

1 Introduction
Sulfoximines are very intriguing sulfur(VI) compounds and can be considered as monoaza analogues of sulfones [1a] (Fig. 1).
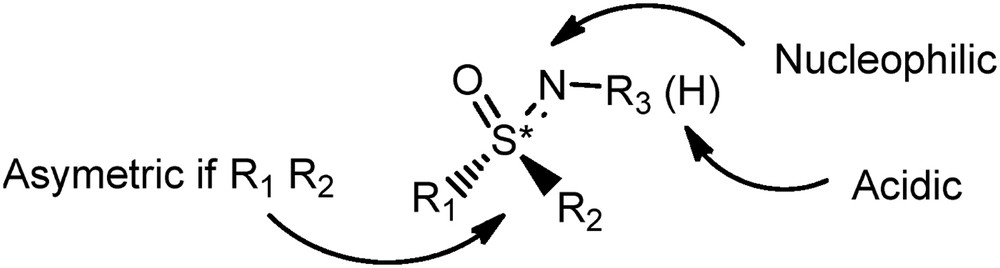
General structure of sulfoximines.
They nevertheless display much more diversity in terms of structure and reactivity than sulfones because of the multiple potential variations offered by the replacement of an oxygen by a nitrogen (Fig. 1). Two major differences may be highlighted. In the case of two different groups R1 and R2, this replacement brings the chirality to the sulfur center and also allows the introduction of a mostly unlimited number of substituents attached to the nitrogen. The variety of possible skeletons has then allowed the fine modulation of the electronic and configuration properties of these original structures, accounting for their use in various applications [1]. Numerous reactions using the chiral pool of the sulfoximines have been described in the field of asymmetric synthesis [2] and ligands for catalysis [3]. To a lesser extent, they also exhibit interesting properties for life science purposes [4] or as building blocks for the preparation of pseudopeptides [5]. Interestingly, a recent article has highlighted this particular heteroatomic group as a “neglected opportunity in life science” [6].
When one of the two groups (R1 or R2) bonded to the sulfur atom is a perfluoroalkyl chain, huge differences immediately appear whether it be for the synthesis or concerning the applications. Most of the synthetic routes reported for the preparation of nonfluorinated sulfoximines start from the corresponding thioethers or sulfoxides [7] and are not effective for the fluorinated series because of the tamed reactivity of the sulfur atom. Therefore, the synthesis of such compounds is a challenge for chemists. In 2009, a three-step methodology for the preparation of NH S-perfluoroalkylated sulfoximines was reported by our group paving the way for an extension of the structural diversity of these sulfur(VI) derivatives [8]. In the last decade, perfluoroalkylated sulfoximines were intensively studied because of their promising properties. They were described as highly electron-withdrawing substituents [9] or as building blocks for liquid crystals [10]. However, the most widespread applications involved their use as reagents for the transfer of the perfluoroalkyl group by electrophilic [11], nucleophilic [12], and very recently radical pathways [13]. In 2014, two reviews, independently written by Bolm et al. [14] and Shen and Hu et al. [15] have made a state in terms of syntheses, properties, and applications of fluorinated sulfoximines. This review will consequently focus on an overview of the new trends in this research area, since 2014. Accordingly, we will discuss the structural diversification of the skeleton of sulfoximines via N-functionalization or ortho-lithiation, their applications in catalytic and organocatalytic systems, their use as perfluoroalkylating reagents, and their potential as biologically active molecules.
2 Synthesis
2.1 N-Functionalization
We have previously demonstrated that the reactivity of the S-perfluoroalkylated sulfoximines depends on the group attached to the nitrogen atom [11f], highlighting thus the need for the development of efficient methods of N-functionalization of sulfoximines.
2.1.1 N-Functionalization of S-perfluoroalkylated sulfoximines
In 2011, our group described the copper-assisted N-arylation of perfluorinated sulfoximines [16]. However, this transformation required the reflux of toluene during several hours as reaction conditions. A slight improvement, using a microwave activation, enabled a significant reduction in the reaction time and an increase in the molecular diversity of the targeted N-arylated sulfoximines 2a–f obtained in 81–92% yield (Scheme 1) [17].
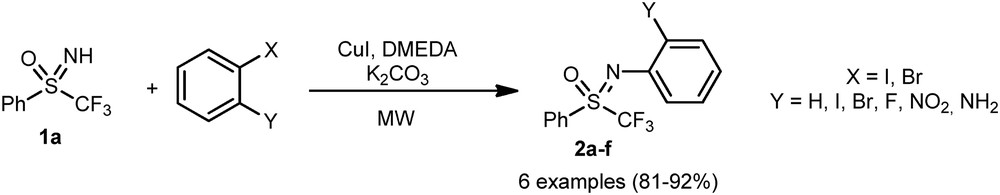
Copper-catalyzed N-arylation of sulfoximines.
The copper-catalyzed preparation of N-alkenyl and N-alkynyl sulfoximines was consequently studied by our group [18]. A wide variety of N-alkenyl trifluoromethyl sulfoximines 3a–f were synthesized in moderate to excellent yields (up to 94%) from the corresponding 1,2-disubstituted vinyl halides, with CuI as a catalyst, K2CO3 as a base, and DMEDA as a ligand (Scheme 2). This procedure was also extended to other perfluoroalkyl chains, in the presence of vinyl bromide, leading to the formation of 3g and 3h in moderate yield. The same reaction conditions, applied to bromophenylacetylene or 1-bromohexyne led to the formation of N-alkynyl sulfoximines 4a and 4b in 98% and 81% yield, respectively. The previous procedure did not succeed with phenylacetylene. Therefore, the reaction conditions were modified to perform the N-alkynylation of sulfoximines, using CuCl2 as a catalyst, pyridine as a ligand, and Na2CO3 as a base, giving rise to products 5a–l with good to excellent yields (55–96%).
