

1 Introduction
The search for new materials has always inspired thinking beings, dating back to the beginning of mankind. As this field became more and more dedicated to “specialists”, in the days of alchemy, most efforts were made in the direction of generating gold from ignoble elements or compounds, or to find the philosopher's stone that promises everlasting youth. One of these alchemists, Hennig Brand, accidentally discovered white phosphorus in 1669, instead of the philosopher's stone, upon cooking and condensing human urine. It was not the material he searched for, but his discovery of the element and its luminescence was a breakthrough from our point of view.
Today, although we try to widely control chemical and materials research and wish, or need, to plan projects and their results, serendipity is still alive and most often provides us with especially beautiful observations. In this regard, recent pleas for fundamental research are more than justified [1], as only this will guarantee original, innovative, and widespread new findings in the long run.
This article is a short survey of a story that evolved from a very simple objective pursued in our group several years ago, went through some systematic developments that branched out into different families of compounds, and finally even led to the generation of new materials with very uncommon structural or physical properties, which were neither predicted nor expected.
The research is based on solid compounds that comprise binary tetrahedral anions of main group elements. These may either be centered tetrahedra with no bonds along the tetrahedral edges or tetrahedral molecules comprising four atoms in a cage-type structure. The first class of anions is found for the elemental combination E14/E16 (E14 = Ge, Sn, Pb; E16 = S, Se, Te), hence for chalcogenidotetrelate anions [E14E164]4− (Fig. 1, left) [2], which are heavier homologs of the ortho-silicate anion [SiO4]4−. The second class of anions is present in binary Zintl anions of group 13–15 (semi-)metal atoms, which accord to the general formula (E13E153)2− or (E142E152)2− (E13 = Ga, In, Tl; E14 = Ge, Sn, Pb; E15 = P, As, Sb, Bi; Fig. 1, right) [3], and represent binary heavier analogs of the P4 molecule. Although these two classes of tetrahedral anions are obviously not highly related in terms of their formal charges, that is, metalate anions with a central (semi-)metal atom in formal oxidation state +IV and ligands with formal oxidation states −II versus metalide anions with no central atom and formal oxidation states −I or −II and ±0, they do have one important thing in common, that is, they represent two types of small anions comprising atoms of two different main groups that allow chemistry in solution. Some larger soluble molecules, like binary 9-atom anions (E147E152)2− (E14/E15 = Ge/P, Ge/As, Sn/Bi, Pb/Bi) [3a,3f,4] or (TlSn8)3− [5], were reported, but none of these have been used for further reactions so far. The only known class of metalate anions composed of group 14 and 15 atoms are the carbonate analogs [TtPn3]5− (Tt = Si, Ge; Pn = P, As) [6], which are absolutely insoluble in common solvents at room temperature. Soluble homologs of nitrido silicates have not been known.

Structures of the anions used in the materials approach discussed in this report, [E14E164]4− (left) and [E142E152]2− (right), with E14 = Si, Ge, Sn, Pb; E15 = P, As, Sb, Bi; E16 = S, Se, Te, and their related homologs [SiO4]4− (center left) and P4 (center right).
Reactions of alkali metal salts of [E14E164]4− anions with transition metal salts or complexes in protic solvent (H2O, methanol) with transition metal (M) salts or complexes served to extend these building blocks into ternary clusters or networks composed of three instead of two components, [MxE14yE16z]q–, which largely affect the structures, the optical absorption properties, and the magnetism of the resulting compounds [2,7]. Such compounds therefore belong to the large family of chalcogenide compounds, which play an important role in the development of materials for new (photo-)semiconducting or thermoelectric devices [8].
