

1 Introduction
Starting from the seminal work of Tamaki et al. [1], the oxalate anion has been widely investigated for the development of new multifunctional molecule-based magnets. The main reason for the infatuation of this ligand lies in its ability to behave as a bis(bidentate) bridge between two metallic ions. In this widely encountered coordination mode, it can build up essentially two types of coordination networks of formula C[M′IIMIII(ox)3], namely three-dimensional (3D) (10,3) networks [2] and 2D (6,3) compounds presenting a honeycomb-layered structure [3]. The dimensionality of the coordination network is mainly controlled by the nature of the template cation C+, although in the case of 2D compounds this templating activity must be reinforced using tris(oxalato)metallate(III) complexes [M(ox)3]3− (M = CrIII, FeIII, RuIII, VIII, MnIII) as ligand. Within the coordination networks, the exchange interaction between the MIII and M′II paramagnetic centers mediated by the oxalate bridge enables the onset of long-range magnetic ordering with critical temperatures up to 45 K [1,4].
For many years, taking advantage of the versatility of this approach, a welfare of the guest cations (photochromic molecules [5], spin transition complexes [6], organic radical cations [7], paramagnetic decamethylferrocenium [8], photochromic [9], organic π-electron donors [10]) has been intercalated between the 2D coordination layers to yield multifunctional materials associating magnetic ordering with another physical property. Along this line, different functional cations such as hyperpolarizable cationic chromophores have been intercalated between the bimetallic oxalato-bridged magnetic layers to yield second harmonic generation, electro-optic devices [5a,11]. But to observe these properties, it is necessary to control the arrangement in a noncentrosymmetric manner, hence the space group in which the hybrid compound is crystallizing. The crystalline structure of layered compounds displaying strong NLO effect has also been solved using XRD on powder [12], but the challenge for breaking the symmetry in such crystals to induce the formation of noncentrosymmetric crystals remains open. For this purpose, chiral tetraalkyl ammonium cations [13] have been used. Through intermolecular interactions between the alkyl chains and the coordination network, such cations allow one to control the absolute configuration within the coordination layers and observe cross effects such as magnetochiral dichroism [14] or magnetization-induced second harmonic generation arising from the coordination network itself. Recently, counterions presenting H-bonding ability were used to introduce properties due to the template cations themselves like ferroelectricity or proton conduction [15]. But, because H-bonds may compete with coordination ones, this introduction dramatically changed the self-assembly process: instead of the usual networks, discrete clusters or coordination networks with original topologies were observed. With bisamidinium cations known to be active H-bonds donors [16,17], we have obtained binuclear [18] or trinuclear [19] complexes but also highly corrugated 2D bimetallic layers [20].
Herein, two dialkyl-substituted monoamidinium cations, namely 1-propyl-2-phenyl-3-methylamidinium (A) and 1-hexyl-2-phenyl-3-methylamidinium (B) (Scheme 1), are used to template the formation of layered oxalate-based assemblies. The cations present a low symmetry whereas the alkyl substitution hinders their H-donating ability: these characteristics will be analyzed here to the light of previous results obtained with more symmetrical H-bond donor cations [18–20]. The synthesis of two 2D-layered oxalate-based compounds A[MnIICrIII(C2O4)3] (1) and B[MnIICrIII(C2O4)3] (2) is presented thereafter. The structures of 1 and 2 are described and discussed in detail. Finally, their magnetic properties are presented.
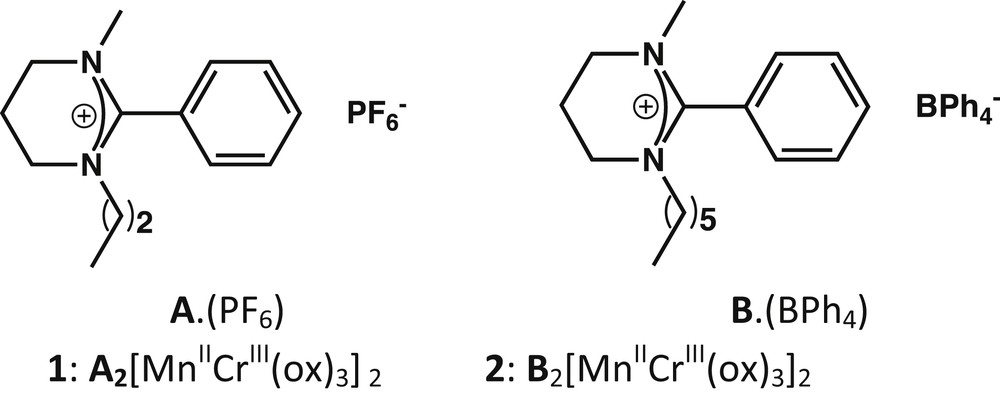
The used monoamidinium cations A+ and B+ for the formation of 1 and 2.