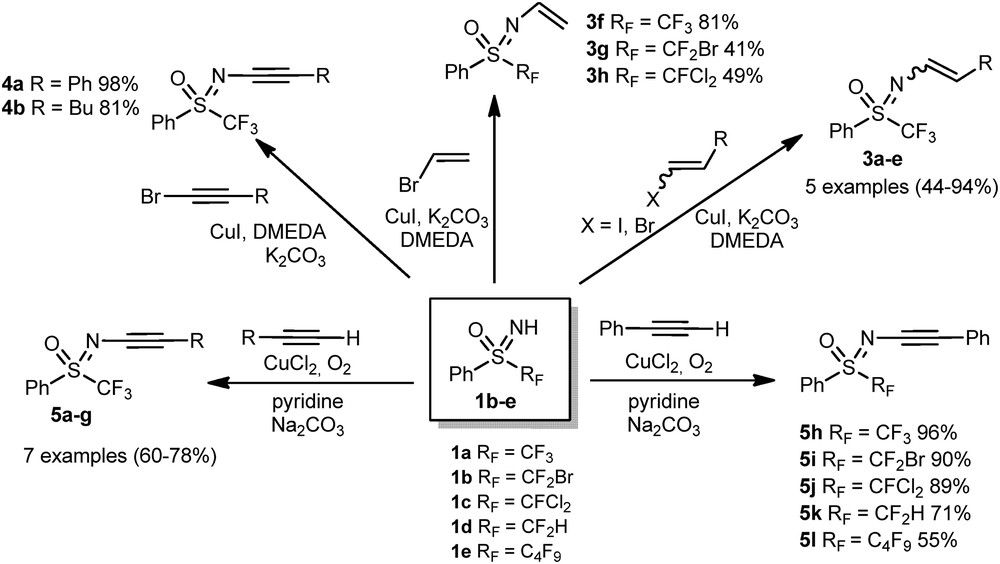
Copper-catalyzed N-alkenylation and alkynylation of sulfoximines.
We also demonstrated that the N-functionalization of sulfoximines could also occur via a nucleophilic pathway providing the use of smooth conditions (Scheme 3). The use of NaH and a catalytic amount of Bu4NBr allowed the N-alkylation of the sulfoximine 1a to give rise to compounds 6a and 6b with excellent yields. The nucleophilicity of the nitrogen atom of the sulfoximine was also underlined by the reaction between 1a and phenylisocyanate to deliver sulfoximine thioureas 7a and 7b.
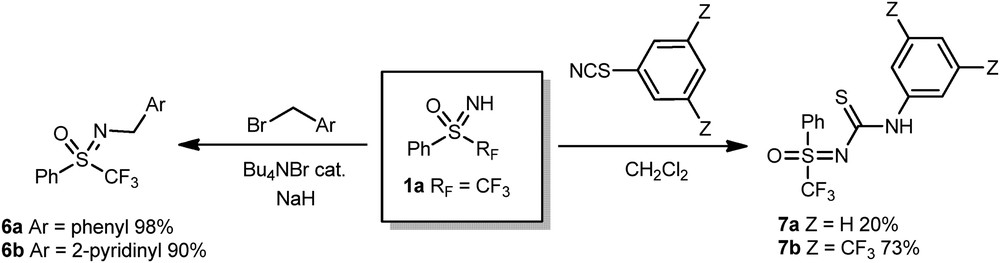
N-functionalization of sulfoximines via a nucleophilic pathway.
The presence of an iodine, bromine, or amino group in ortho-position in compounds 2 enabled a postfunctionalization (Scheme 4). Bis-sulfoximines 8a and 8b were thereby synthesized with 76% and 81% yields, respectively, starting from dibromo- or diiodobenzene and the N-arylated sulfoximine 2a. A two-step reductive amination delivered the molecule 10 in 97% yield, whereas the treatment of compound 2c with isothiocyanate allowed the preparation of sulfoximine thioureas 9a and 9b in good yields.
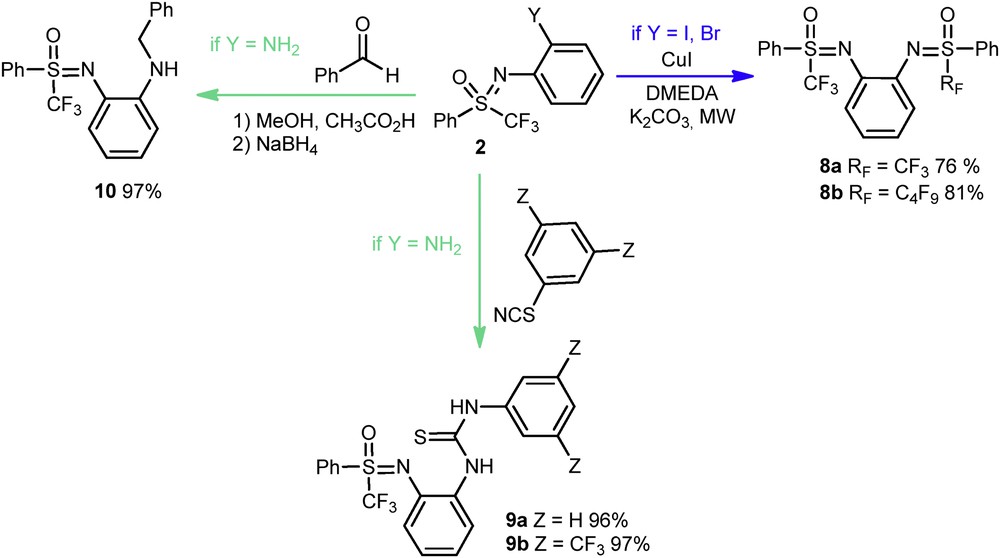
Postfunctionalization of N-substituted sulfoximines.
