

1 Introduction
Supported rhodium(0) nanoparticles efficiently catalyze the hydrogenation of aromatic rings under rather soft temperature and pressure conditions [1]. Main applications include, for example, obtaining cyclohexane from benzene, the decrease in the fraction of aromatic compounds in gasolines, or the one-step preparation of substituted cyclohexane derivatives with stereoselectivity challenges. However, rhodium is particularly expensive. Efforts must be made to minimize the metal loading on the catalyst supports. Porous carbonaceous and oxide materials with large specific surfaces are privileged as well as protocols incorporating a large number of active atoms. Usually, strategies implemented to increase the dispersion of rhodium nanoparticles rely on the use of controlled reduction processes after the deposition of the metal precursor or before (colloidal approach [2–5]). In the first approach, better dispersions are more easily obtained for carriers bearing either intrinsic [6] or added [7–13] anchoring sites.
As far as mesostructured supports are concerned, the introduction of the metal source in the synthesis gel has also been implemented using either inorganic salts [6,14,15] or metal soaps [16]. Often, structural and textural parameters of the resulting solids are seriously degraded. For materials of the MCM-41 type, another strategy affording supports with structural and textural parameters of high quality as well as the incorporation of significant amounts of active phases uses silicate species (I−) on the internal surface of the preformed as-synthesized solid as anchoring sites. Indeed, the metal cationic precursor is expected to exchange with the template (cetyltrimethylammonium ion (CTA+) denoted as S+) leading to the metal uptake and also to a cleaner removal of organics as compared to calcination protocols. Such a pathway was used for the first time to prepare Mn–MCM-41 [17]. Since this so-called template-ion exchange (TIE) method has been extended to prepare MCM-41 type materials incorporating Cr, Fe, Co, V, Ti, Ni, and so forth [18–38]. Usually the resulting solids are calcined and the heteroelements introduced are tested in oxidation or acid catalysis tests. Fewer studies deal with the use of this strategy to get zerovalent metal particles and locate them within the mesopores [34,35].
In the present article, rhodium(0) nanoparticles useful for the hydrogenation of aromatic substrates under mild conditions were prepared by this “TIE” approach with or without any additional reducing agent. Parameters influencing the amount and location of Rh(0) in the final materials were studied. The structural and textural properties of the resulting rhodium-containing mesoporous silicas, the size and the dispersion of the metal nanoparticles, and their catalytic performances were compared to those of Rh0–MCM-41 prepared through the incorporation of Rh(III) ions in the synthesis gel of MCM-41 [6].
2 Experimental section
2.1 Synthesis of Rh0–MCM-41 materials
2.1.1 From the as-synthesized MCM-41 and ethanolic rhodium solutions
Pure silica MCM-41 (MCM-41) was synthesized according to the method reported by Brühwiler and Frei [39] using cetyltrimethylammonium bromide (CTAB) as a templating agent and tetraethoxysilane (TEOS) as a silicon source. Amounts of silicon, carbon, and nitrogen in the as-synthesized MCM-41 (as-MCM-41), that is, 29.6, 21.6, and 1.3 wt %, respectively (elemental analysis) are consistent with the presence of CTAB (C/N = 19.4 instead of 19) as the main organic component. In the present article, the amount of template (CTA+) in the as-MCM-41 is considered to be ca. 26 wt %. Full extraction of CTA+ was also performed for blank experiments using repeated extractions of the as-MCM-41 in the presence of NH4NO3 aqueous solutions [12,40]. The corresponding surfactant-free support was denoted as ext-MCM-41.
Targeted materials Rh0–MCM-41/t(y), named Rh0–M/t(y), were prepared in ethanol using RhCl3·xH2O (Rh = 40.79 wt % corresponding to x = 2.25; Strem chemicals) as the rhodium source. The influence of the contact time (t) between as-MCM-41 and RhCl3·xH2O and of the nominal Si/Rh molar ratio (y) was first tested. Hence, as-MCM-41 (1.0 g) was added to 40 mL of an ethanolic solution of rhodium(III) chloride. The resulting mixture was stirred vigorously at 60 °C for t = 3, 6, or 24 h and y value was varied from 135 to 35. Resulting RhIII–M/t(y) solids were recovered through a filtration step, then washed with distilled water, dried at 40 °C for 24 h, and calcined in air, first at 300 °C for 2 h and then at 550 °C for 12 h (heating rate, 2 °C min−1). For the sake of comparison, two blank materials, M/6(∞) (y = ∞, t = 6 h) and M/24(∞) (y = ∞, t = 24 h), were also prepared in the absence of rhodium.