2 Materials and methods
2.1 Synthesis
MnCl2·4H2O was purchased from commercial sources and used as received. (NH4)3[Cr(ox)3]·3H2O [21], A.(PF6), and B.(BPh4) were prepared following the published procedures [22].
2.1.1 Synthesis of 1 and 2
A2[MnIICrIII(ox)3]2 (1): (NH4)3[Cr(ox)3]·3H2O (85 mg, 0.2 mmol) and MnCl2·4H2O (40 mg, 0.2 mmol) in 8 mL methanol was introduced in a 1.5 cm diameter tube and layered with 10 mL CH3OH. A methanolic solution (5 mL) containing A(PF6) (73 mg, 0.2 mmol) was then layered. After 2–3 days of slow evaporation, violet crystals were formed at the interface. Elemental analysis calculated (%) for C40H42Cr2Mn2N4O24: C 40.83%, H 3.59%, N 4.76%; found: C 40.47%, H 3.65%, N 4.71%; IR: ν = 3195, 3069 (C–H), 1627, 1458, 1434 cm−1 (C–O) (see Fig. S1, Supplementary Material).
B2[MnIICrIII(ox)3]2 (2): (NH4)3[Cr(ox)3]·3H2O (52 mg, 0.125 mmol) and MnCl2·4H2O (25 mg, 0.125 mmol) in 8 mL methanol was introduced in a 1.5 cm diameter tube and layered with 10 mL CH3OH/CH3CN solution. A CH3CN solution (5 mL) containing B(BPh4) (73 mg, 0.125 mmol) was then layered. After 2–3 days of slow evaporation, violet crystals were formed at the interface. Elemental analysis calculated (%) for C46H54Cr2Mn2N4O24: C 43.82%, H 4.31%, N 4.41%; found: C 43.42%, H 4.21%, N 4.54%; IR: ν = 3120, 2950 (C–H), 1627, 1434, 1324 cm−1 (CO) (see Fig. S2, Supplementary Material).
2.2 Characterization techniques
FT-IR spectra were recorded using a PerkinElmer ATR spectrometer.
Elemental analyses were performed by the Service de Microanalyses de la Fédération de Recherche Chimie of the University de Strasbourg.
Thermogravimetric analysis (TGA) measurements have been performed on polycrystalline compounds on Pyris 6 TGA Lab System (PerkinElmer), using a N2 flow of 20 mL/min and a heat rate of 4 °C/min.
Variable temperature (2.0–300 K) magnetic susceptibility measurements were carried out on polycrystalline samples checked by powder XRD (PXRD) using a MPMS SQUID magnetometer by applying a 1000 G external magnetic field.
2.3 Structural studies
2.3.1 Powder diffraction studies
PXRD diagrams were collected on polycrystalline samples, on a Bruker D8 diffractometer using monochromatic Cu Kα radiation with a scanning range between 3.8° and 40° using a scan step size of 2°/min.
2.3.2 Single-crystal studies
Data were collected at 173(2) K using a Bruker APEX8 CCD Diffractometer equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo Kα (λ = 0.71073) radiation. The structures were solved by direct methods and refined by full-matrix least squares techniques based on F2. The non-H atoms were refined with anisotropic displacement parameters. Calculations were performed using SHELX-2013 crystallographic software package. All hydrogen atoms were generated geometrically [23].
CCDC 1890603–1890604 contain supplementary crystallographic data for the two compounds. They can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/datarequest/cif.
3 Results and discussion
3.1 Synthesis
Compounds 1 and 2 were obtained as crystalline materials suitable for XRD by slow evaporation at room temperature of a methanolic solution containing the ammonium salt of tris(oxalato)chromate(III), chosen for its solubility in methanol, manganese(II) chloride, and the hexafluorophosphate salt of the amidinium cation A+ for 1 and tetraphenylborate salt for amidinium cation B+ for 2. For 1 and 2, PXRD analysis of the polycrystalline samples (Fig. 1) demonstrated that only one crystalline phase is obtained during the crystallization processes, even if discrepancies in intensity were observed between the observed and simulated patterns, owing to the preferential orientations of the microcrystalline powders.