2.1.2 N-fluoroalkylation of sulfoximines
In 2015, Bolm et al. [19] reported that nonfluorinated sulfoximines can be successfully N-trifluoromethylated using a silver catalysis (Scheme 5). Twenty N-CF3 sulfoximines 12 were synthesized by treatment of the corresponding NH-sulfoximines 11 with Ag2CO3 as the catalyst, 1,10-phenanthroline as the ligand, and TMSCF3 as a fluorine source, with yields ranging from 33% to 85%.
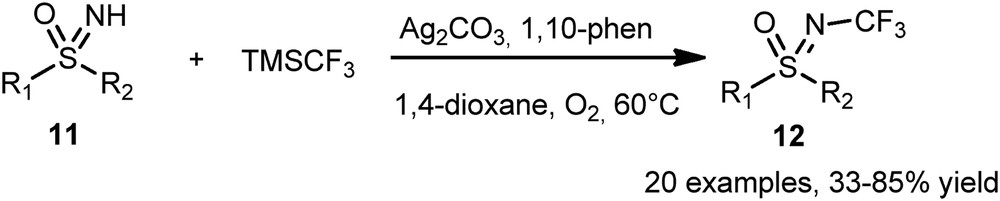
Silver-mediated N-trifluoromethylation of sulfoximines.
Because of radical scavengers inhibiting the reaction, Bolm et al. proposed a radical pathway for this transformation (Scheme 6).
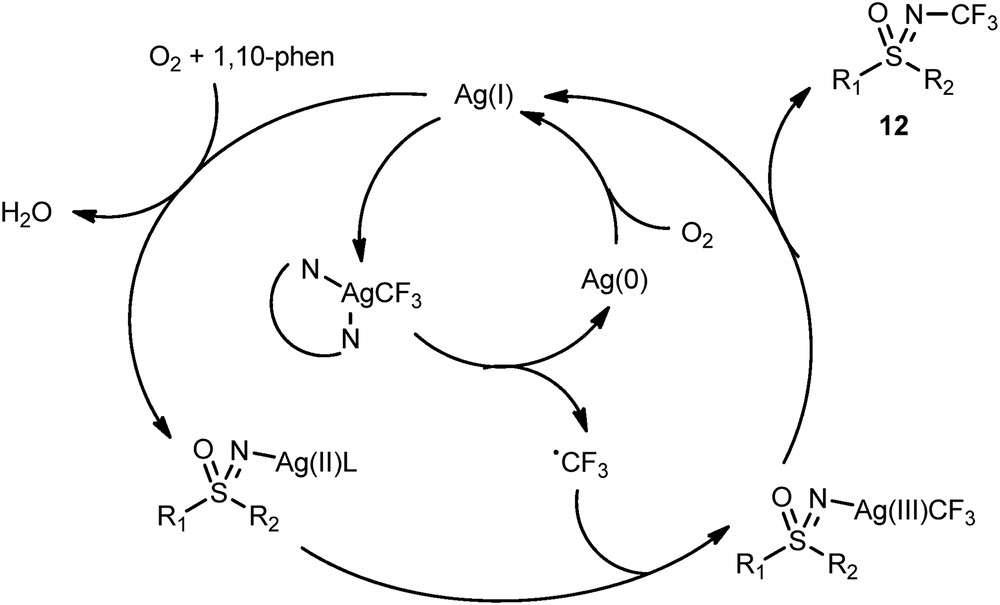
Mechanism of the N-trifluoromethylation of sulfoximines.
The same group also achieved the N-trifluoromethylthiolation starting from N-bromosulfoximines 13a–l [20]. Treated by AgSCF3 in acetonitrile, the latter gave rise to a new class of N-SCF3 sulfoximines 14a–l in good to excellent yields (51–98%) (Scheme 7).
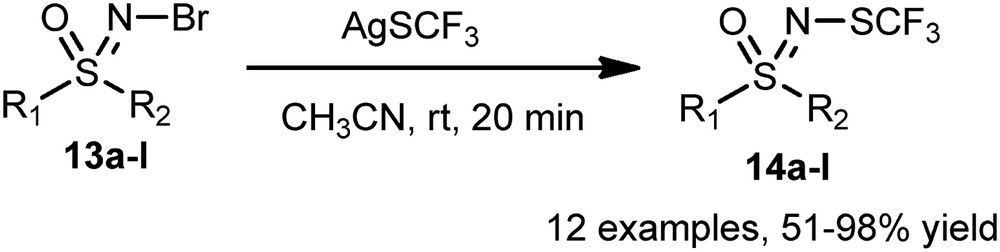
N-Trifluoromethylthiolation of nonfluorinated sulfoximines.
The specific behavior of the sulfoximine 1a was emphasized in this work. A one-pot cascade was set up to ensure the preparation of the N-SCF3 sulfoximine 14n with moderate yield (Scheme 8).

One-pot N-trifluoromethylthiolation of 1a and 13m.
2.2 Perfluorinated sulfoximines as ortho-directing metalation group
Another challenge was the late functionalization of the aromatic ring directly bonded to the sulfur atom. In 2016, our group disclosed the first use of NH S-trifluoromethyl sulfoximines as an ortho-directing group [21]. The sulfoximine 1a was treated with n-BuLi to form an ortho-lithiated intermediate, which was quenched with various electrophiles to deliver the functionalized sulfoximines 15a–m with moderate to very good yields (Scheme 9). This reaction demonstrated a broad substrate scope as it enabled the introduction of halogens (15a–c), alcohol function (15m), azido group (15h), and allyl group (15e). Sulfur derivatives were also successfully synthesized (15i–k), as well as stannylated (15f), borylated (15l), and silylated (15g) molecules. Most of the isolated compounds possess reactive functions paving the way to further functionalization of the aromatic part by cross-coupling reaction. It is noteworthy that no N-functionalization was observed.

Ortho-functionalization of 1a.
Nevertheless, thanks to the proper choice of an electrophile, this N-functionalization occurred and cyclic sulfoximines 16a–d isolated (Scheme 10).