The corresponding reduced materials of the first series, Rh0–M/t(y), were obtained through a dihydrogen treatment. In a typical experiment [6], the RhIII–M/t(y) solid was preheated under oxygen flow (1.4 mL s−1) from 25 to 300 °C (ramping rate, 60 °C h−1), then 2 h under vacuum at 300 °C to obtain RhOx species. After cooling, H2 gas (0.83 mL s−1) was passed through the sample while heating from 25 to 220 °C (ramping rate, 12 °C h−1). The temperature was maintained at 220 °C for 1 h and hydrogen gas was then evacuated. Finally, the sample was further heated from 220 to 400 °C (ramping rate, 60 °C h−1) and kept at 400 °C for 12 h before cooling to room temperature under vacuum.
Three other samples were obtained through the reduction of rhodium using boiling ethanol. Among them, Rh0–M/6(Et,78,2)/- and Rh0–M/6(Et,78,2)/c materials were prepared by a 2-h treatment of the intermediate solid, here RhIII–M/6(70), either by increasing the temperature from 60 to 78 °C (case of Rh0–M/6(Et,78,2)/-) or by treating all the isolated calcined form of RhIII–M/6(70) (corresponding to 1 g of as-MCM-41) by 40 mL of ethanol at 78 °C (case of Rh0–M/6(Et,78,2)/c). The third material, Rh0–M′/6(Et,78,6), was prepared as Rh0–M/6(70) but using ethanol at 78 °C instead of 60 °C for 6 h. Conditions for the preparation of all the solids described in this study are summarized in Table 1.
Rh-M/t(y) samples prepared from as-synthesized MCM-41 and ethanolic solution.
| Samples | Sample preparation method | Reduction treatment | Nominal Si/Rh molar ratio | Contact time t (h) |
| Rh0–M/6(70) | TIE | H2 | 70 | 6 |
| Rh0–M/24(70) | TIE | H2 | 70 | 24 |
| Rh0–M/6(130) | TIE | H2 | 130 | 6 |
| Rh0–M/6(35) | TIE | H2 | 35 | 6 |
| Rh0–M(I)/120 | DHT [6] | H2 | 70 | – |
| Rh0–M(Et,78,2)/- | TIE | EtOH | 70 | 6 (60 °C) No isolation +2 (78 °C) |
| Rh0–M/6(Et,78,2)/c | TIE | EtOH | 70 | 6 (60 °C) Isolation, calcination +2 (78 °C) |
| Rh0–M′/6(Et,78,6) | TIE | EtOH | 70 | 6 (78 °C) |
2.1.2 By a direct hydrothermal synthesis method
The reference material obtained by direct hydrothermal synthesis, Rh0–M(I)/120, was prepared as described previously [6]. Hence, CTAB (2.2 g) was dissolved under slight warming (35 °C) in a mixture of 52 mL of distilled H2O and 24 mL of 30 wt % aqueous ammonia. Rhodium chloride hydrate was introduced into the solution (6 × 10−4 mol), which was stirred for 15 min until it became clear. Then, 10 mL of TEOS was slowly added. The resulting gel was aged for 3 h at room temperature, then transferred into a Fluorinated Ethylene Polymer (FEP) bottle and was hydrothermally treated at 110 °C for 48 h. The solid product was filtered, washed with distilled water, dried at 60 °C for 24 h, calcined in air, first at 300 °C for 2 h and then at 550 °C for 12 h (heating rate was 2 °C min−1), and reduced by H2 using the procedure described in Section 2.1.1.
2.2 Characterization
The silicon, carbon, nitrogen, and rhodium compositions of the various materials were determined by Inductively Coupled Plasma - Atomic Emission Spectroscopy (ICPAES) in the “Centre national de la recherche scientifique” (CNRS) at Vernaison (France). Fourier transform infrared (FTIR) spectra were collected using a Bruker Vector 22 spectrophotometer (resolution of 2 cm−1) with KBr pellets (4 wt % in KBr). CTA+ were quantified in the different uncalcined samples (RhIII–M/t(y), M/6(∞), and M/24(∞) solids) by monitoring the area of ν(CH) bands at 2854 and 2925 cm−1 normalized against that of the ν(SiOSi) of the support at 800 cm−1. Measurements performed on the materials before (as-MCM-41) and after the incorporation of rhodium were used to estimate the loss of surfactant (ΔS). The quantity (in moles) of surfactant removed per mole of rhodium incorporated (ΔS/Rh) was calculated by coupling IR data (ΔS) and elemental analyses of as-MCM-41 and Rh0–M/t(y) samples. X-ray diffraction (XRD) patterns were recorded between 1° and 7° (2θ) using a Bruker Advance D8 diffractometer with the Cu Kα radiation (λ = 0.15418 nm) with steps of 0.02° and a count time of 6 s at each point.
