

1 Introduction
Conjugated organic small molecules and polymers are a class of functional materials that have been widely used in optoelectronic devices including organic light-emitting diodes (OLEDs) [1,2], organic photovoltaics (OPVs) [3–5], organic field effect transistors (OFETs) [6,7], electrochromic devices [8,9], chemo-/bio-sensors [10,11], and so on. Their properties such as thermotics, light absorption and emission, electronic energy level structures, charge transport, and so on can be tailored by modifying their chemical structures, which is beneficial for realizing high-performance organic electronic devices.
Among the building blocks for conjugated organic semiconductors, fluorene has drawn great attention because of its advantages such as low cost and high reactivity, and a mass of fluorene-based small molecules and polymers has been developed and applied to organic optoelectronic devices. For instance, in OLED field, polyfluorenes (PFs) have emerged as the most attractive blue emitters resulting from their high luminescent efficiencies [12–14]. However, one of the most serious drawbacks for 9,9-dialkylated PFs is that they often formed fluorenone defects after thermal annealing or during the long-term device operation, as a result of thermal, photo-oxidative, or electro-oxidative degradation [15,16]. The fluorenone defects red-shifted the electroluminescence spectra and subsequently reduced the spectral stability of OLEDs. Importantly, it has been confirmed that bis-alkoxyphenyl substitution at the 9-position of fluorene could effectively improve the structural stability of molecules incorporating fluorene, leading to higher device performance [17–19]. For instance, our group got highly efficient and spectrally stable blue-light-emitting PFs via introducing two 4-(2-ethylhexyloxy)phenyl groups into the 9-position of fluorene to suppress the generation of fluorenone defects [15].
Sulfur (S)-containing heterocyclic aromatic units such as thiophene [20], benzothiophene [21], dibenzothiophene [22], and benzodithiophene [23] have been a series of efficient bricks for organic semiconductors that have greatly promoted the development of organic electronic devices (e.g., OPVs [5,24–31] and OFETs [6,24,32–35]). For instance, Zhao et al. [25] synthesized a polymer donor and a small molecule acceptor, which were both based on various S-containing aromatic scaffolds, and applied them as the active layer components to fullerene-free OPV, during which a record power conversion efficiency as high as 13.1% was achieved. Our group developed a p-type small molecule based on benzothiophene-fused indolocarbazole, and the OFET devices incorporating this molecule displayed high hole mobility of 0.17 cm2 V−1 s−1 and excellent stability with little roll-off of hole mobility in air [6].
Herein, we present the design and synthesis of two novel S-containing polycyclic aromatic small molecules: 9,9-bis(4-(2-(2-methoxyethoxy)ethoxy)phenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (FBTPhO3) and 9,9-bis(4-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)phenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (FBTPhO4). As compared with the compound (FBT) reported by our group previously [36], the introduction of phenyl at the 9-position of fluorene cores is expected to increase the thermal stability and inoxidizability toward light and electricity, which may be beneficial for improving the performance of optoelectronic devices. Their thermal, optical, electrochemical properties, and morphology as well as theoretical computation were investigated in detail. This class of compounds may be promising building blocks for high-performance optoelectronic functional materials.
2 Experimental section
2.1 Measurement and characterization
1H NMR and 13C NMR measurements were carried out using a Bruker AV-500 spectrometer (Bruker, Rheinstetten, Germany) operating at 500 MHz (for 1H NMR) and 75 MHz (for 13C NMR) with tetramethylsilane as the internal reference. Atmospheric pressure chemical ionization-mass spectrometry (APCI-MS) analyses were obtained from an Acquity Waters UPLC equipped with a Waters Acquity TQ detector (Thermo Finnigan LCQ Fleet system; Waters, Milford, MA). Elemental analyses were performed using a Vario EL elemental analysis instrument (Elementar Co., Langenselbold, Germany). Melting points (mp) were tested on a micro melting point apparatus (Beijing Tech Instrument Co. Ltd., Beijing, China). Thermogravimetric analysis (TGA) tests were conducted using a NETZSCH TG 209 at a heating rate of 10 °C min−1 under nitrogen flow. Differential scanning calorimetry (DSC) tests were measured using a Netzsch DSC 204 under nitrogen flow at a heating rate of 10 °C min−1 and a cooling rate of 20 °C min−1. UV–vis absorption spectra were recorded using an HP 8453 spectrophotometer. Photoluminescence (PL) spectra were recorded using an Instaspec IV CCD spectrophotometer (Oriel Co., Stratford, CT). Cyclic voltammetry (CV) measurements were characterized on a CHI600D electrochemical workstation with a standard three electrode cell based on a Pt wire counter electrode and a Pt working electrode, against a saturated calomel electrode (SCE) as the reference electrode at a scan rate of 50 mV s−1 within a nitrogen-saturated anhydrous solution of 0.1 mol L−1 tetrabutylammonium hexafluorophosphates in acetonitrile versus ferrocene/ferrocenium (Fc/Fc+) as the internal reference. The theoretical computation was carried out using Gaussian 09 at the B3LYP/6-31G(d) level. Atomic force microscope (AFM) studies were carried out using a Digita Instrumental DI Multimode Nanoscope IIIa in taping mode.