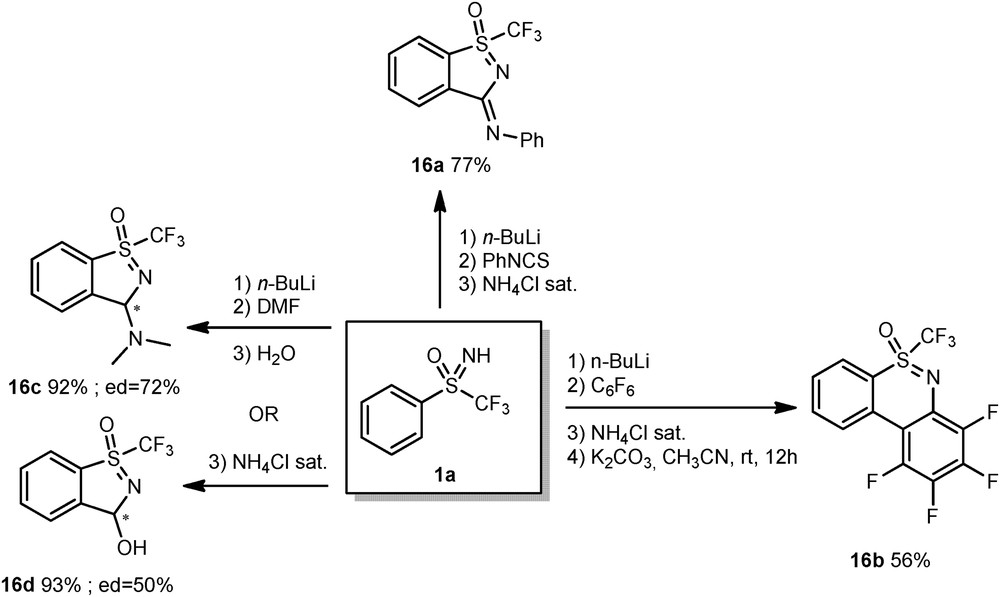
Synthesis of cyclic sulfoximines 16.
The deprotonated sulfoximine 1a reacted with phenylisocyanate to give 16a in 77% as sole diastereoisomer, the structure of which was confirmed by an X-ray analysis. When hexafluorobenzene was used as an electrophile, an additional treatment by K2CO3 was necessary to complete the full cyclization and give compound 16b in 56% yield. Finally, the use of DMF as an electrophile delivered two different cyclic derivatives, depending on the final treatment of the reaction. Thus, final addition of water produced the sulfoximine 16c, whereas a quench by a saturated solution of NH4Cl led to the compound 16d.
2.3 Isolation of enantiomerically pure fluorinated sulfoximine
As mentioned in Section 1, the sulfoximine entity becomes chiral when R1 is different from R2 (Fig. 1). Nonfluorinated enantiomerically pure sulfoximines were extensively used in asymmetric synthesis as ligand, as pool chiral, or as chiral leaving group [1a,22]. Unfortunately, all the methods described for the preparation of the enantiopure sulfoximines failed to be adapted to the S-perfluoroalkyl series. To date, and to the best of our knowledge, no direct preparation of optically active S-perfluoroalkyl sulfoximines has been described. However, we succeeded in the separation of the two enantiomers of sulfilimine 18 [23] by supercritical fluid chromatography [24] (Scheme 11). They proved to be stable and no epimerization was detected even after several weeks at room temperature. The relative configuration has been secured, thanks to an X-ray analysis.
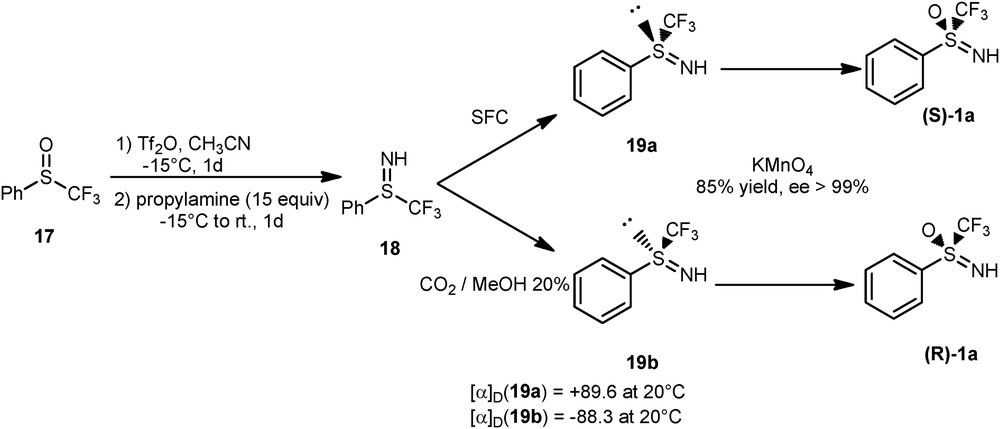
Preparation of enantiomerically pure sulfoximines. SFC, supercritical fluid chromatography.
Further oxidation of each enantiomer of the sulfilimines 19a and 19b with KMnO4 proceeded with retention of the relative configuration to give rise to the two enantiomers of 1a. The scope of this procedure was then explored to other fluoroalkylated compounds and enabled the isolation of optically pure 1b and 1c. The enantiomerically pure version of the Shibata reagent 21 [11a] was also prepared from (S)-1a with an 85% global yield (Scheme 12).

Preparation of the enantiopure Shibata reagent 21.
3 Application
3.1 Application of perfluorinated sulfoximines in catalytic systems
Some bis-sulfoximines and sulfoximine thioureas prepared by our group [17] (Section 2.1.1) were investigated through model reactions, already described with nonfluorinated sulfoximine: a hetero Diels–Alder (HDA) reaction [25], a Mukaiyama-type aldol reaction [26], and a Biginelli reaction [27]. The HDA reaction was performed between cyclohexadiene 22 and the α-ketoester 23 catalyzed by a copper catalyst in the presence of 8a as ligand (Scheme 13). The adducts 24a and 24b were isolated in 51% and 55% yield, respectively. They are slightly lower to those obtained with the nonfluorinated bis-sulfoximine 25 (62% and 95% yield).