The specific surface area, pore volume, and pore diameter of calcined samples were determined from N2 sorption data obtained at −196 °C using an ASAP 2010 Micromeritics instrument. Before the analysis, the samples were outgassed at 200 °C until a stable static vacuum of 2.10−3 Torr was reached. The pore diameter and specific pore volume were calculated according to the Barrett–Joyner–Halenda (BJH) model. The specific surface area was obtained by using the Brunauer-Emmett-Teller (BET) equation. Transmission electron analyses were performed using a JEOL 100CXII transmission electron microscope operated at an acceleration voltage of 100 kV. In some cases, the materials were included in a resin (AGAR 100) and heated for 48 h at 60 °C, then thin (70 nm) sections cut with a Leica microtome (Ultracut UCT) were collected on carbon-coated copper grids.
2.3 Catalysis tests
Rh-containing samples were tested as catalysts in the hydrogenation of aromatic substrates at room temperature under atmospheric pressure (styrene) or at 1 MPa (disubstituted arene derivatives) of dihydrogen. Solids were first dispersed in hexane (10 mL) under magnetic stirring for 15 min before the introduction of the substrate (100 equiv metal−1). Products were quantified with n-decane as an internal standard using a Delsi Nermag DN 200 gas chromatograph equipped with an SPB-5 capillary column and a flame ionization detector.
3 Results and discussion
As-MCM-41 used as the starting material was estimated to contain 9 × 10−4 mol g−1 of CTA+ (see Section 2) with thus a maximum exchange capacity toward Rh3+ of 3 × 10−4 mol g−1. Under these conditions, a minimum Si/Rh molar ratio expected was estimated to be 35.
3.1 Variations in the contact time (t) between as-MCM-41 and RhCl3·xH2O and in Si/Rh molar ratio (y)
First, three materials with nominal Si/Rh values (y) equal to 35, 70, and 130 were prepared by contacting as-MCM-41 and RhCl3 6 h in ethanol at 60 °C (see RhIII–M/6(y) solids in Table 2). Resulting solids were characterized in their Rh(III) form (FTIR) and at the zerovalent state, that is, after calcination and H2 treatment (XRD, N2 sorption, and transmission electron microscopy [TEM]). For the discussion, they have also been compared with Rh0–M/24(70) resulting from a longer contact time (24 h) and two blank samples, M/6(∞) and M/24(∞), obtained in the absence of rhodium.
Physicochemical properties of Rh0–M/t(y) samples synthesized in ethanol.
| Samples | Si/Rha | Rh (wt %) | Yieldb (%) | ΔS/Rhc | SBET (m2 g−1) | Vpores (cm3 g−1) | Dp (nm) |
| Si-MCM-41 | – | – | – | – | 800 | 0.70 | 2.6 |
| Rh0–M/6(70) | 113 | 1.35 | 60 | 7 | 775 | 0.69 | 2.7 |
| Rh0–M/24(70) | 136 | 1.11 | 50 | 6 | 740 | 0.66 | 2.6 |
| Rh0–M/6(130) | 444 | 0.34 | 30 | 17 | 745 | 0.66 | 2.6 |
| Rh0–M/6(35) | 123 | 1.30 | 28 | 8 | 780 | 0.71 | 2.7 |
| Rh0–M/6(70/70)d | 100 | 1.55 | 35 | 7.5 | 770 | 0.68 | 2.7 |
| Rh0–M(I)/120 | 120 | 1.30 | 58 | – | 720 | 0.63 | 2.7 |
a Si/Rh experimental molar ratio.
b Rh incorporation yield = [(Si/Rh nominal)/(Si/Rh exp)] × 100.
c Mole of surfactant removed per mole of metal introduced.
d As-MCM-41 was contacted twice for 6 h with an ethanolic solution of RhCl3 (y = 70).
