

1 Introduction
The evolution and continuous discovery of the Pd-catalyzed cross-coupling reactions [1] greatly helped to form and synthesize highly arylated heterocycles. As a result, heterocyclic scaffolds with various functions, which can be alternated into a variety of functionalized heterocycles by using regioselective and efficient reactions, have become more attractive [2].
In the search for novel biocompatible heterocycles with nitrogens, pyridopyrimidine appears to be a very important molecule. Inclusion of this heterocycle in more complex structures has guided to a diverse interval of molecules that can be useful as antibacterial [3], antifolate [4], anti-inflammatory [5], antiviral [6], antimicrobial [7] and anticancer agents [8]; antihypertensives [9]; antileishmanials [10]; anticonvulsants [11]; potassium diuretics and preservatives [12]; antiaggressives [13]; and tyrosine kinase [14] and antitumor derivatives [15] with selective proapoptotic activity [16]. This shows the interest of chemists in developing versatile methods for obtaining and functionalizing this heterocycle [17].
Because of our experience with these heterocycles [18], we decided to use this methodology to access the trisubstituted pyrido[2,3-d]pyrimidines at positions C2, C4, and C6 in the isomeric position by Suzuki–Miyaura coupling reactions (Scheme 1). Herein, we report the preparation of new trihalogen derivatives 6 and 7 and both regioselectivity functionalized by two different classes of reactions.
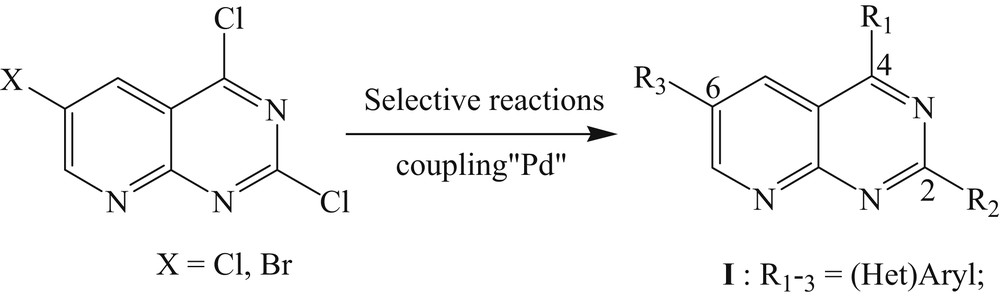
Strategy.
2 Results and discussion
The trichlorination of heterocycles has been observed with pyrido[3,2-d]pyrimidine [19] and quinazoline [20,21]; however, to our knowledge, the direct trichlorination in a single step of pyrido[2,3-d]pyrimidine has not yet been described. We, therefore, performed a halogenation of nicotinic acid before obtaining the final trihalogenated intermediate by cyclization and chlorination.
The halogenation of 2-aminonicotinic acid is now well known [22]. It involves the reaction of chlorine or bromine with 2-aminonicotinic acid in acetic acid medium (Scheme 2) [23].
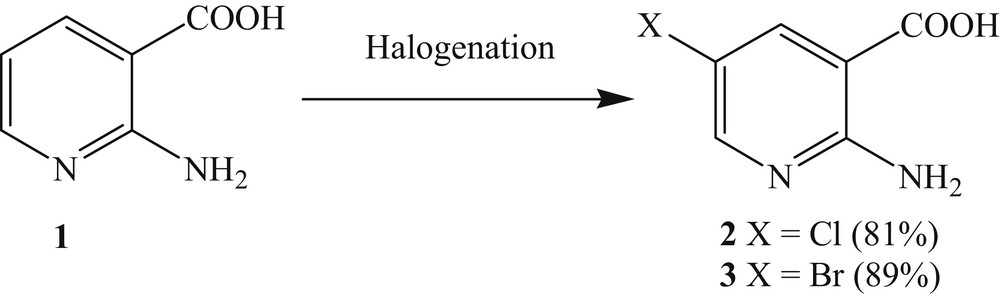
Halogenation of 2-aminonicotinic acid at position C5.
The cyclization and chlorination of the halogenated 2-aminonicotinic acid has been previously described by reaction with urea for cyclization and POCl3 for chlorination [18c].
Products 2 and 3 were heated to 280 °C in the presence of urea to yield compounds 4 and 5 with good yields [24]. Chlorination at positions C2 and C4 was then performed using phosphorus oxychloride and a few drops of DMF at reflux to give intermediates 6 and 7 in good yields (Scheme 3) [25].
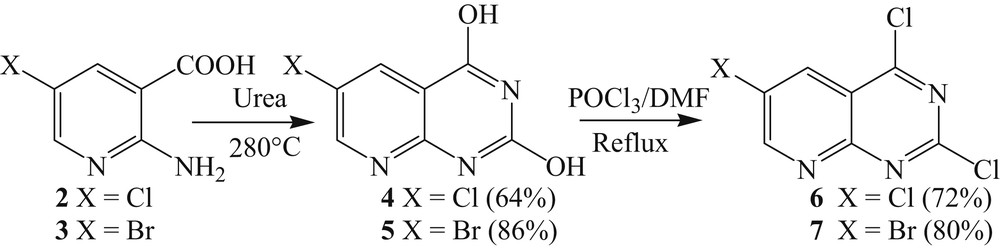
Cyclization and halogenation of compounds 2 and 3.