2.2 Materials
All reagents, unless otherwise specified, were purchased from Aladdin Chemical Co., Sigma-Aldrich Chemical Co., Alfa Aesar Chemical Co., and J&K Chemical Co. and were used as received. The solvents were further purified by normal procedures and distilled before use. All air and water sensitive synthetic manipulations were performed under dry argon or nitrogen atmosphere. 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaboralan-2-yl)-9,9-bis(4-(2-ethylhexyloxy)phenyl)fluorene (1) [15], 1-bromo-2-(methylsulfinyl)benzene (2) [36], 2-(2-methoxyethoxy)ethyl 4-methylbenzenesulfonate (7) [37], and 2-(2-(2-methoxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (8) [37] were synthesized according to the previously published methods.
2.2.1 2,7-Bis(2-(methylsulfinyl)phenyl)-9,9-bis(4-(2-ethylhexyloxy)phenyl)fluorene (3)
Compound 1 (2.48 g, 3.0 mmol), 2 (1.64 g, 7.5 mmol), Bu4N+Br− (50 mg), K2CO3 aqueous solution (2 M, 6.0 mL), Pd(PPh3)4 (0.347 g, 0.30 mmol), and 30 mL of toluene were mixed together under argon. The reaction was heated to 85 °C and stirred overnight with argon protection. Then the mixture was cooled to room temperature (r.t.) and water was added. The aqueous layer was extracted three times with CH2Cl2. The combined organic layer was washed with brine and then dried over anhydrous MgSO4. The organic solvent was evaporated and the crude product was purified by column chromatography on silica gel using ethyl acetate/petroleum ether (1/4, v/v) as eluent to afford 3 as a colorless oil (1.35 g, 53%). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.11–8.09 (d, J = 10 Hz, 2H), 7.87–7.85 (d, J = 10 Hz, 2H), 7.62–7.59 (m, 2H), 7.54–7.51 (m, 2H), 7.40–7.37 (m, 4H), 7.34–7.33 (d, J = 10 Hz, 2H), 7.16–7.06 (m, 4H), 6.84–6.69 (m, 4H), 3.83–3.71 (m, 4H), 2.32 (s, 6H), 1.72–1.65 (m, 2H), 1.43–1.24 (m, 16H), 0.93–0.84 (m, 12H). 13C NMR (75 MHz, CDCl3) δ (ppm): 158.41, 153.09, 143.83, 139.44, 139.27, 137.56, 130.72, 130.30, 128.94, 128.90, 128.83, 128.58, 126.97, 123.53, 120.86, 114.27, 70.43, 64.39, 41.40, 39.35, 30.47, 29.05, 23.80, 23.01, 14.07, 11.09. MS (APCI) calculated for C55H62O4S2: 851.2, found: 852.3. Elemental analysis calculated for C55H62O4S2: C, 77.61; H, 7.34; S, 7.53, found: C, 77.44; H, 7.68; S, 7.18.