The evolution of the areas of the ν(CH) bands of CTA in as-MCM-41 and uncalcined RhIII–M/t(y), MCM-41/6, and MCM-41/24 materials is presented in Fig. 1. From comparisons with as-synthesized silica MCM-41, it is clear that no significant release of CTA+ was observed in the absence of rhodium (case of M/6(∞) and M/24(∞), Fig. 1(b) and (c)). In fact, loss of CTA+ over a period of 24 h (MCM-41/24) was less than 7 wt %. In the presence of rhodium, with RhIII–M/6(130) and RhIII–M/6(70) samples (Fig. 1(d) and (e)), the normalized area of the ν(CH) bands decreased with an increase in the nominal amount of metal. However, additional quantities of rhodium (RhIII–M/6(35)) did not lead to further surfactant removal (Fig. 1(g)). No significant difference between rhodium loadings (about 1.3 wt %) was also found for Rh0–M/6(70) and Rh0–M/6(35) samples, thus assessing a relationship between CTA+ release and Rh3+ insertion. Regardless of the amount of rhodium, values of Rh loadings were lower than nominal ones. Incorporation yields of the metal vary between 30% and 60% (Table 2) with the maximum reached for Si/Rh = 70. Surfactant losses versus rhodium incorporation (expressed as ΔS/Rh values) in RhIII–M/6(y) samples were always superior to 3, the value expected for a true ion exchange between one Rh3+ and three CTA+. Indeed ΔS/Rh values varied from 17 for the smallest nominal metal loading (RhIII–M/6(130)) to 7.5 for highest ones (RhIII–M/6(35)). For y = 70, a longer contact time was not helpful because of affording a lower rhodium incorporation yield (50% for RhIII–M/24(70)) as compared to 60% for RhIII–M/6(70). These results are in agreement with the existence of a nonhomogeneous population of CTA+ in as-MCM-41, most of them being released with the help of rhodium ions and, part of them, being unavailable for exchanges, may be due to stronger interactions with native silica.
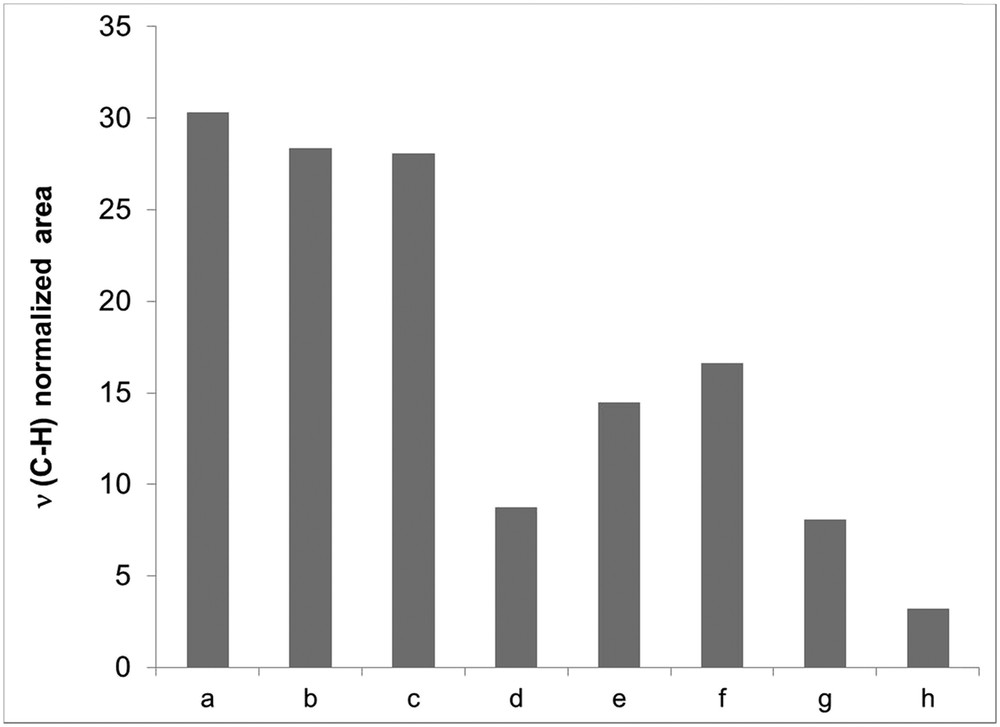
Normalized ν(CH) area for the as-synthesized (a) MCM-41, (b) M/6(∞), (c) M/24(∞), (d) RhIII–M/6(130), (e) RhIII–M/6(70), (f) RhIII–M/24(70), (g) RhIII–M/6(35), and (h) RhIII–M/6(70/70).
Another sample, RhIII–M/6(70/70), was prepared according to two consecutive exchange steps using Si/Rh = 70 in each one. Such synthesis strategy corresponding to a nominal molar Si/Rh ratio of 35 was tested to increase the incorporation yield of rhodium as compared to that of RhIII–M/6(35). From FTIR measurements, residual surfactant molecules were still removed from the intermediate solid, RhIII–M/6(70), in the second step accompanied by some incorporation of rhodium. However, similar ΔS/Rh values were obtained for Rh0–M/6(70/70) and Rh0–M/6(35) (7.5 and 8, respectively) and, globally, the rhodium incorporation yield in Rh0–M/6(70/70) was only slightly higher than in Rh0–M/6(35) (35% vs 28%), which remains very low. Again, it seems that one third of CTA+ molecules embedded in as-MCM-41 is very tightly bound to the surface.