Recent modifications and extension of the first studies led to the following directions. (1) By admixture of main group chalcogenides under solid-state conditions, quaternary solids are obtained that contain two different types of molecular tetrahedral anions and show superion conductivity. (2) A transfer to low-temperature melts, the so-called “ionic liquids”, as alternative reaction media, and the use of supertetrahedral adamantane anions [E144E1610]4−, formal condensation products of four of the original tetrahedra, allowed for obtaining very unusual cluster architectures. (3) The adamantane unit is also the base for small ligand-protected organotetrel clusters of the type [(RE14)4E166] (R = organic group), which are obtained in a slightly varied way and may be further extended regarding both the inorganic part or the ligand shell of such hybrid compounds. (4) The use of the alternative type of tetrahedral anions, the binary Zintl anions, in reactions with transition metal compounds leads to the formation of intermetalloid clusters [MxE13/14yE15z]q− and hence nanoscale monodisperse ligand-free metal particles, which may be regarded as molecular models of doped metals or intermetallic phases [9].
In all of these cases, it was initially necessary to perform a large portion of exploratory work, as there have not been any documents on reaction patterns, mechanisms, or pathways starting out from such anions. In the following section, some examples shall be given here to illustrate these four directions.
2 Materials from small binary molecules
2.1 Superionic conductors
In the first example, we reacted the simple lithium or sodium salts Li4[SnS4] or Na4[SnS4] with P2S5. By choosing a suitable ratio of the reactants, the lithium superion conductor Li10SnP2S10 or the sodium superion conductor Na11Sn2PS12 was obtained [10]. Both belong to the best-known sulfide-based solid-state ion conductors, with ion conductivities on the order of magnitude of liquid electrolytes [11]. In contrast to the liquid materials, the solid-state ion conductors do not bear the risk of inflammation. However, the use of such materials still requires optimization of electrode contacting and the electrochemical stability, which is currently actively investigated worldwide. Somewhat better Li+ ion conductivities were obtained for the homolog Li10GeP2S10 [12], but this compound is more expensive (by a factor of 2–3), owing to the presence of Ge instead of Sn. The price of the sodium conductor would be again lower, in accordance with the larger abundance of sodium versus lithium in the earth crust. The crystal structures of the said compounds and their conductivities are illustrated in Fig. 2.

Crystal structures (top and center) of Li10Sn2PS12 (left) and Na11SnP2S12 (right). Arrhenius plots of the conductivity (grain, grain boundary, total) in comparison with that of Na3PS4 (bottom).
As mentioned above, although the ion conductivity properties of such quaternary salts are excellent, their electrochemical stability is limited and they may undergo chemical reactions with electrode materials, which currently inhibit their use in commercially available batteries. One possible way to overcome this drawback, for instance, is the developing taylor-made interphases (solid electrolyte interphases) between the electrode and the electrolyte [13].
2.2 Supersphere cages
The second example addresses reactions in ionic liquids. These are—as per definition—salts that melt (markedly) below 100 °C, many of them even below or around room temperature, as the combination of cations (mostly organic) and anions (mostly weakly coordinating) does not allow for a sufficiently large lattice energy of the crystal structure [14]. Such melts have very good solvent properties, yet without the disadvantageous properties of common solvents, such as low boiling points and high vapor pressure, which causes the release of (toxic) vapor. Moreover, many ionic liquids are readily synthesized and may be easily varied by the choice of cations and anions. This is why ionic liquids are often referred to as “designer solvents” that enable, for instance, the synthesis of new solid compounds at relatively low temperatures between room temperature and moderately elevated temperatures (≤200 °C), whereas solid materials have been traditionally accessed via high-temperature approaches (≥700 °C).