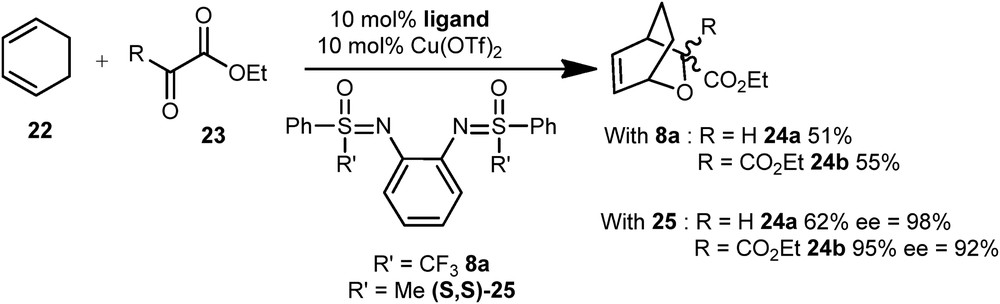
HDA reaction copper-catalyzed with bis-sulfoximine 8a as ligand.
The sulfoximine 10 associated with Cu(OTf)2 proved to be efficient to catalyze a Mukaiyama-type aldol reaction. The compound 28 was isolated with 50% yield, which is comparable to the one obtained with the nonfluorinated bis-sulfoximine 29 (Scheme 14).
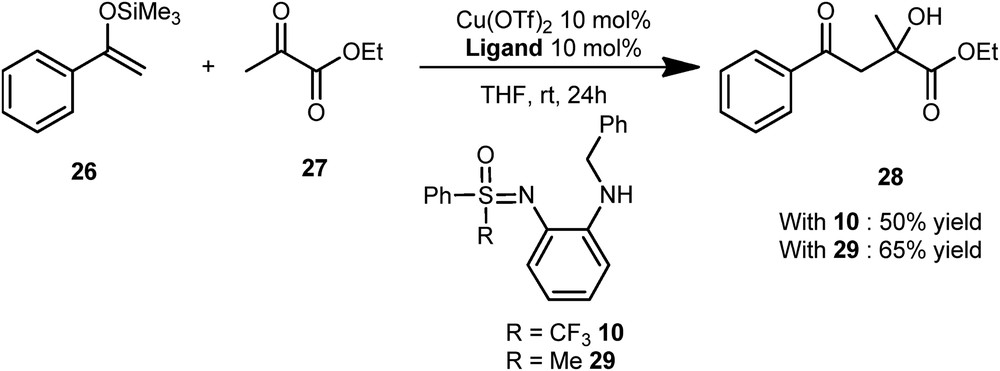
Mukaiyama-type aldol reaction.
The potential of 9a and 9b as organocatalysts was also compared to their nonfluorinated analogues in a Biginelli reaction between the benzaldehyde, the β-ketoester 30, and the urea 31 (Scheme 15). This reaction enabled to set out 32 with a good yield.
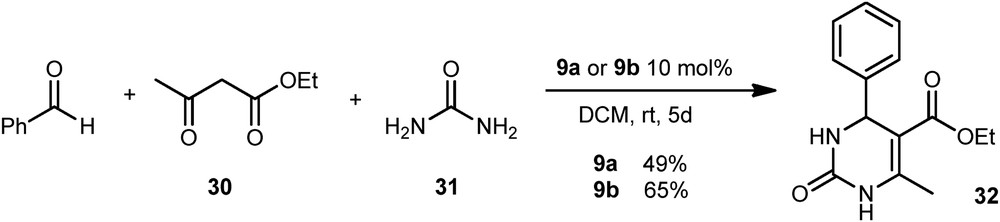
Biginelli's reaction.
Finally, the efficiency of these ligands was performed with a Friedel–Crafts reaction between the indole 33 and the β-(E)-nitrostyrene 34 (Scheme 16). Without catalyst, the product 35 was obtained in a 34% yield, compared to a 65% yield, in the presence of sulfoximine 9b, demonstrating for the very first time the catalytic effect of a perfluoroalkylated sulfoximine for such a transformation. The same reaction with the enantiomerically pure sulfoximine (R)-(+)-9b delivered any asymmetric induction.
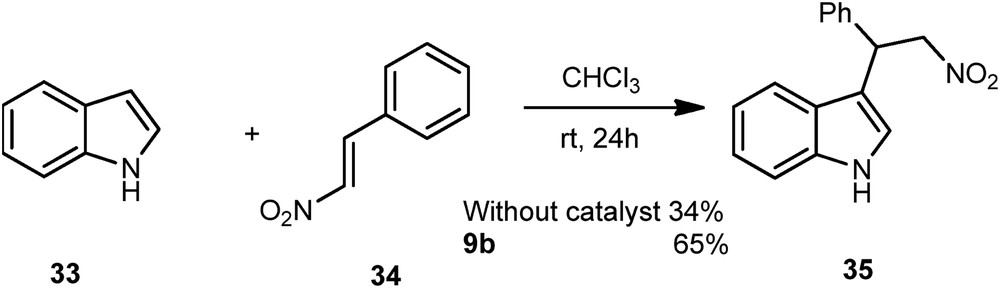
Friedel–Crafts reaction.