To emphasize the importance of the surfactant molecule in rhodium insertion, two blank experiments were carried out with Rh(III) (nominal Si/Rh = 70) in ethanol at 60 °C using a surfactant-free support resulting from the pre-extraction of CTA+, that is, ext-MCM-41 instead of as-MCM-41. Clearly, in the absence of CTA+, no rhodium was incorporated after 6 h. However, addition of CTAB (0.26 g) to a mixture of ext-Si-MCM-41 (1 g) and Rh(III) in ethanol at 60 °C gave rise to a metal uptake (1.3 wt %) similar to that obtained for the Rh0–M/6(70) sample. Rhodium apparently needs CTAB help to be incorporated in resulting RhIIIM(6)/y materials.
With three well-resolved diffraction peaks, XRD patterns of all Rh0–M/6(y) samples were consistent with the (100), (110), and (200) lattice planes of the expected hexagonal structure of MCM-41 (Fig. 2). Increasing amounts of Rh seem to lead to the decrease in the intensity of the (100) diffraction peak without modifying tremendously its half-width value meaning that the grain size of the support is maintained. However, values of the structural parameters of Rh0–M/6(35) would be slightly lower than those of calcined MCM-41 and other Rh0–M/6(y) samples as evidenced by the shift in the (100) diffraction peak of Rh0–M/6(35) toward higher values of 2θ. The half-width of the (100) peak of Rh0–M(I)/120 prepared by a direct hydrothermal treatment is a little bit larger than others.
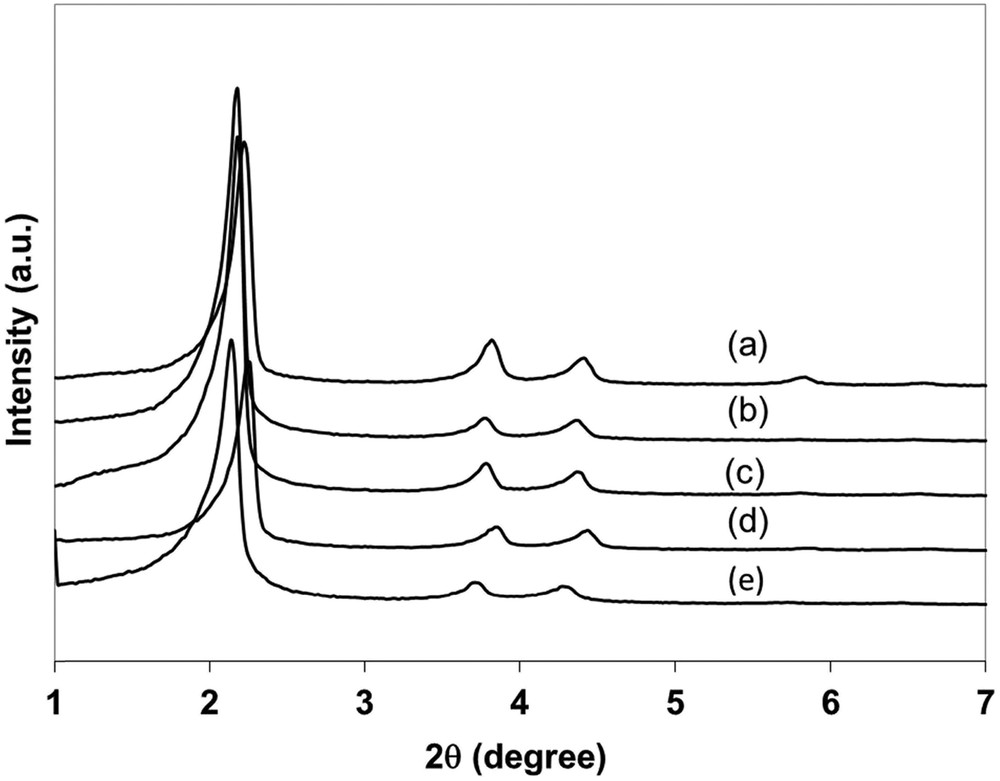
XRD patterns of (a) calcined MCM-41, (b) Rh0–M/6(130), (c) Rh0–M/6(70), (d) Rh0–M/6(35), and (e) Rh0–M(I)/120 samples.
Nitrogen adsorption–desorption isotherms (not shown here) of the different reduced solids prepared with various Rh contents (from 0.34 to 1.4 wt % Rh) were also characteristic of materials of the MCM-41 type. Textural properties listed in Table 2 tend to show that Rh(0)-containing materials have specific surface area, pore volume, and average pore diameter values slightly inferior to those of calcined MCM-41. Textural parameters of Rh(0)-containing materials could not help to conclude about the location of the nanoparticles due to the relatively low amount of metal loaded on the support.