We use ionic liquids for the synthesis of novel chalcogenidometalates from a diversity of chalcogenidometalate salts based on [E14E164]4− tetrahedra [15] or their molecular extension into an adamantane-type [E144E1610]4− anion [16]. Our studies also indicated the possibility for a dimensional reduction of three-dimensional solids [17], such as the known phase K2Sn2Se5 [18]. By choosing suitable reaction conditions in terms of the nature of the ionic liquid, the nature and amount of an auxiliary amine, and the temperature, it is possible to produce salts of a supersphere anion, [Ge24Sn36Se136]24−. It is the largest known main group element polyanion, which has a spherical ball-type structure surrounding a large inner void space of more than 800 Å3 [16]. The reactant K4[Ge4Se10]·3H2O is reacted with SnCl4·5H2O in different ionic liquids of the general type [Cn(C1)C1Im]X, with Cn representing an alkyl chain with n = 4, 6, 8, or 10 carbon atoms, C1 representing methyl groups on the imidazolium (Im) cation, and X representing a 1:1 mixture of Cl− and [BF4]− anions. In the presence of a small amount (0.1 mol %) of the auxiliary amine 1,3-dimethylmorpholine, large red crystals of salts of the supersphere anion are selectively observed, whereas larger amounts of the auxiliary serve to form different, non-molecular chalcogenidometalate structures. Depending on the nature of the ionic liquid cation, the ball-shaped anions are arranged differently in the crystal structures, with the cations located in between them—in some cases also occupying the void space. The compounds are capable of trapping small molecules like I2 and to heterolytically cleave the I–I bond. Fig. 3 illustrates the synthesis of this compound, its crystal structure, and the behavior towards a solution of I2 in cyclohexane. Besides these hints toward molecular activation, the anion and the corresponding crystal structures have attracted attention by their mere aesthetics.

Synthesis of the compound [BMIm]24[Ge24Sn36Se132] by ionothermal reaction of K4[Ge4Se10] and SnCl4 in the ionic liquid [BMIm][BF4] = 1-butyl-4-methylimidazolium-tetrafluoroborate in the presence of a small amount of the auxiliary amine 1,3-dimethylmorpholine (top left). Packing of the anions in the crystal structure and molecular structure of the supersphere anion (top right), with gray spheres = Sn atoms, blue spheres = Ge atoms, red spheres = Se atoms, and the yellow ball inside the molecule representing the inner void space with a diameter of approximately 1.2 nm. Photograph of the single crystals (center left). Interaction of crystalline [BMIm]24[Ge24Sn36Se132] with a cyclohexane solution of I2 (photographic tracking of the color fading over a 3 hour period, and cartoon of the process, bottom left). Thermogravimetric analyses (TGA) indicating the uptake of up to 11 equivalents of I2 by pristine [BMIm]24[Ge24Sn36Se132] (center right). Optical absorption spectrum of an ethanol suspension of the I2-loaded, brown crystal (see inset photograph), indicating the formation of I− and, finally, I3− (bottom right).
2.3 White-light generators
As briefly outlined above, another extension of the basic concept is the replacement of one of the chalcogenide ligands by an organic group. Small organotetrel chalcogenide clusters of the general formula [(RE14)4E166] or [(RE14)3E164]+ are obtained by a reaction of organotetrel trihalides RSnX3 (X = Cl, Br) with A2E16 (A = alkali metal, SiMe3). Dependent on the nature of the organic group R, the structure of the [(RE14)4E166] clusters is either adamantane-type (thus related to the previous example) or “double-decker”–type, with two coplanar (E142E162) four rings that are bridged by two E16 atoms [19]. For R comprising a donor atom (O, N), back-coordination to the E14 occurs, which leads to the preferred formation of the double-decker–type motif with five-coordinate E14 atoms, whereas organic ligands without donor atoms stabilize the adamantane-type architecture with four-coordinate E14 atoms [20]. The inorganic cores of these small clusters may be further modified by a reaction with transition metal compounds; if the organic ligands bear reactive groups, an extension of the ligand shell with complementarily functionalized organic compounds is also possible [21]. However, even the small clusters themselves show uncommon properties, which were observed only recently.
As indicated in Fig. 4 (left), the reaction of an organotin trichloride with a styryl ligand affords an air-stable amorphous powder. Its composition was determined by means of mass spectrometry as [(StySn)4S6] (Sty = styryl, C6H4CHCH2; Fig. 4, right). As the molecular structure within amorphous compounds cannot be determined by X-ray diffraction, quantum chemistry was applied, indicating the cluster to adopt the adamantane-type structure (Fig. 4, center). This structure type lacks molecular inversion symmetry, which prompted us to investigate potential nonlinear optical properties. A typical nonlinear phenomenon is second harmonic generation, in which monochromatic light is converted into light with exactly doubled frequency (i.e., with half of the original wavelength) upon transmission of a material. Although this was what we expected to find, [(StySn)4S6] does not exhibit second harmonic generation properties. Instead, and most notably, the amorphous powder is capable of transforming infrared laser light from an inexpensive continuous wave (CW) GaAs laser diode into white laser light without loss of the directionality, as illustrated in Fig. 5 [22]. In the meantime, further amorphous compounds comprising adamantane-based clusters [(RE14)4E166] (R = phenyl, styryl; E14 = Si, Ge, Sn; E16 = S, Se) were identified that show the same characteristics [23].