3.2 Biologically active compound
To increase the chemical diversity of the family of glucokinase-glucokinase regulatory protein (GK-GKRP) disruptors, the sulfoximine group was introduced as a bioisostere of the trifluoromethyl carbinol group of the bioactive compound 36 [28]. First, the carbinol moiety was replaced with sulfones and sulfonamides because of their three-dimensional features, important for the interactions with the targeted protein GKRP. A wide range of derivatives such as molecules 37a and 37b were synthesized, but revealed only weak to moderate disruptor properties (Fig. 2). The sulfoximine moiety was afterward evaluated. Its mildly basic nitrogen function and chiral sulfur center could indeed mimic the chiral trifluoromethyl carbinol group of compounds 36. Although the nonfluorinated sulfoximine 38a gave disappointing results, the NH-S-trifluoromethyl sulfoximine 38b was approximately seven times more potent than the parent molecules 36. The (S)-NH-S-trifluoromethyl sulfoximine (S)-38b was found to have favorable in vivo pharmacokinetic properties in rodents and produced a significant reduction in blood glucose levels (about 40–58%, 6 h after dose at 100 mg/kg) when dosed orally in db/db mice.
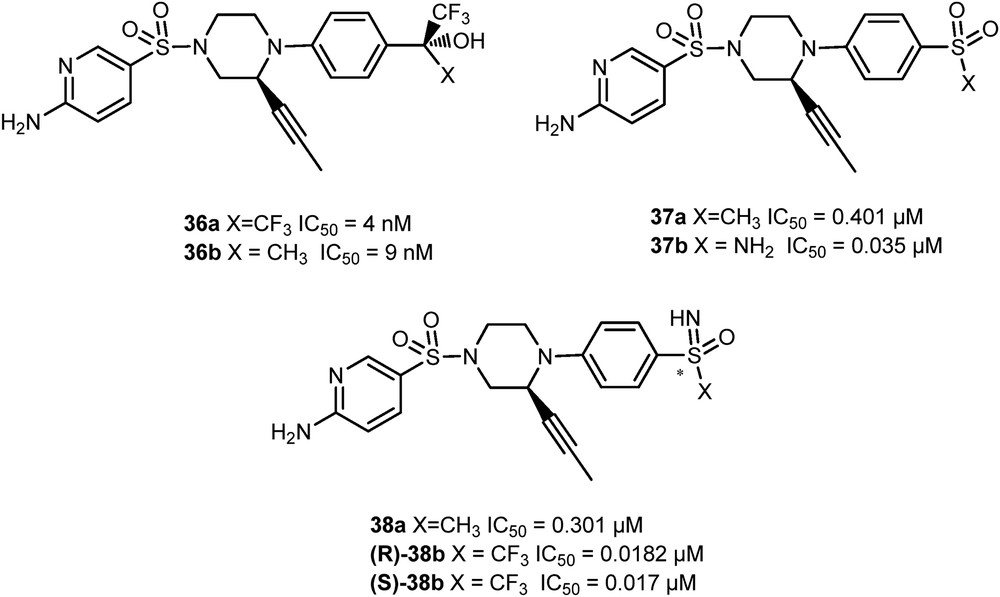
Analogues of GK-GKRP disruptors.
3.3 Perfluorinated sulfoximines as perfluoroalkylating reagent
The introduction of fluoroalkyl groups into molecules is a modern and very important challenge. In the past decades, fluorinated sulfoximines have emerged as promising and efficient fluoroalkylating reagents through the work of Shibata and co-workers [11a,c,g], Hu and co-workers [11b,12], and Prakash et al. [11e] and also by our group [11f] (Fig. 3).
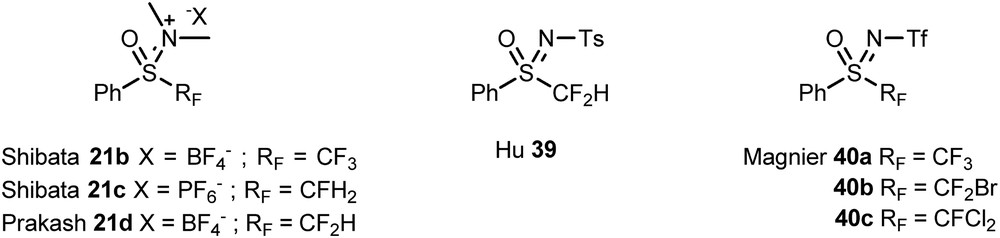
Overview of sulfoximines as fluoroalkylating reagents.
3.3.1 Electrophilic pathway
Sulfoximines were described as electrophilic perfluoroalkylating reagent [11]. Our group proposed recent extension. We reported the first example of electrophilic chlorofluoromethylation [29], thanks to a new sulfoximine 41 reagent (Fig. 4).

New electrophilic reagent 41.
Its electrophilic activity was compared to Hu's reagent 39 in the perfluoroalkylation of the β-ketoester 42. In the reaction conditions previously described by Shibata et al. [11c], Hu's reagent 39 gave a mixture of C-alkylated 43a and O-alkylated 44a derivatives whereas reagent 41 reacted to give the C-alkylated compound 43b as the sole product with 40% yield (Scheme 17). It is noteworthy that a study of this C/O-selectivity was carried out in 2014 by Shibata et al. [11d].
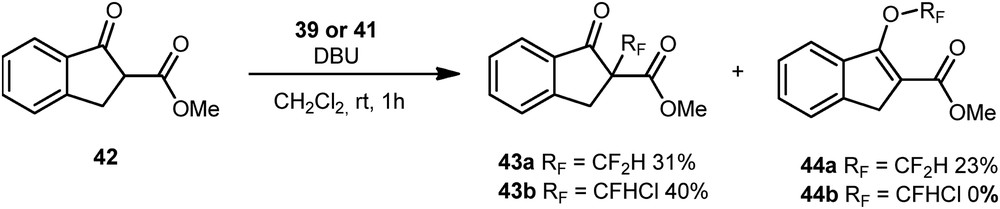
Reactivity of 39 and 41 toward β-ketoesters.
We proposed a carbenic pathway for this transformation based on previous mechanistic studies from Hu's group and our own (Scheme 18).