TEM analysis of the most interesting sample in terms of Rh incorporation yield, that is, Rh0–M/6(70), confirmed the well-ordered pore structure with the presence of parallel channels (longitudinal view) (Fig. 3). More importantly, most of the observed nanoparticles were characterized by a small average diameter (2–3 nm) and appeared to be spatially distributed in the direction of the channels, which is strongly consistent with their inclusion in the mesopores [41]. Nevertheless larger Rh nanoparticles (6–7 nm) could also be observed on the external surface.

TEM images of (a) Rh0–M/6(70), (b) Rh0–M/6(Et,78,2)/-, (c) Rh0–M/6(Et,78,2)/c, and (d) Rh0–M′/6(Et,78,6) samples.
3.2 Reduction by ethanol
Alcohols with high boiling points like glycols are well-known reducing agents used for the preparation of Rh(0) colloidal dispersions in ranges of temperatures between 100 and 150 °C [42]. We have checked that no Rh(0) formation was observed in EtOH in the aforementioned samples prepared at 60 °C (RhIII–M/t(y) samples). In fact, reported uses of ethanol in the literature correspond to reflux temperature, 78 °C [43]. It was then tempting to look in this work at ethanol as both a solvent and a reducing agent under more appropriate conditions. Three sets of experimental conditions were tested with a nominal molar ratio Si/Rh = 70 (Table 3).
Physicochemical properties of Rh0–M/6 samples reduced by ethanol.
| Samples | Treatment | Rh (wt %) | Si/Rh exp. | SBET (m2 g−1) | Vpores (cm3 g−1) | Dp (nm) | ||
| Step 1 | Calcination | Step 2 | ||||||
| Si-MCM-41 | – | – | – | 800 | 0.70 | 2.6 | ||
| Rh0–M/6(Et,78,2)/- | 60 °C 6 h | No | 78 °C 2 h | 0.94 | 130 | 687 | 0.48 | 2.1 |
| Rh0–M/6(Et,78,2)/c | 60 °C 6 h | Yes | 78 °C 2 h | 0.72 | 179 | 861 | 0.75 | 2.7 |
| Rh0–M′/6(Et,78,6) | 78 °C 6 h | No | – | 1.02 | 123 | 591 | 0.45 | 2.2 |
Rh3+ ions were either reduced
- • in a second step (78 °C, 2 h) with or without isolation and calcination of the intermediate solid (RhIII–M/6(70)) affording Rh0–M/6(Et,78,2)/c or Rh0–M(Et,78,2)/- samples, respectively, or
- • simultaneously (78 °C, 8 h) with its introduction onto the support (Rh0–M′/6(Et,78,6) sample).
As expected from the study of the influence of the contact time (Table 2), longer treatments of the materials by ethanol (8 h in all instead of 6 h: Rh0–M/6(Et,78,2)/c and Rh0–M(Et,78,2)/-) lead to a decrease in the Rh uptake as compared to that of Rh0–M/6(70) obtained after H2 reduction. Lower values of the textural parameters of uncalcined samples (Rh0–M/6(Et,78,2)/- and Rh0–M′/6(Et,78,6)) as compared to those of Rh0–M/6(Et,78,2)/c and Rh0–M/6(70) solids are due to the presence of residual amounts of the template not removed by a calcination step.
TEM images of Rh0–M(Et,78,2)/- showed a very poor dispersion of rhodium particles. About 50–100 nm metal aggregates were formed on the external surface (Fig. 3(b)), which means that Rh precursors linked to the surface are quite mobile. Such a strong reorganization was not observed after isolation and calcination of the intermediate solid before its reduction by dihydrogen (Rh0–M/6(70), Fig. 3(a)) or ethanol (Rh0–M(Et,78,2)/c, Fig. 3(c)) or when Rh insertion and reduction were carried out simultaneously (Rh0–M′/6(Et,78,6) (Fig. 3(d)). In these latter cases, particles are smaller but observed 6–7 nm diameter values indicate that all of them are not included in the channels of the MCM-41 support.