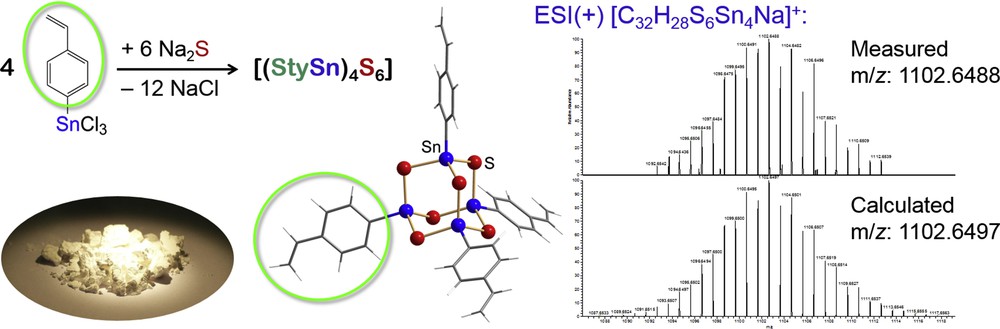
Synthesis and photograph of the amorphous powder of [(StySn)4S6] (Sty = styryl, C6H4–CH=CH2; left), along with the molecular structure that was calculated by means of DFT calculations (center; blue spheres = Sn atoms, red spheres = S atoms, gray wires = styryl groups). Mass spectrum of the molecular peak (right) with the experimentally found (top) and calculated (bottom) isotope pattern around m/z = 1102.6.
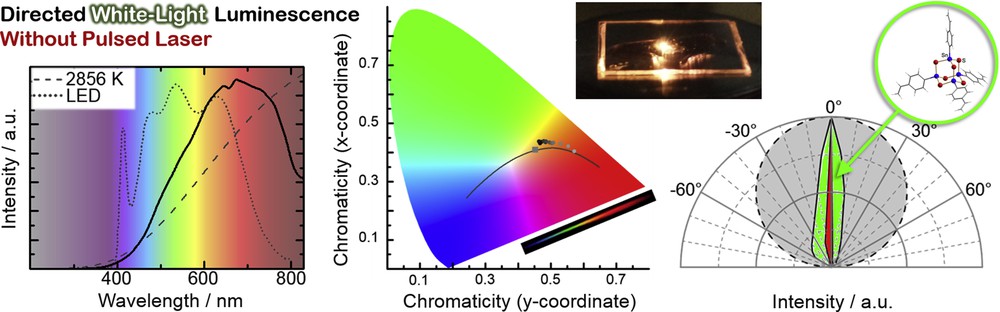
Photoluminescence spectrum (left) received upon irradiation of the amorphous powder of [(StySn)4S6] with a commercially available continuous-wave infrared laser diode (solid line), in comparison with the spectrum of a black body radiator at 2856 K, that is, the color temperature of a common tungsten halogen bulb (dashed line), and the spectrum of a commercially available white-light LED (dotted line). Chromaticity of a black body radiator (center, solid line with gray square indicating the color at T = 2856 K) and chromaticity upon irradiation of [(StySn)4S6] (gray dots indicating low excitation fluencies and black dots indicating large excitation fluencies) and photographs of the warm white light impression (top) as well as the rainbow spectrum obtained by scattering at a prism (bottom). Highly directional spatial emission pattern (right) of the white-light spectrum (green area) and of the excitation laser at 980 nm (red area) in comparison with the intensity distribution of a perfect Lambertian emitter (gray area).
Supercontinuum generation processes in general have been already described for many systems, however only upon use of very expensive pulsed lasers, and not necessarily along with directionality. This indicates that the new findings may revolutionize the generation of directed white-light, which may be useful for projection or medical purposes. The exact mechanism behind this new type of supercontinuum generation is not known so far, but apparently it is dependent on missing long-range order in the material and the presence of aromatic ligands for electron delocalization.