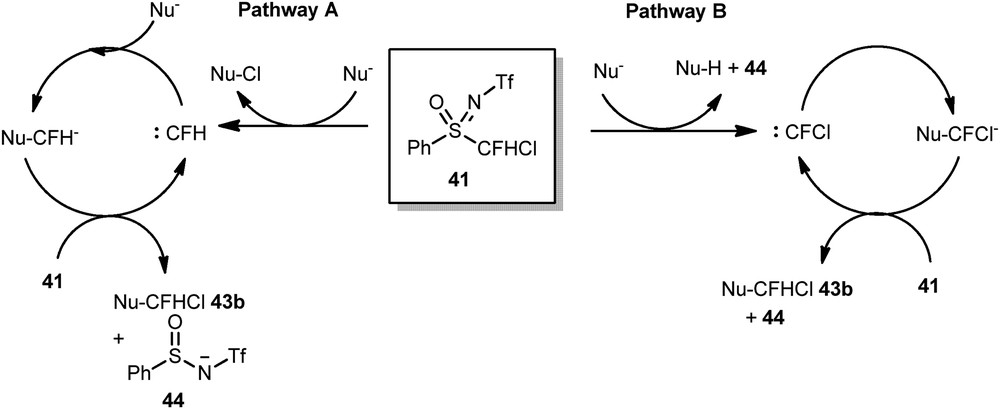
Proposed mechanism.
The carbenic pathway A was proved to be the most plausible, even if the pathway B could not be totally excluded.
3.3.2 Nucleophilic pathway
In 2014, Hu et al. [30] developed a new efficient and regioselective nucleophilic ring-opening difluoromethylation of epoxides using a difluoromethyl sulfoximine 46. This strategy was successfully applied to a wide variety of three- and four-membered oxacycles 45 as substrates, leading to compounds 47 in good to excellent yields (Scheme 19). This regioselective reaction took place at the less hindered position of the oxacycles.
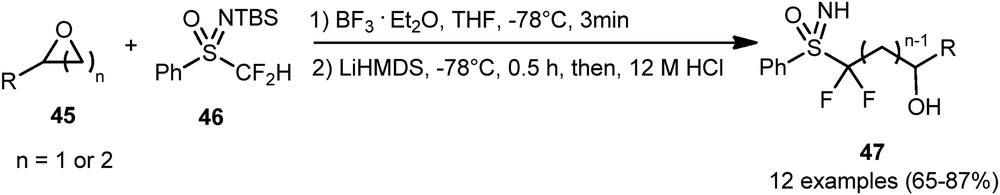
Nucleophilic difluoromethylation of three- and four-membered oxacycles.
The difluorinated species 47 can be engaged in further transformation to form β-difluoromethyl alcohol 48 by reductive desulfoximination or β-difluoromethylenyl alcohol 49 by elimination process (Scheme 20).

Synthetic utility of ring-opened compounds.
More recently, Hu et al. described the preparation of a wide range of arylfluoroepoxides 52 (Scheme 21) using perfluoroalkyl sulfoximines [31]. These fluoroepoxides could be isolated without significant decomposition and were next engaged in a Brønsted acid–catalyzed 1,2-fluorine migration.
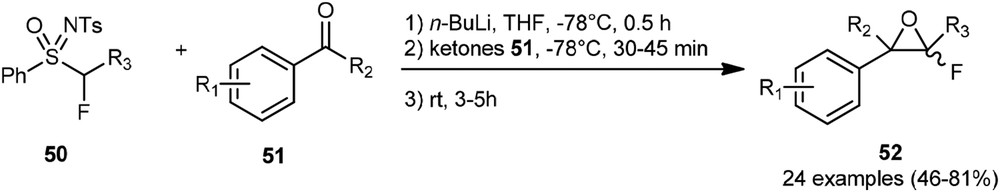
Synthesis of arylfluoroepoxides.
In 2017, the same group reported a stereoselective synthesis of terminal monofluoroalkenes [32] through a Julia-Kocienski–type reaction. A wide variety of aromatic carbonyl compounds 54 were successfully transformed into monofluoroalkenes 55 in the presence of the sulfoximine 53 (Scheme 22).
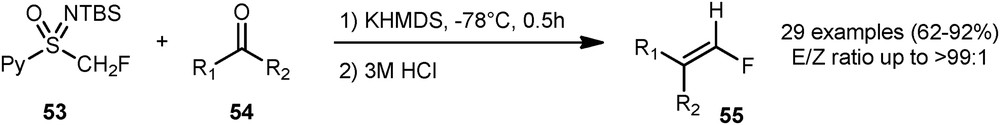
Stereoselective synthesis of terminal monofluoroalkenes.
The reaction tolerated many substituents on the aromatic ring, such as bromine, methoxy, benzoxy, methylthio groups, and heteroaromatic groups.
3.3.3 Radical—Advent of the photoredox-catalyzed fluoroalkylation
In recent years, the visible light photoredox catalysis has emerged as one of the most promising strategies for the introduction of fluorinated moiety into organic molecules. In 2016, Akita et al. described the direct hydroxydifluoromethylation of alkenes by photoredox catalysis [13] using Hu's reagent 39 at room temperature with visible light irradiation (Scheme 23).
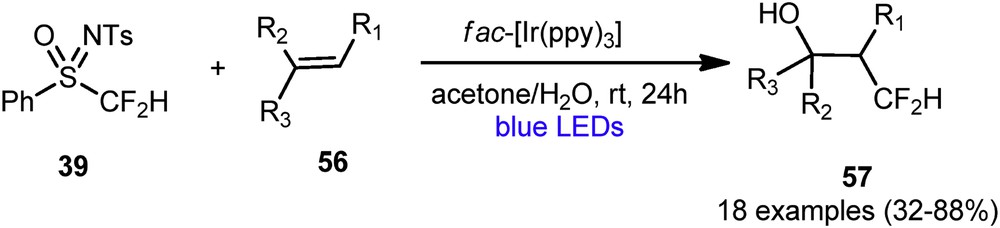
Hydroxydifluoromethylation of alkenes via photoredox catalysis.