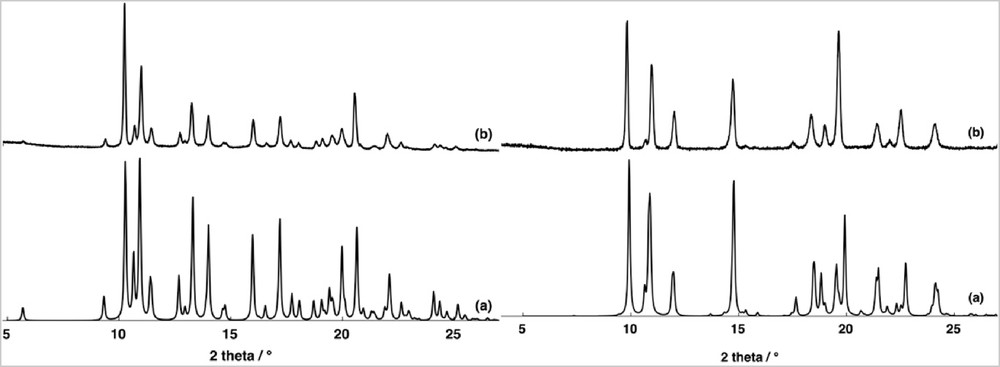
Comparison of PXRD patterns for 1 (left) and 2 (right) (a) calculated and powdered recorded (b).
TGA measurements (Fig. S3) confirm that the products are pure and single phased. In particular, there are no traces of the ammonium-templated 3D bimetallic networks previously synthesized [15], indicating a stronger templating activity of the amidinium cations. Moreover, the abruptness of the TGA traces (Fig. S3) gives evidence that for both compounds there are no solvent molecules inside the unit cell. This conclusion is supported by the single-crystals XRD analysis.
3.2 Single-crystal XRD
XRD on single crystals showed that compounds 1 and 2 crystallize in the polar Pc and chiral P21 space groups, respectively (Table 1). In both compounds, the asymmetric unit is composed of two amidinium monocationic (A+ or B+) and a [Mn2Cr2(ox)6]2− dianionic moiety, leading to the formulas (C14H21N2)2[Mn2Cr2(C2O4)6] for 1 and (C17H27N2)2 [Mn2Cr2(C2O4)6] for 2, evidencing the absence of solvent molecule in the unit cell (Fig. 2a and b).
Crystallographic parameters for compounds 1 and 2 recorded at 173 K.
| 1 | 2 | |
| Formula | C40H42Cr2Mn2N4O24 (Cr2Mn2C12O24)(C14H21N2)2 | C46H54Cr2Mn2N4O24 (Cr2Mn2C12O24)(C17H27N2)2 |
| Molecular weight | 1176.66 | 1260.81 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | Pc | P21 |
| a (Å) | 16.033(3) | 9.3876(2) |
| b (Å) | 9.4755(19) | 17.8144(4) |
| c (Å) | 17.770(4) | 16.2318(4) |
| β (deg) | 104.56(3) | 90.6230(10) |
| V (Å3) | 2613.0(9) | 2714.36(11) |
| Z | 2 | 2 |
| Color | Violet | Purple |
| Dcalc (g cm−3) | 1.496 | 1.543 |
| F(000) | 1200 | 1296 |
| μ (mm−1) | 0.960 | 0.930 |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Number of data meas. | 25,554 | 18,508 |
| Number of data with I > 2σ(I) | 10773 | 10257 |
| Final R1a, wR2b [I > 2σ(I)] | 0.0963, 0.1003 | 0.0914, 0.1002 |
| R1a, wR2b (all data) | 0.2460, 0.2519 | 0.2459, 0.2566 |
| GOF | 1.028 | 1.062 |
| Largest peak in final difference (eÅ−3) | −0.582 and 1.391 | −1.081 and 1.017 |
| Flack parameter | 0.18(3) | 0.00(4) |
a R1 = ∑||Fo| − |Fc||/∑|Fo|
b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2; w = 1/[σ2(F02) + (aP)2 + bP], where P= [max(F02, 0) + 2Fc2]/3.