2.4 Multimetallic clusters
The fourth and final type of compound is taken from the mentioned reactions of the P4 analogs (E142E152)2− or (E13E153)2−, with formally charged (E14)− or (E13)2− atoms being isoelectronic with neutral (E15) atoms. We note that, for the elemental combination of E14 and E15, such anions also represent heavier homologs of a hypothetical tetrahedral dimer of cyanide (CN)−, being isoelectronic with the lighter P4 homolog N2. The chemistry behind these studies belongs to the contemporary chemistry of Zintl ions, which themselves have been known for more than a century, but the post-synthesis reactions of which have only recently been investigated. Besides organic derivatization and linkage of clusters [24] or their oxidative degeneration to form oligomers, polymers, opal structures, or, finally, new elemental modifications [25], a third direction of how to modify Zintl anions is the formation of heterometallic clusters upon reactions with transition metal compounds [9]. In most cases, the obtained clusters represent so-called “intermetalloid” clusters, that is, main group atom cages that embed one (or several) interstitial transition metal atom(s). Given suitable electron counts, such systems may be also viewed as superatoms [26]. Intermetalloid clusters are very reactive and do not only readily undergo oxidation, they may also deprotonate (basic) solvents and they further seem to support C–C coupling reactions [27]. This is one of the reasons, why we are interested to find out more about these heterometallic molecules, besides very fundamental objectives, such as their intriguing structures and uncommon bonding situations.
The concept of using binary anions as precursors has two significant consequences. First, it enables the introduction of many different binary combinations of group 13–15 elements into such systems, as the (E142E152)2− or (E13E153)2− anions have been synthesized and isolated as their [K(crypt-222)]+ salts (crypt = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane, C18H36N2O6) for many elemental combinations, namely Ga/Bi, In/Bi, Tl/Bi; Si/P, Ge/P, Ge/As, Ge/Sb, Sn/Sb, Sn/Bi, Pb/Sb, Pb/Bi [3]. Second, the structural variety is naturally larger than upon use of homoatomic systems, in which the adjustment of negative charges to fit with the requirements of the structure and crystallization cannot be done by simply choosing between (E14)−, (E13)2−, or (E15) atoms, respectively, for a specific atomic position. Consequently, a large variety of ternary clusters with tunable electronic properties have been accessed.
Fig. 6 gives an overview of the clusters that were obtained with binary Zintl anions of the named elemental combinations and different types of transition metal atoms [3a,3b,3f,4,27,28]. Some elemental combinations do not lead to the formation of small tetrahedral anions but to larger molecules instead, because of a distinct mismatch of atomic sizes. An example is (Ge4Bi14)4−, which shows an element segregation on the molecular level, in accordance with the immiscibility of Ge and Bi solids [29].
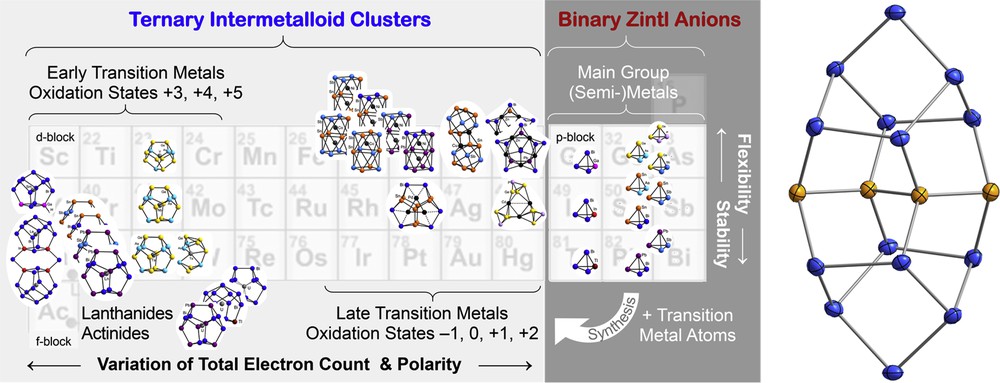
Overview of ternary intermetalloid clusters (left) obtained by reactions of binary tetrahedral Zintl anions [E142E152]2− or [E13E153]2− (center), indicating different families of clusters to be formed with certain types of transition metal, lanthanide, or actinide metal, respectively. Because of a mismatch of their atomic sizes, elemental combinations like Ge and Bi do no form tetrahedral binary anions, but larger molecules, like (Ge4Bi14)4− (right; yellow spheres = Ge atoms, blue spheres = Bi atoms).