The reaction conditions were successfully applied to a wide range of styrenes. Various functional groups, such as halogens, boronic acid esters, and cyanomethyl- and Boc-protected amino groups, are well tolerated delivering a set of alcohols with yields up to 88%. The reaction was also carried out on the aliphatic dicyclohexylethene leading to the corresponding alcohol 57 with a 32% yield. Moreover, the hydroxydifluoromethylation of internal and trisubstituted alkenes was also possible with this photocatalytic reaction. Alcohols and carboxylic acids ROH were tested as nucleophiles, leading to the CF2H-containing ethers 58a,b and esters 58c (Scheme 24).

Oxydifluoromethylation of styrenes.
Akita et al. proposed a radical pathway for this transformation (Scheme 25). The photocatalyst is excited by blue light to the species *IrIII able to reduce Hu's reagent 39 delivering then the CF2H radical. Its addition, alkene 56 generates an intermediate, which is then oxidized by IrIV. The resulting carbocationic intermediate is finally trapped by a nucleophile to give rise to the oxydifluoromethylated products 57 or 58.
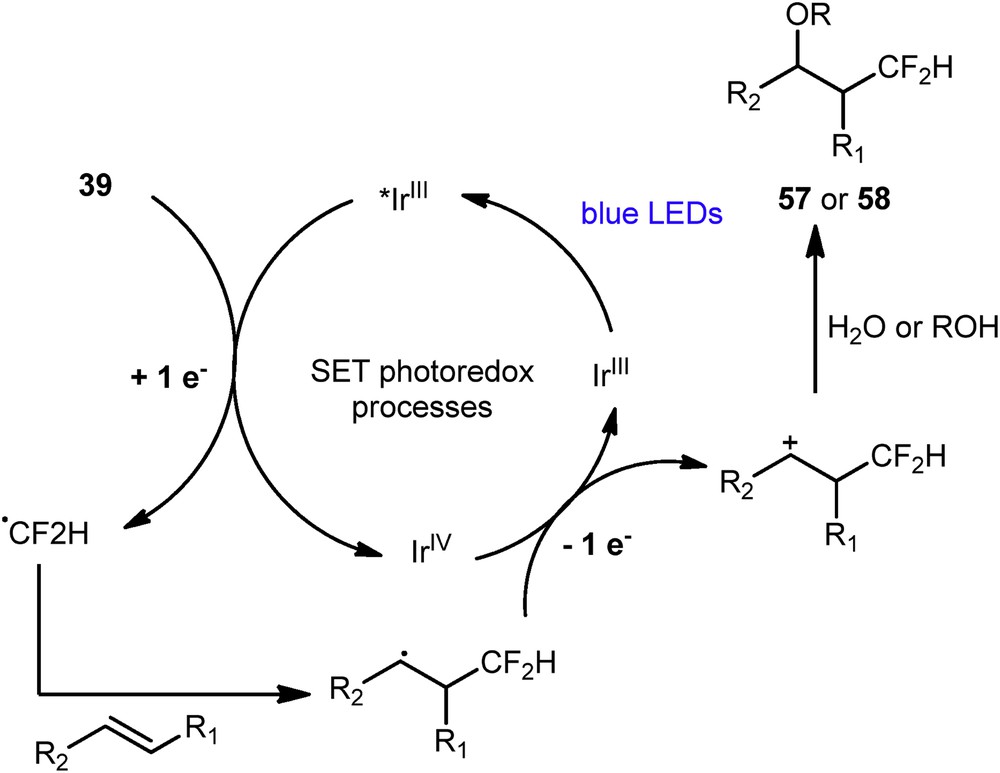
Proposed mechanism for the oxydifluoromethylation.
The same year, Akita et al. extended the previous reaction to the synthesis of CF2H-spiroethers 60, using Hu's reagent 39 and fac-[Ir(ppy)3], under similar reaction conditions [33]. These CF2H-spiroethers were synthesized in moderate yield, but with good diastereoselectivities in an anti-fashion (Scheme 26).
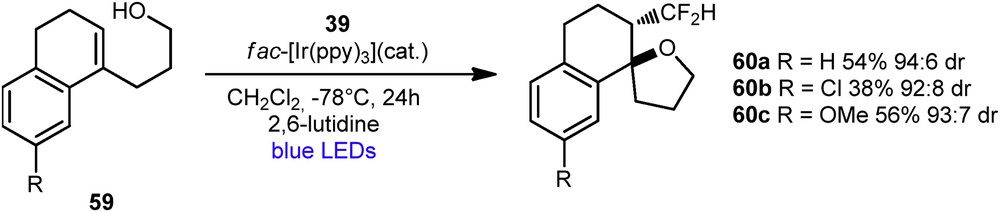
Synthesis of CF2H-containing spiroethers.
The authors proposed that the alcohol attacks in an anti-fashion because of the electronegativity of the fluoromethyl group.
More recently, our group has developed a new oxyperfluoroalkylation method by photoredox catalysis using sulfoximines 21a and 40a as sources of trifluoromethyl radicals [34]. With optimized reaction conditions, using fac-Ir(ppy)3 as the photocatalyst and methanol as both solvent and nucleophile, under blue LED irradiation, methoxyfluorinated compound 62 have been synthesized with moderate yield (Scheme 27). During the same work, we demonstrated the potential of sulfoximines 40b and 40c for the methoxy mono- and di-fluoromethylation of vinylnaphthalene.

Methoxyperfluoroalkylation of vinylnaphthalene 61.
4 Conclusions
After a long period of latency, the chemistry of S-perfluoroalkylated sulfoximines is rising up. This could be explained by both the extended availability of the starting materials, thanks to the progress realized for their preparation, and by the increasing applications offered by these uncommon moieties. We have summarized in this short review all the recent developments, and we are fully convinced that they will exponentially grow in close future.
Acknowledgments
The authors are grateful to the CNRS and the French Ministry of Research (grant; A.L.B.) for financial support. The French Fluorine Network is also acknowledged for its support.