2.2.2 Sulfonium salt intermediate (4) and 9,9-bis(4-(2-ethylhexyloxy)phenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (5)
Compound 3 (5.10 g, 6.0 mmol) and P2O5 (2.0 g) were mixed with 100 mL of CF3COOH, and the reaction was stirred at r.t. for 24 h. The reaction mixture was poured into ice water and subsequently filtered. The resulting yellow residue (crude 4) was dried under vacuum at r.t. and then mixed with 100 mL of pyridine. The mixture was heated to 115 °C and stirred for 24 h. After being cooled to r.t., the reaction was neutralized with dilute HCl aqueous solution. The resulting aqueous layer was extracted three times with CH2Cl2. The combined organic layer was washed with brine before drying over anhydrous MgSO4. The organic solvent was distilled and the crude product was purified by column chromatography on silica gel using CH2Cl2/petroleum ether (1/20, v/v) as eluent to afford 5 as a white solid (2.69 g, 57%). Mp 165 °C [recrystallization solvents: tetrahydrofuran (THF)/ethyl alcohol (1/5, v/v)]. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.27 (s, 2H), 8.11 (s, 2H), 8.07–8.06 (m, 2H), 7.84–7.82 (m, 2H), 7.44–7.39 (m, 4H), 7.28–7.26 (d, J = 10 Hz, 4H), 6.79–6.77 (d, J = 10 Hz, 4H), 3.78–3.76 (m, 4H), 1.69–1.64 (m, 2H), 1.48–1.26 (m, 16H), 0.89–0.85 (m, 12H). 13C NMR (75 MHz, CDCl3) δ (ppm): 158.26, 149.95, 139.86, 139.18, 138.94, 138.14, 135.74, 135.48, 129.23, 126.62, 124.33, 122.85, 121.66, 119.15, 114.23, 114.18, 70.28, 63.59, 39.38, 30.50, 29.06, 23.83, 23.02, 14.07, 11.10. MS (APCI) calculated for C53H54O2S2: 787.1, found: 788.3. Elemental analysis calculated for C53H54O2S2: C, 80.87; H, 6.91; S, 8.15, found: C, 80.78; H, 7.03; S, 8.06.
2.2.3 9,9-Bis(4-hydroxyphenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (6)
Compound 5 (3.15 g, 4.0 mmol) was dissolved in 30 mL of CH2Cl2, after which BBr3 (2.23 mL, 24 mmol) was added slowly at 0 °C under argon. The reaction was stirred overnight, and water was dropped in carefully to quench the reaction. The aqueous layer was extracted three times with ethyl acetate. The combined organic layer was washed with brine before drying over anhydrous MgSO4. The organic solvent was distilled and the crude product was purified by column chromatography on silica gel using ethyl acetate/petroleum ether (1/3, v/v) as eluent to afford 6 as a white solid (1.89 g, 84%). Mp 364 °C [recrystallization solvents: THF/n-hexane (1/1, v/v)]. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.31 (s, 2H), 8.64 (s, 2H), 8.40–8.38 (d, J = 10 Hz, 2H), 8.34 (s, 2H), 8.03–8.01 (d, J = 10 Hz, 2H), 7.52–7.45 (m, 4H), 7.14–7.12 (d, J = 10 Hz, 4H), 6.67–6.65 (d, J = 10 Hz, 4H). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 156.54, 150.59, 139.42, 139.06, 138.67, 136.94, 135.59, 135.41, 129.51, 127.53, 125.23, 123.55, 122.76, 119.90, 115.48, 115.30. MS (APCI) calculated for C37H22O2S2: 562.7, found: 564.1. Elemental analysis calculated for C37H22O2S2: C, 78.98; H, 3.94; S, 11.40, found: C, 78.90; H, 3.85; S, 11.57.
2.2.4 9,9-Bis(4-(2-(2-methoxyethoxy)ethoxy)phenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (FBTPhO3)
Compound 6 (0.563 g, 1.0 mmol) and NaH (72.0 mg, 3.0 mmol) were mixed with 5 mL of THF under nitrogen. The reaction was stirred at r.t. for 0.5 h. After compound 7 (0.822 g, 3.0 mmol) was added under nitrogen, the mixture was stirred overnight. Water was added to the reaction slowly, and the aqueous layer was extracted three times with ethyl acetate. The combined organic layer was washed with brine before drying over anhydrous MgSO4. The organic solvent was distilled and the crude product was purified by column chromatography on silica gel using ethyl acetate/petroleum ether (1/4, v/v) as eluent to afford FBTPhO3 as a white solid (0.614 g, 80%). Mp 136 °C [recrystallization solvents: THF/methanol (1/5, v/v)]. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.26 (s, 2H), 8.10 (s, 2H), 8.08–8.07 (m, 2H), 7.84–7.82 (m, 2H), 7.44–7.40 (m, 4H), 7.27–7.25 (d, J = 10 Hz, 4H), 6.82–6.80 (d, J = 10 Hz, 4H), 4.09–4.07 (t, J = 5 Hz, 4H), 3.82–3.80 (t, J = 5 Hz, 4H), 3.69–3.67 (m, 4H), 3.54–3.52 (m, 4H), 3.36 (s, 6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 157.64, 149.80, 139.86, 139.24, 138.92, 138.63, 135.74, 135.44, 129.27, 126.65, 124.36, 122.84, 121.67, 119.12, 114.43, 114.21, 71.93, 70.69, 69.69, 67.34, 63.56, 59.06. MS (APCI) calculated for C47H42O6S2: 767.0, found: 768.2. Elemental analysis calculated for C47H42O6S2: C, 73.60; H, 5.52; S, 8.36, found: C, 73.48; H, 5.66; S, 8.41.