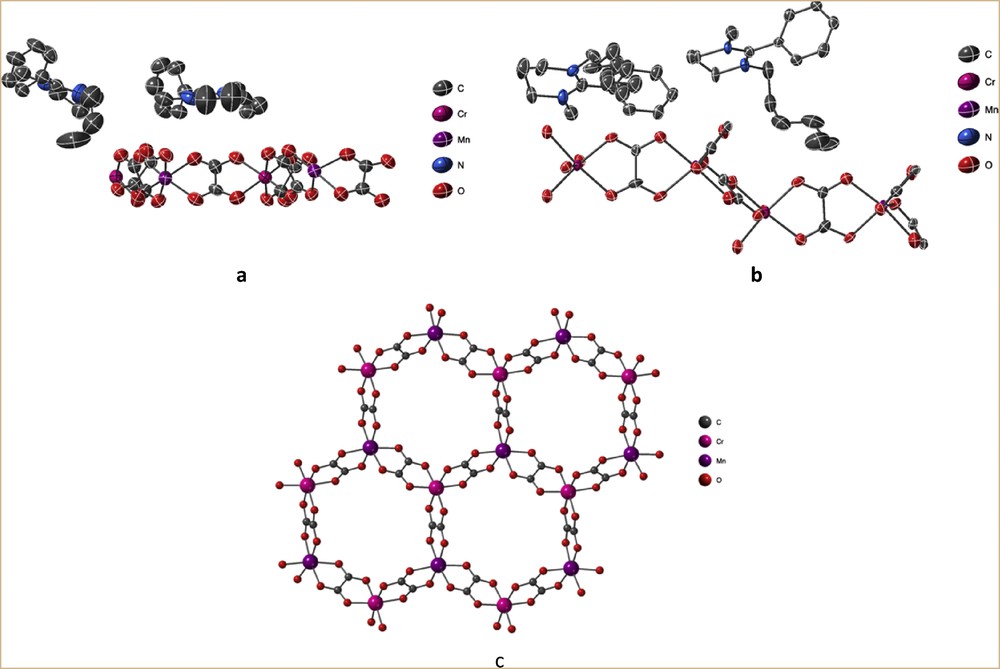
Asymmetric unit based on monocationic (A+ or B+) and [Mn2IICr2(ox)6]2− dicationic species forming 1 (a) and 2 (b) and their thermal ellipsoid representations, together with the representation of the 2D bimetallic network observed in both compounds (c).
Both compounds are based on anionic 2D bimetallic oxalate–based networks [MnIICrIII(ox)3]−, with monocations inserted between the anionic bimetallic layers (Fig. 2c). In contrast to the strong corrugation observed in the networks templated by bis(amidinium) cations [20], in 1 and 2, the coordination layers are essentially flat (Fig. 3a and b). Moreover, these anionic layers are stacked above one another (Fig. 3c and d) rather than staggered as observed in most of the 2D oxalate-based compounds.
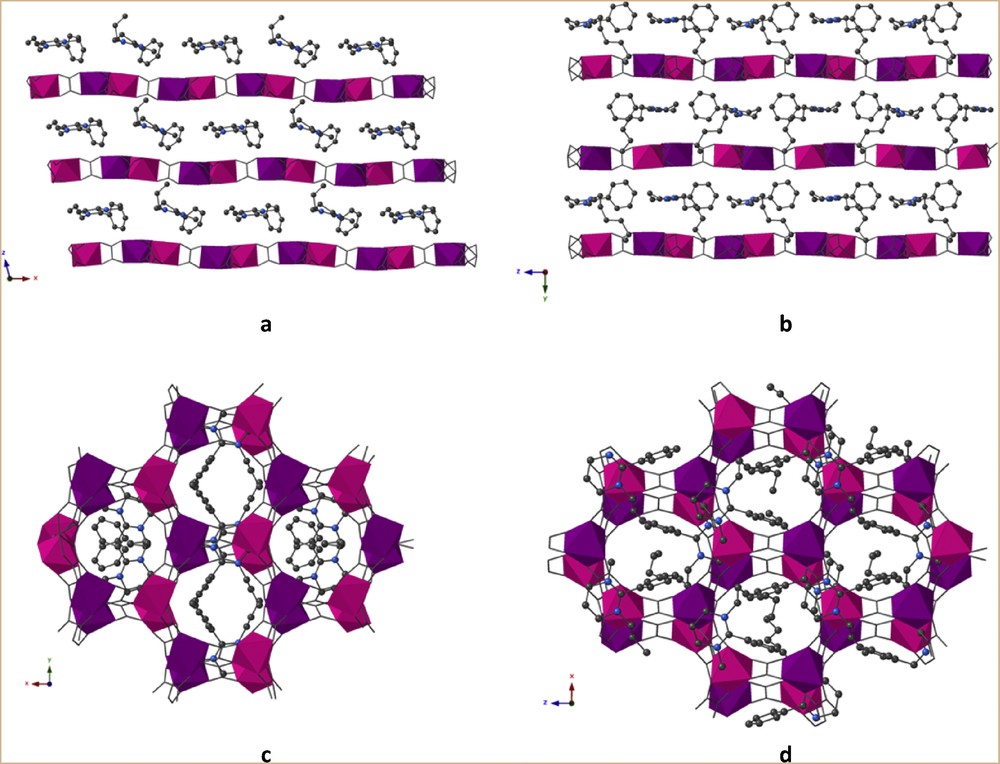
Location of the cations between the anionic bimetallic planes: A+ in 1 (a and c, view along the c and b axis, respectively) and B+ in 2 (b and d, view along the a and c axis, respectively). The disorder in the alkyl chain for 2 has been omitted for clarity.
In both 1 and 2, the CrIII and MnII ions are surrounded by three bidentate oxalate ligands forming slightly deformed octahedral environments with Cr–O and MnO bond lengths and the OCrO and OMnO angles in a narrow range (Table 2). According to the heterochiral nature of the 2D bimetallic oxalate-bridged compounds [24], within a layer, the absolute configuration of the helical tris(bischelated) CrIII and MnII metal centers is alternating. In 1, there is an alternation of the absolute configurations of CrIII and MnII from one layer to the other. On the contrary, in 2, each metal ion exhibits the same absolute configuration throughout the crystal (Δ for CrIII and Λ for MnII for the described single crystal). Accordingly, a spontaneous resolution occurred during the formation of 2, leading to the formation of crystals of the two enantiomers in equal proportions, that is, to a racemate.