3.3 Comparison between direct hydrothermal synthesis and TIE Procedures
Mesoporous siliceous materials with MCM-41 pore architecture with up to 1 wt % of rhodium(0) were previously prepared in our group via a template directed hydrolysis–polycondensation of TEOS in the presence of aquachlororhodium(III) complexes followed by the calcination of the template (CTAB) and the reduction of Rh(III) species by a H2 treatment. Therefore, we were interested to determine if a better inclusion of metal nanoparticles in the mesopores could be obtained via the TIE route. Such a possibility would be based on (1) the evidence for a relationship between the incorporation of Rh and the release of CTA+ from as-Si-MCM-41 and (2) the fact that the largest proportion of CTA+ in as-Si-MCM-41 is located in the mesopores. Of course, such hypothesis also implies that Rh(0) and Rh(III) locations are related together (influence of the reduction treatment).
This part is focused on the comparison of Rh0–M/6(70) (this work) and Rh0–M(I)/120 [6], that is, two samples containing 1.3 wt % of rhodium(0) issued from similar H2 reduction steps (220 °C after calcination under air flow). Both materials are characterized by similar textural parameters (Table 1) and are well structured (Fig. 4). XRD patterns of Rh0–M/6(70) are almost the same as those of pure Si-MCM-41 due to the preformation of the support in the TIE method (Fig. 2). In contrast, the lattice parameter of Rh0–M(I)/120 is greater than those of Rh0–M/6(70) and Si-MCM-41 as expected due to the addition of Rh3+ ions during the condensation of silica.

TEM images (microtomy) of the Rh0–M(I)/120 sample.
Both insertion methods of rhodium seem to have some influence on the location, dispersion, and particle size of the resulting metal nanoparticles. Hence, microtomy TEM images of Rh0–M(I)/120 showed that most of the observed nanoparticles fit with a size distribution with a mean value of 2.8 nm, which is close to the pore aperture (Fig. 4). Moreover, most of these nanoparticles were observed inside the pore channels of Rh0–M(I)/120, which was not the case for Rh0–M/6(70) obtained by TIE. Thus, using MCM-41 synthesis protocols both derived from Brühwiler and Frei [39] and using exactly the same metal precursors, reduction modes and experimental metal loadings, it was clearly demonstrated that the TIE route does not lead necessarily to a better inclusion of rhodium(0). In both cases, the mean size value of included nanoparticles was similar and ultimately depends on the size of the mesopores.
3.4 Catalytic performances of the materials
Rh(0) particles prepared onto mesoporous supports of the MCM-41 type were tested as catalysts in the hydrogenation of styrene at atmospheric hydrogen pressure. Three materials were considered, that is, Rh0–M/6(70), Rh0–M/6(Et,78,2)/c, and Rh0–M/6(Et,78,2)/-, with the aim to compare in particular the influence of the nature of the reductant on the catalytic performances. The reaction was carried out with similar substrate/Rh molar ratio (100) at room temperature in hexane. Whatever the catalyst considered, the external double bond of styrene was more easily reduced than the aromatic ring of the molecules. Ethylcyclohexane (EC) and the intermediate, ethylbenzene (EB), were the only products detected. Histograms in Fig. 5 show the yields of EB and EC versus time for the different tested materials. Styrene was converted to EB and EC in less than 3 h in the presence of Rh0–M/6(70) and Rh0–M/6(78,2)/c samples. The reaction was completed within 24 h in all cases. Quasi-quantitative yields of EC (90–100%) were obtained in less than 6 h for Rh0–M/6(70) and Rh0–M/(78,2)/c. Bad performances of Rh0–M/6(78,2)/- are linked to the presence of Rh(0) aggregates located on the external surface of the MCM-41 support.
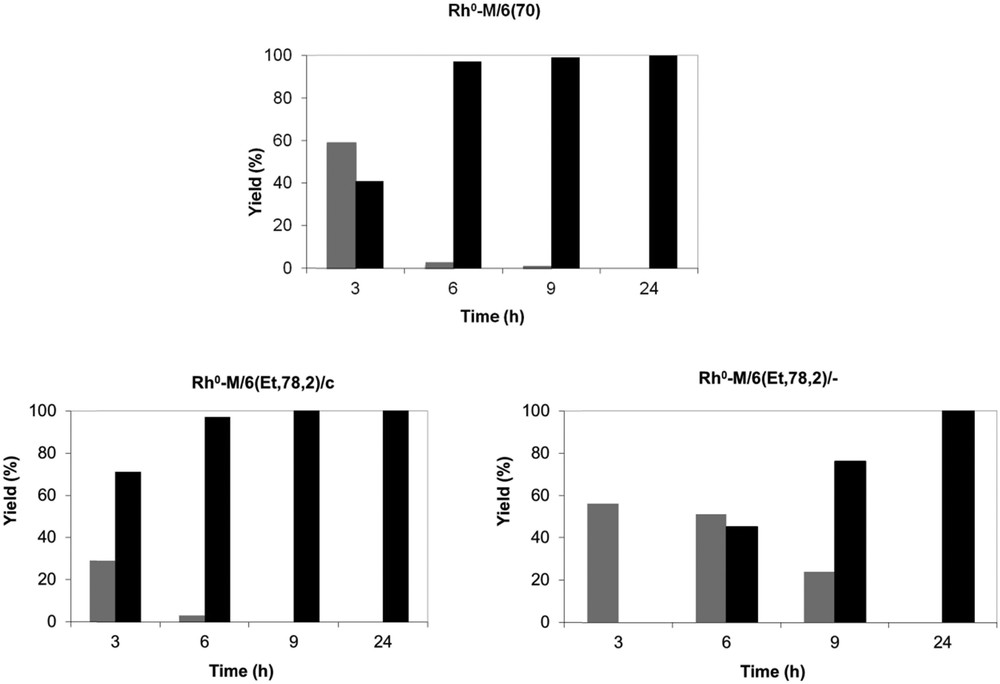
Hydrogenation of styrene catalyzed by Rh0–M samples (substrate/Rh = 100) at room temperature under atmospheric pressure of dihydrogen (EB, grey; EC, black).