2.2.5 9,9-Bis(4-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)phenyl)fluorene bis[2,3-b;6,7-b]benzo[d]thiophene (FBTPhO4)
The synthetic procedure was identical to FBTPhO3, finally FBTPhO4 was obtained as a white solid (0.667 g, 78%). Mp 109 °C [recrystallization solvents: THF/methanol (1/5, v/v)]. 1H NMR (500 MHz, CDCl3) δ (ppm): 8.25 (s, 2H), 8.15 (s, 2H), 8.09–8.07 (m, 2H), 7.82–7.80 (m, 2H), 7.42–7.38 (m, 4H), 7.33–7.31 (d, J = 10 Hz, 4H), 6.86–6.84 (d, J = 10 Hz, 4H), 4.10–4.08 (t, J = 5 Hz, 4H), 3.83–3.81 (t, J = 5 Hz, 4H), 3.72–3.69 (m, 4H), 3.66–3.62 (m, 8H), 3.53–3.51 (m, 4H), 3.35 (s, 6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 157.72, 149.84, 139.89, 139.29, 138.96, 138.72, 135.78, 135.45, 129.35, 126.70, 124.41, 122.86, 121.70, 119.16, 114.51, 114.30, 71.94, 70.80, 70.66, 70.57, 69.71, 67.41, 63.64, 59.04. MS (APCI) calculated for C51H50O8S2: 855.1, found: 856.2. Elemental analysis calculated for C51H50O8S2: C, 71.64; H, 5.89; S, 7.50, found: C, 71.79; H, 5.80; S, 7.59.
3 Results and discussion
3.1 Synthesis
The synthetic route for these small molecules is shown in Scheme 1. The common Pd(0)-catalyzed Suzuki cross-coupling reaction between compounds 1 and 2 offered compound 3 that was purified by column chromatography on silica gel. Under the treatment with CF3COOH and P2O5 at r.t., the intramolecular electrophilic coupling reaction took place within compound 3, which generated the sulfonium salt intermediate 4. The reaction mixture was poured into ice water and subsequently filtered. The resulting yellow residue (crude 4) was dried under vacuum at r.t. and directly used for demethylation to afford 5 without further isolation. In the previous reports, CF3SO3H [6,36,38–41] and CF3COOH [42] have been used widely to prepare the sulfonium salt intermediates. Herein, CF3COOH was used as the strong acid, and it was found that CF3SO3H is useless for the transformation from 3 to 4, which may be associated with the degradation of oligo(ethylene glycol) (OEG)-substituted phenyl groups in CF3SO3H. The chemical structure of 5 was verified by 1H NMR and 13C NMR (Fig. S1–S2, ESI), MS (APCI), and elemental analysis. Then, compound 6 was synthesized via the dealkylation of 5 in the presence of BBr3, and the target compound FBTPhO3 (or FBTPhO4) was acquired via the reaction between 6 and 7 (or 8) with NaH as the base. The chemical structures of FBTPhO3 and FBTPhO4 were confirmed by 1H NMR and 13C NMR (Fig. S3–S6), MS (APCI), and elemental analysis. The resulting molecules can be readily dissolved in common organic solvents such as chloroform (CF), THF, and dioxane because of their long OEG side chains.
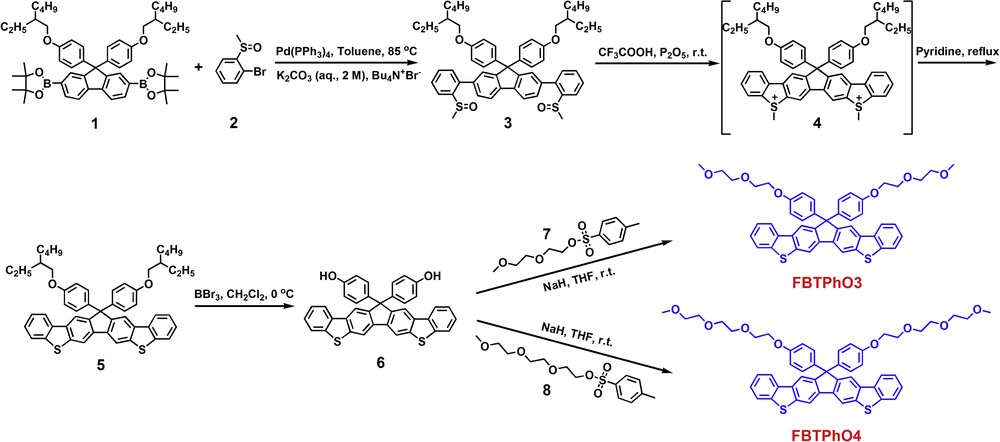
Synthetic route for FBTPhO3 and FBTPhO4.