Compounds 6 and 7 were first engaged in a Suzuki coupling reaction [26]. Then, we carried out a regioselective arylation at C4, by reacting the dichloride compound 6 with 1 equiv of p-methoxyphenylboronic acid using 5 mol % of Pd(PPh3)4, potassium carbonate, and toluene as solvent under reflux (Scheme 4). After completion of the reaction, the monoarylated derivative 8 was obtained in a good yield (83%) (Table 1, entry 1).
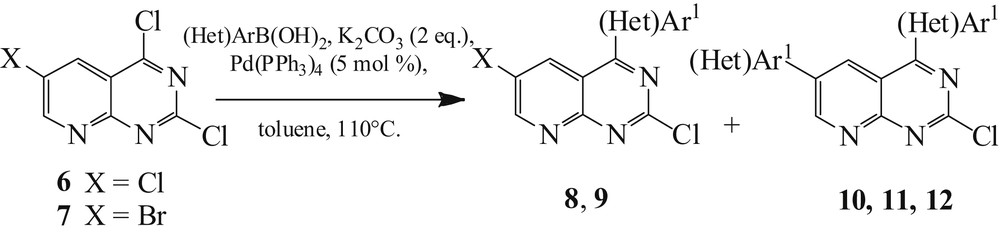
Trihalogenated pyridopyrimidines 6 and 7 (Het)arylation.
Reactivity of halogenated products 6 and 7 with different boronic acids.
| Entry | X | Ar1 | Amount of boronic acid (equiv) | Substitution (yields, time)a | |
| At position C4 | At positions C4 and C6 | ||||
| 1 | Cl | Image 2 | 1 | 8 (83%, 3 h) | – |
| 2 | Br | 1 | 9 (7%, 3 h) | 10 (36%, 3 h) | |
| 3 | Br | 2 | – | 10 (76%, 3 h) | |
| 4 | Br | Image 3 | 2 | – | 11 (71%, 3 h) |
| 5 | Br | Image 4 | 2 | – | 12 (75%, 3 h) |
a Yield of isolated product. Reaction conditions: Ar1B(OH)2, K2CO3 (1.5 equiv), Pd(PPh3)4 (5 mol %), toluene, 110 °C.
For compound 7, a mixture of monoarylated 9 and biarylated 10 was obtained in a relatively low global yield (43%) (Table 1, entry 2). Therefore, we chose to treat compound 7 with 2 equiv of boronic acid to produce the diarylated products (Table 1, entries 3–5) [27]. The heteroaryl (Het)Ar1 was introduced and the diarylated products were successfully obtained after purification on silica gel using column chromatography. Compounds 10–12 were obtained after 3 h in good yields (71–76%) (Table 1).
Chlorine at position C2 of compounds 10–12 was then engaged in a Suzuki coupling and the last (Het)aryl was introduced in the presence of a second boronic acid (Scheme 5, Table 2) [28].

Reactivity of halogenated products 10–12 via Suzuki cross-coupling reaction.
Heteroarylation of compounds 13–15.
| Entry | (Het)Ar1 | (Het)Ar2 | Products | Yielda (time) |
| 1 | Image 2 | Image 4 | 13 | 71% (3 h) |
| 2 | Image 3 | Image 2 | 14 | 67% (2 h) |
| 3 | Image 4 | Image 2 | 15 | 72% (2 h) |
a Yield of pure product. Conditions of reaction: Ar2B(OH)2 (1.05 equiv), Na2CO3 (2 equiv), Pd(PPh3)4 (5 mol %), toluene/ethanol, 110 °C.
The expected products 13, 14, and 15 were obtained after purification on silica gel using column chromatography in good yields of 71%, 67%, and 72%, respectively (Table 2).
We then engaged compound 8 in a second heteroarylation at the position C2 without any influence on the chlorine atom at position C6. For this second arylation we used ethanol as cosolvent and sodium carbonate as base to achieve the best yield (Scheme 6) [29]. The 3-thienylboronic acid reacted with compound 8 after 2 h to obtain the di(arylated) compound 16 in good yield of 83%.

Reactivity of halogenated product 24 via Suzuki coupling reaction.
We then engaged the compound 16 in a third arylation at the C6 position. The best result was obtained in the presence of sodium carbonate and ethanol as cosolvent (Scheme 7) [29].