Rh0–M(I)/120 and Rh0–M/6(70) samples were compared in the hydrogenation of four disubstituted aromatic substrates: m-xylene, m-cresol, 3-methylanisole, and tetraline (Table 4). Reactions were performed at room temperature under 1 MPa hydrogen pressure. In all of the cases considered, total hydrogenation of the aromatic ring was obtained in less than 2 h and led to the formation of cis and trans isomers (Table 4).
Hydrogenation of different disubstituted aromatic substrates.
| Substrate | Products | Rh0–M(I)/120 | Rh0–M/6(70) | ||||
| Yield (%) | Time (h) | cis/trans | Yield (%) | Time (h) | cis/trans | ||
| Tetraline | 100 | 1.7 | 89/11 | 100 | 1.0 | 91/09 | |
| m-Xylene | 100 | 0.5 | 80/20 | 100 | 0.5 | 85/15 | |
| m-Cresol | 100 | 2.5 | 63/37 | 100 | 1.75 | 60/40 | |
| 3-Methylanisole | 95 | 1.5 | 56/44 | 100 | 1.2 | 61/39 |
The cis compound was obtained predominantly (up to 90% in the case of decalin). For both catalysts, the cis/trans selectivity depends first on the structure of substrate molecules and follows the order: decalin >1,3-dimethylcyclohexane > 3-methylcyclohexanol > 1-methoxy-3-methylcyclohexane. With the exception of m-cresol, cis selectivity is consistently higher for tests carried out with the material prepared by the TIE method. The case of m-cresol is particular. Indeed, it is the sole compound among those tested where significant amounts of the intermediate resulting from the addition of two moles of H2 per mole of the substrate (3-methylcyclohexanone here) could be detected in the reaction medium [44]. Such observations suggest that the high stability of 3-methylcyclohexanone allows a “roll-over” type mechanism [45–47] to proceed affording a significant formation of the trans isomer of 3-methycyclohexanol.
4 Conclusions
Impregnation of the as-MCM-41 silica by ethanolic rhodium(III) chloride solutions allowed the incorporation of the metal with loadings of up to 1.5 wt % of the final calcined material. In ethanol, it was shown that the templating agent was still removed for longer contact times without any incorporation of additional quantities of Rh. Control experiments have shown, however, that the initial introduction of rhodium in alcohol requires the presence of the CTA+ cations.
The reducing action of ethanol was compared with that of dihydrogen. Clearly, whatever the reductant used, the results indicated that a strong association of Rh(III) precursors to the surface of the support through a drying and a calcination treatment of the sample before the reduction step leads to good dispersions of Rh(0). Some few nanoparticles with a size of 2–3 nm were observed, others with diameters of about 6–7 nm have also been identified for materials calcined before reduction treatments by H2 or alcohol.
From a textural point of view, there were no significant differences between the solids obtained by direct synthesis procedures and those prepared in this study. However, structural differences allowed us to distinguish the impregnation approach (lattice parameter similar to that of the pure calcined silica) from the direct synthesis one. In this latter case, partial incorporation of Rh(III) in the walls justifies the higher value of the lattice parameter. In the hydrogenation of disubstituted aromatic compounds, the best cis/trans selectivities obtained with the materials resulting from impregnation also seemed to indicate that differences exist in the environment of the particles depending on whether they were introduced by an impregnation or a direct synthesis strategy.
Acknowledgments
The authors would like to thank Professor A. Roucoux and Dr. A. Denicourt for their help to carry out hydrogenation tests in their laboratory. M. Lin is also gratefully acknowledged for complementary experiments.