3.2 Thermal properties
The thermal properties of FBTPhO3 and FBTPhO4 were investigated by TGA and DSC as shown in Fig. 1. Both compounds own single thermal decomposition process, which is attributed to the cleavage of OEG side chains. The 5% weight loss temperature (Td) is about 398 °C for FBTPhO3 and 384 °C for FBTPhO4. The TGA results mean that these molecules own higher thermal stability than the previously reported FBT (with a Td of 350 °C) [36], which is beneficial for constructing high-performance organic semiconductors. During the DSC tests, although FBTPhO3 melted at about 139 °C during the first heating process, no crystallization upon cooling or melting upon reheating could be observed. Instead, it exhibited obvious glass transition at about 72 °C during both the first and second heating processes. The DSC studies revealed the amorphous state of FBTPhO3 clearly. For FBTPhO4, no glass transition emerged during the first or second heating, probably because of its longer side chains and thus higher flexibility than FBTPhO3. Similar to FBTPhO3, no crystallization upon cooling or melting upon reheating was seen for FBTPhO4 although it melted at 109 °C during the first heating, which uncovered its amorphous state.
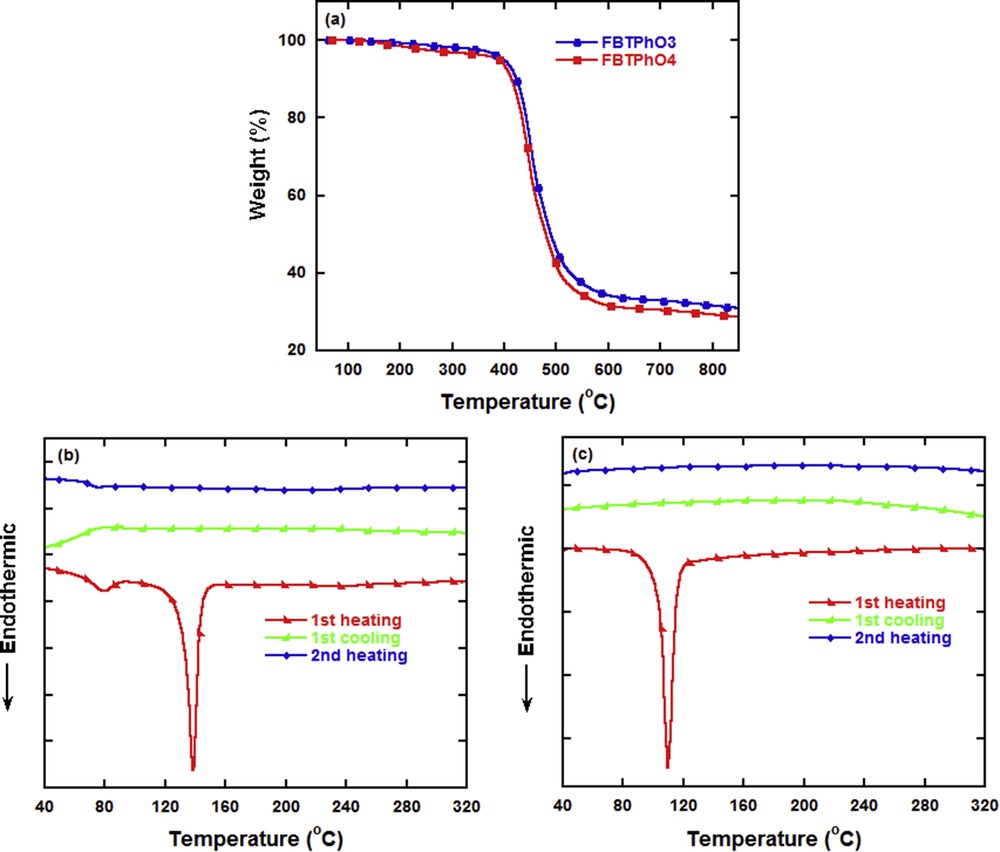
(a) TGA curves of FBTPhO3 and FBTPhO4 and DSC curves of (b) FBTPhO3 and (c) FBTPhO4.