The bonding situation and structures of most of the shown clusters can be rationalized by one of two well-known concepts. Electron-precise clusters, like the binary tetrahedral precursors and the aforementioned (Ge4Bi14)4− anion, or ternary clusters enclosing early transition metal or lanthanide/actinide ions, comply with the pseudo-element concept by Zintl [30a], Klemm [30b], and Busmann [30c]. Here, the number of bonds per atom follow the 8-N rule, with N being the number of valence electrons plus the formal (negative) atomic charge of the respective atom. An electron-precise interpretation also applies for ternary clusters with interstitial Co2− ions or Ni atoms that can be regarded from the viewpoint of fulfilling the 18-electron rule at the transition metal atom. Pseudo-element situations are also found in ligand-like adducts of otherwise unchanged homoatomic or binary Zintl anions to transition metal ions like Cu+, Zn2+, or Cd2+. Electron-deficient clusters with their typical deltahedral shape are formed with interstitial Cu+ or Zn2+ ions and are in accordance with Wade–Mingos rules [31]. In this regard, ternary transition metal–main group element clusters [9d] do not differ from their binary transition metal–main group element cluster cousins [9a,9b,9c]. However, in both the binary and ternary intermetalloid/heterometallic clusters, there are examples known that cannot be understood in terms of any of these concepts. Examples of corresponding binary clusters are [M@Ge10]3− (M = Fe, Co) [32] and the paramagnetic [Ru@Ge12]3− anion [33]. These clusters feature three-bonded Ge atoms within non-deltahedral 10-atom or 12-atom cages that, however, cannot be simply regarded as comprising pseudo-group 15 atoms. Instead, the anions have to be viewed as entities comprising delocalized cluster orbitals (with the unpaired electron of the latter example residing in a Ge12-derived molecular orbital). This finding is in contrast to a ternary (and diamagnetic) analog of the Ru/Ge cluster, [Ta@Ge83− = [Ta5+@(Ge−)8As4]3− [3a], in which the pseudo-element concept is perfectly fulfilled. At first glance, the binary cluster [U@Bi12]3− [3h], in which a doughnut-shaped Bi12 ring surrounds a U atom is a relatively simple case, consisting of an interstitial U3+ ion and a macrocyclic (Bi12)6− ligand. However, the magnetic behavior and the reproduction of structural features by means of DFT calculations indicated [U4+@(Bi12)7−]3− to be the correct formulation, including a highly unexpected paramagnetic (Bi12)7−• unit. Another extraordinary example is the ternary cluster anion [Pd3@Sn8Bi8]4− [28e], which accords to neither an electron-precise model nor a typical Wade–Mingos-type electron-deficient description. Again, the help of sophisticated quantum chemistry was required to show that a neutral Pd3 ring is enclosed within an uncommon (Sn8Bi6)4− shell.
Among the known cases of ternary intermetalloid/heterometallic cluster anions (Fig. 6), one recognizes the predominance of certain structural motifs for certain types of interstitial atoms, which indicates the latter to act as a template during the formation of the clusters. This is obvious for the 12-atom, 13-atom, or 14-atom cages that are formed with interstitial lanthanide, actinide, or group five metal ions, for which a clear correlation between cage size and ionic radii of the inner atom is observed [28b,28i]. Hence, we asked ourselves how such clusters might form around the d-block or f-block metal atoms. On the basis of the isolation of an intermediate cluster, [Ta@Ge6As4]3−, which crystallized upon the extraction of a quaternary K/Ta/Ge/As solid mixture, and the observation of the known Ge104− anion in the reaction solutions, we proposed the following formation cascade to occur at the formation of such nondeltahedral clusters (Fig. 7) [3a].

