloxybenzoylaminophenyl)-2-cyano-ethenyl]carbazole (CN-DDEC2). The synthetic method of CN-DDEC2 was similar to that of CN-ODEC1 using 2-(4-(4-dodecyloxybenzoylamino)phenyl)-1-acetonitrile 4 (0.68 g, 1.63 mmol), 2B (0.12 g, 0.357 mmol), and NaOH (25 mg) as reagents. Yield: 0.2 g, 47% Rf: 0.52; mp > 200 °C. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.65 (s, 2H, Ar–H), 8.52 (s, 1H, Ar–H), 8.31 (s, 1H, Ar–H), 8.22 (d, 2H, Ar–H), 8.00 (s, 1H, N–H), 7.89 (d, 7H, Ar–H + N–H), 7.76 (d, 8H, Ar–H), 7.50 and 7.56 (s, 2H, vinyl-H), 7.01 (t, 2H, Ar–H), 4.37 (t, 2H, CH2N), 4.06 (t, 4H, CH2O), 1.86 (t, 6H, CH2), 1.30 (m, 54H, CH2), 0.91 (t, 9H, CH3). FT-IR (KBr, cm−1): 3349 (N–H), 2925, 2850, 2214 (–CN), 1655 (CO), 1607, 1514, 1467, 1408, 1324, 1244, 1174. MALDI-TOF MS m/z: 1197.9 [M + H]+.
Acknowledgments
This work was supported by the Ministry of Science and Technology of China (2013CB834505), the National Natural Science Foundation of China (21374041, 20901081, 51073068, 51172245, and 91127005), Postdoctoral Program for African Researchers of China–Africa Science and Technology Partnership Program (CASTEP), and the 100 Talents Program of the Chinese Academy of Sciences.