3.3 Optical properties
The UV–vis absorption and PL spectra of FBTPhO3 and FBTPhO4 in different solvents and film are shown in Fig. 2, and the corresponding optical parameters are summarized in Tables 1 and 2, respectively. In THF, FBTPhO3 displayed absorption peaks at 343 and 368 nm, which was assigned to the π–π* transition of the conjugated backbone. When altering the testing solvents, the absorption profiles and peaks showed no obvious difference: specifically, the absorption maximal peaks were 368, 370, 368, 367, and 370 nm in toluene, CF, dimethyl formamide (DMF), methanol, and dimethyl sulfoxide (DMSO), respectively. In contrast, the absorption spectrum of FBTPhO3 in film exhibited obviously bathochromic shift and broadening (the peaks were located at 348 and 377 nm), which resulted from the intermolecular aggregation in the film state. For the absorption spectra of FBTPhO4 in various polar solvents and film, the similar variation trend as FBTPhO3 could be observed. For instance, the absorption maximal peaks were at 370, 371, 368, 368, 366, 370, and 376 nm in toluene, CF, THF, DMF, methanol, DMSO, and film, respectively. The optical band gap (Egopt) values calculated from the absorption onset in the film are 3.20 and 3.17 eV for FBTPhO3 and FBTPhO4, respectively.
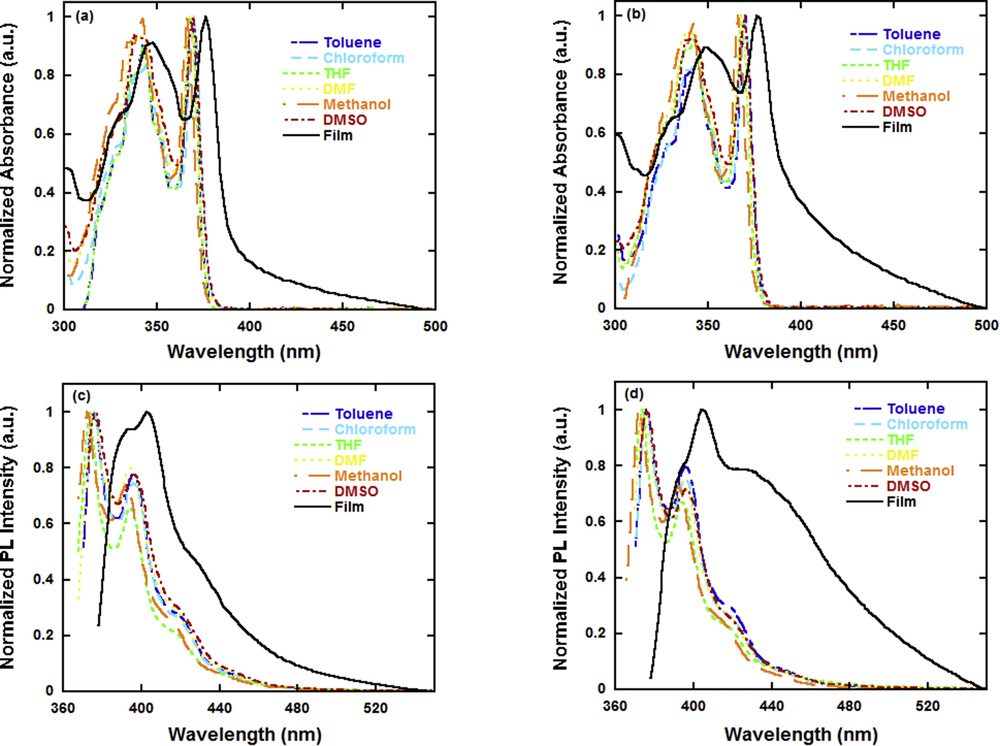
UV–vis absorption spectra of (a) FBTPhO3 and (b) FBTPhO4 and PL spectra of (c) FBTPhO3 and (d) FBTPhO4.