Proposed formation cascade of the ternary intermetalloid clusters [Ta@Ge8As6]3− and [Ta@Ge8As4]3− (top) from the binary Zintl anion (Ge2As2)2−, the homoatomic Zintl anion Ge102−, and Ta metal (bottom), passing through binary (Ge7As2)2− and the intermediate [Ta@Ge8As6]3− (black spheres = Ta atoms, yellow spheres = Ge atoms, blue spheres = As atoms). Colored structure figures correspond to species isolated as single crystals and confirmed by electrospray ionization mass spectrometry (ESI-MS). The assignment of Ge and As atomic sites was done by means of first-order perturbation theory. Structure figures drawn in gray scale refer to calculated species. The occurrence of (Ge3As)3− (right) was supported by ESI-MS. The shown pathway is balanced regarding atoms and electrons, with the exception of a 2-electron oxidation step at the outset, which is anticipated to feed reductive side reaction (not indicated here).
As observed very recently, some of the clusters seem to be in the position to activate small molecules. As monitored by electrospray ionization mass spectrometry, the cluster [{Ru(cod)}Tl2Bi6]2− (cod = cycloocta-1,5-diene) can form an O2 adduct upon release of the cod ligand. Quantum chemical calculations indicated the activation of an O2 molecule attached to the Ru atoms, as can be seen from the significant elongation of the O–O bond (Fig. 8) [28g]. Further studies regarding the potential use of such species for redox catalysis are underway.
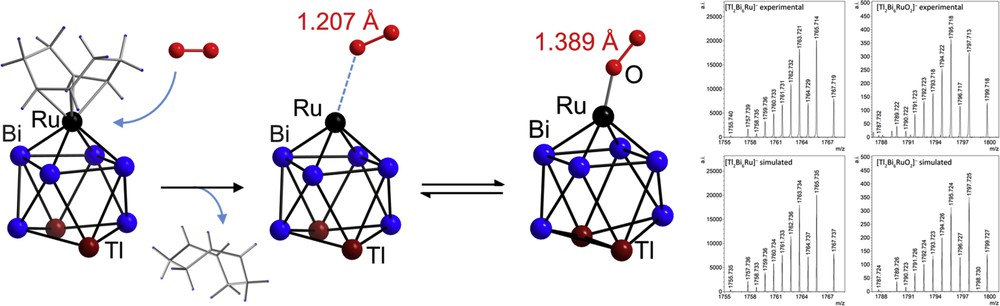
Experimental structure of [Tl2Bi6{Ru(cod)}]2− (left), theoretical prediction of reversible O2 attachment upon detachment of 1,5-cyclooctadiene (cod) to form [Tl2Bi6RuO2]2−, which comprises activated O2 molecules (center; DFT minimum structure shown). Mass spectrometric proof of the [Tl2Bi6Ru] unit without and with O2, as given by the excellent agreement of experimental (top) and simulated (bottom) mass spectra of [Tl2Bi6Ru]− (second from right) and [Tl2Bi6RuO2]− (rightmost graphic).
3 Conclusions
This short overview of the chemistry with binary tetrahedral molecules comprising main group element atoms served to illustrate the large variety of target compounds that result from a very simple general approach, according to the variation in the involved starting compounds and the reaction conditions, and their respective material properties. The spectrum of new materials ranges from sulfide-based lithium and sodium superion conductors and (photo-)semiconducting chalcogenides including compounds with superspheric anionic cages to white-light generators based on organotetrel chalcogenide clusters and, finally, all-metallic ternary cluster molecules, so-called intermetalloid clusters, that may be used as homogenous catalysts in the near future.
Acknowledgments
We acknowledge generous support by the Deutsche Forschungsgemeinschaft (DFG), the Alexander von Humboldt Stiftung, the Marburg University Research Academy (MARA), the Fonds der Chemischen Industrie (FCI), the Friedrich Ebert Stiftung, and the Bundesministerium für Bildung und Forschung (BMBF).