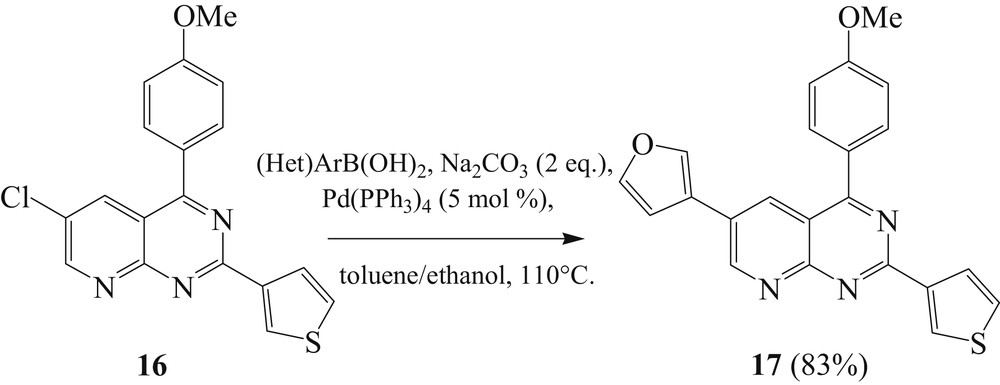
Reactivity of halogenated product 16 via Suzuki coupling reaction.
The diarylated compound 16 reacted in the presence of 3-furylboronic acid to afford the tri(arylated) compound 17 in only a few hours with good yield of 83%.
Usually the Pd-catalyzed site selective arylation reactions occur first at the more electron-deficient site. The selectivity of the site can be clarified by the fact that position C2 of 2,4,6-trihalogenopyrido[2,3-d]pyrimidine is less electron deficient than position C4, and position C6 is less electron deficient than position C2 (Fig. 1) [21,30].
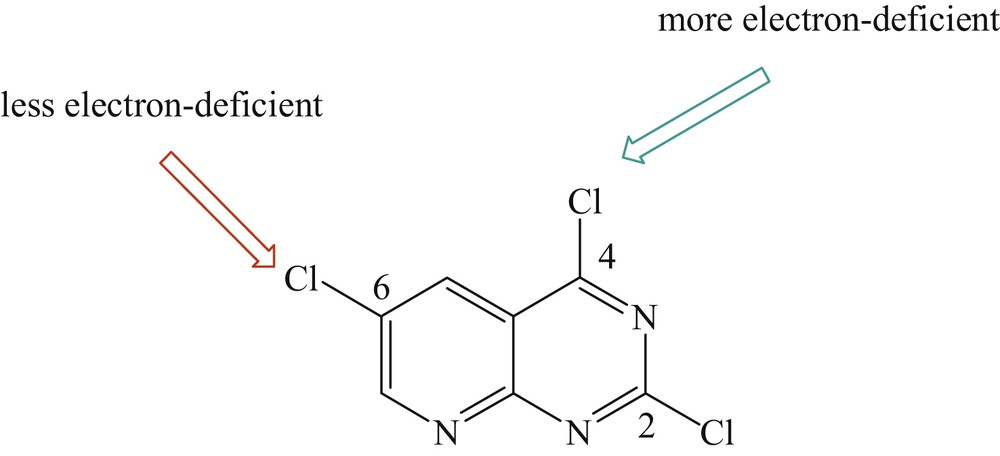
The site selection process of the palladium-catalyzed coupling reactions of 2,4,6-trihalogenopyrido[2,3-d]pyrimidine.
Based on the studies performed by Handy and Zhang [31], we can justify the order and site of the cross-coupling reaction using the 1H NMR chemical shift values of the parent nonhalogenated heterocyclic compound. Indeed, we found that Suzuki coupling reactions were performed successfully in the order C4, C2, and finally C6 (Fig. 2), which means that the carbon with the most deshielded proton is the most reactive carbon.
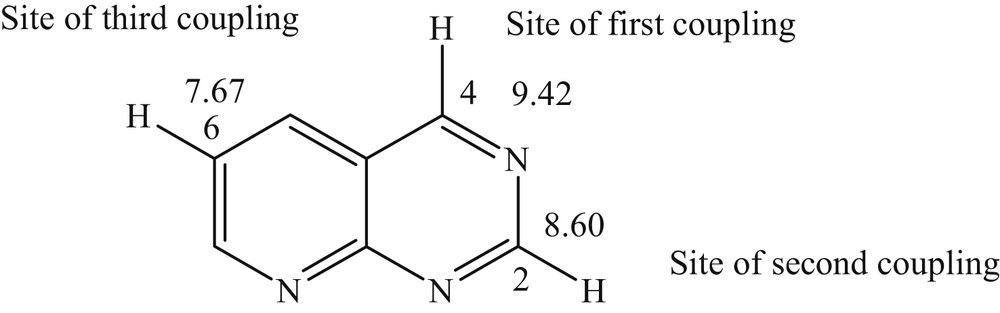
Suzuki cross-coupling order based upon the 1H NMR chemical shift values.
In this article, we have developed the first access to 2,4-dichloro-6-halogeno-pyrido[2,3-d]pyrimidine and its use to prepare two series of substituted pyridopyrimidine using an efficient and novel strategy.
Next, we reported an efficient and simple cross-coupling method for highly trisubstituted-pyrido[2,3-d]pyrimidines, which can help in the orchestration of regioselective palladium-catalyzed cross-coupling reactions for the synthesis of focused libraries of biologically active scaffold.
Acknowledgments
The authors thank the “Volubilis” Hubert Curien Program for financial support.