UV–vis absorption parameters of FBTPhO3 and FBTPhO4.
| Molecule | λabs,toluene (nm) | λabs,CF (nm) | λabs,THF (nm) | λabs,DMF (nm) | λabs,methanol (nm) | λabs,DMSO (nm) | λabs,film (nm) | Egopt (eV) |
| FBTPhO3 | 343 | 344 | 343 | 343 | 342 | 339 | 348 | 3.20 |
| 368 | 370 | 368 | 368 | 367 | 370 | 377 | ||
| FBTPhO4 | 343 | 344 | 343 | 343 | 343 | 342 | 349 | 3.17 |
| 370 | 371 | 368 | 368 | 366 | 370 | 376 |
PL parameters of FBTPhO3 and FBTPhO4.
| Molecule | λPL,toluene (nm) | λPL,CF (nm) | λPL,THF (nm) | λPL,DMF (nm) | λPL,methanol (nm) | λPL,DMSO (nm) | λPL,film (nm) |
| FBTPhO3 | 376 | 376 | 374 | 375 | 373 | 377 | 393 |
| 396 | 396 | 394 | 394 | 393 | 396 | 403 | |
| FBTPhO4 | 376 | 376 | 374 | 375 | 373 | 377 | 405 |
| 395 | 397 | 394 | 394 | 393 | 397 |
To get more insightful understanding into the optical properties of these molecules, their PL spectra were also recorded. As shown in Fig. 2c, the polarity of solvents did not affect the PL spectra of FBTPhO3 markedly, and the emission dominant peaks were located at 376, 376, 374, 375, 373, and 377 nm in toluene, CF, THF, DMF, methanol, and DMSO, respectively. This phenomenon might be attributed to the lack of intramolecular charge transfer (ICT) interaction within the conjugated backbone of FBTPhO3, leading to weak solvation effects between these solvents and FBTPhO3 [36,43]. As compared to the PL spectra of FBTPhO3 in solutions, in film that was dramatically red-shifted because of the intermolecular stacking in solid state. On the other hand, for the PL spectra of FBTPhO4 in various polar solvents and film, the similar changing trend as FBTPhO3 was observed (Fig. 2d). Moreover, it could be found that FBTPhO3 and FBTPhO4 showed very close absorption and emission peaks in the same solvent, implying that the OEG side chains do not affect the electronic structures of these two molecules in an isolated state, which is consistent with the previous report [37].
3.4 Electrochemical properties
CV was used to investigate the electrochemical properties of FBTPhO3 and FBTPhO4 as shown in Fig. 3. The oxidation potential of Fc/Fc+ was measured to be 0.42 V using SCE as a reference electrode under the same experimental condition. Because the has an absolute energy level of −4.80 eV to vacuum energy level [44], the highest occupied molecular orbital (HOMO) energy level (EHOMO) and lowest unoccupied molecular orbital (LUMO) energy level (ELUMO) values could be calculated according to the following empirical equations: EHOMO = −e (Eox + 4.8 − 0.42) (eV), ELUMO = −e (Ered + 4.8 − 0.42) (eV), where Eox and Ered are the onset oxidation and onset reduction potentials versus SCE, respectively. The Eox was tested as 1.29 V for FBTPhO3 and 1.30 V for FBTPhO4y. Hence, the EHOMO values were calculated to be −5.67 and −5.68 eV for FBTPhO3 and FBTPhO4, respectively. Their deep-lying EHOMO values suggest that they own good inoxidizability. In addition, FBTPhO3 and FBTPhO4 exhibit ELUMO of −2.31 and −2.32 eV, respectively, which were estimated from their Ered of −2.07 and −2.06 V, respectively. The similar electronic energy level structures of FBTPhO3 and FBTPhO4 are assigned to their same conjugated backbones in spite of their different OEG side chains.

CV curves of FBTPhO3 and FBTPhO4 in a film.
3.5 Theoretical computation
To understand the spatial distributions of frontier molecular orbitals of FBTPhO3 and FBTPhO4, density functional theory calculation was performed to analyze these compounds using the Gaussian 09 program at the B3LYP/6-31G(d) level. Because OEG side chains have a little impact on the energy level structures of these conjugated molecules, the OEG side chains within FBTPhO3 and FBTPhO4 were replaced with methoxy groups during calculation. As shown in Fig. 4, the HOMO was spread out over all of the aromatic rings including the phenyl groups at the 9-position of the fluorene core without distinct localization and the LUMO just sprawled in the conjugated backbone excluding the phenyl groups. The calculated distribution of HOMO and LUMO confirmed the lack of ICT interaction within the conjugated backbone, which resulted in weak solvation effects. The calculated EHOMO is −5.45 eV and ELUMO is −1.58 eV.
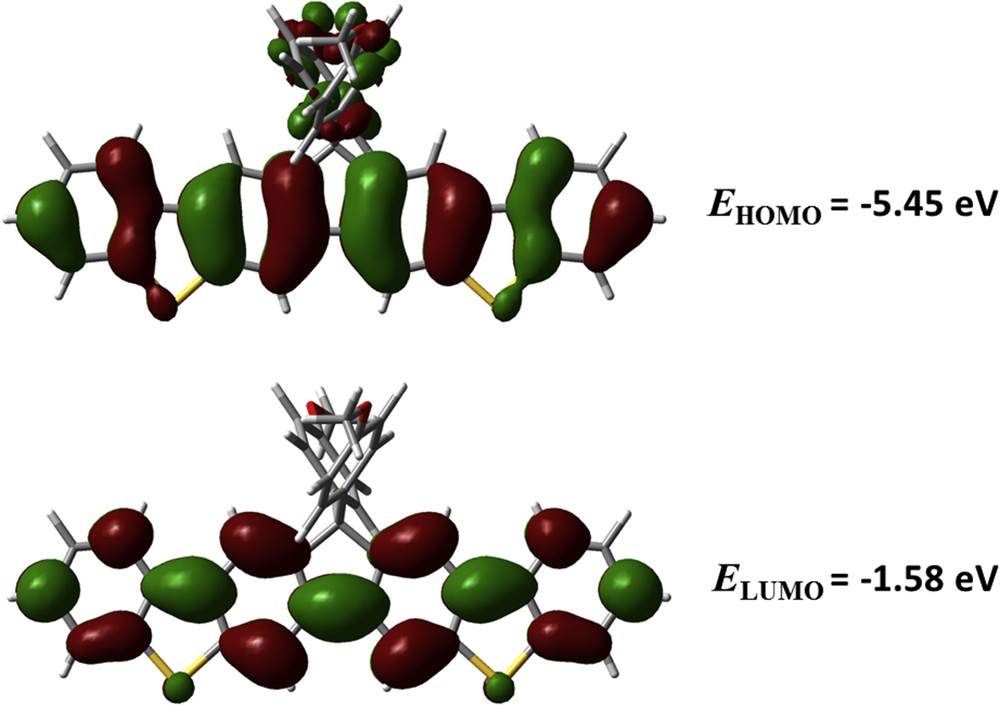
Spatial distributions of HOMO (top) and LUMO (bottom) of the resulting compounds calculated by density functional theory.
3.6 Morphology
AFM was used to analyze the surface morphology of FBTPhO3 and FBTPhO4 films (the films were spin-coated on top of indium tin oxide substrates), and the images are shown in Fig. 5. FBTPhO3 film exhibited a relatively rough surface with root-mean-square (RMS) roughness of 12.3 nm. In contrast, the RMS roughness of the FBTPhO4 film drastically decreased to 2.79 nm, which was probably attributed to the longer OEG side chains and thus better film-forming ability for FBTPhO4. It is believed that the film-forming properties can be further improved after structural modifications toward these molecules (e.g., preparing conjugated polymers based on them), and the derivatives may be useful for organic electronic devices.

Surface topographic AFM images (2.3 μm × 2.3 μm) of (a) FBTPhO3 and (b) FBTPhO4 films.
4 Conclusions
To sum up, two novel S-containing polycyclic aromatic compounds (FBTPhO3 and FBTPhO4), whose fluorene cores are functionalized with phenyl groups substituted by OEG chains, were designed and synthesized through an intramolecular electrophilic cyclizing reaction in the presence of CF3COOH and subsequent demethylation induced by pyridine. TGA demonstrated that they both own excellent thermal stability. DSC tests revealed their amorphous state in nature. As compared to FBTPhO3, FBTPhO4 does not exhibit glass transition before melting probably because of its longer OEG side chains and thus higher flexibility. From the PL spectra in different solvents, it was found that FBTPhO3 and FBTPhO4 both display deep blue light emission and weak solvation effects because of the lack of ICT interactions within their conjugated backbones. From the CV measurements, the ELUMO values are −2.31 and −2.32 eV and the EHOMO values are −5.67 and −5.68 eV for FBTPhO3 and FBTPhO4, respectively, which mean that they both own good inoxidizability and close energy level structures regardless of their difference in the OEG chain length. Moreover, AFM tests suggested that as compared with FBTPhO3, better film-forming properties can be realized by FBTPhO4 because of its longer OEG side chains. These molecules may be promising building blocks for high-performance conjugated organic small molecules and polymers.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (nos. 51273069, 91333206), Guangdong Provincial and Municipal Co-construction Program for Jiaying University (No. 252B0101), Guangdong Natural Science Foundation (No. 2017A030307019), and Science and Technology Program of Guangdong, China (No. 2014A020216050).