Distances and angles around the Cr(III) and Mn(II) metallic centers for compounds 1 and 2.
| 1 | 2 | |
| CrO (Å) | 2.033(5)–2.070(6) | 2.035(7)–2.082(6) |
| MnO (Å) | 2.097(6)–2.144(6) | 2.069(7)–2.120(7) |
| OCrO (deg) | 81.2(2)–95.2(2) 170.8(2)–172.4(2) | 80.0(2)–95.8(3) 168.0(2)–171.2(3) |
| OMnO (deg) | 78.9(2)–98.56(19) 166.40(19)–171.7(2) | 79.3(2)–97.1(2) 167.4(3)–172.1(3) |
In both compounds 1 and 2, there are two independent monocationic units located between the anionic layers. In 2, there is a disorder in the alkyl chain for one of the two cations. The amidinium-phenyl axis of both cations is pointing in two different directions and there is no specific orientation of the cations toward the hexagonal cavities formed by the honeycomb-like network for 1 and 2. Together with the noncentrosymmetry of the space groups, the situation compares well with the one encountered in the multiferroic oxalate–based compound [15a].
There are no hydrogen bonds in 1 and 2 but short van der Waals contacts between the pendant alkyl chains of the cations and the bimetallic coordination network as shown in Fig. 2a and b. The absence of H-bonds and/or any directional intermolecular interactions avoids the modification of the coordination environment of the two metallic ions as previously observed with H-donating bisamidinium cations [18–20]. For the 2D compound [20], the H-bond between the cation and an oxalate ligand led to a weakening of the coordination ability of this oxalate toward MnII and to the coordination on this metal ion of a methanol molecule. The seven coordination of the MnII induced a strong corrugation of the layers that is not observed here for compounds 1 and 2.
3.3 Physical properties
The magnetic susceptibility for 1 and 2 has been measured on microcrystalline powder between 2 and 300 K. The thermal variations of χMT and χM−1 for 1 are shown in Fig. 4a (for 2, see Fig. S4a). Following the structural similarity of the coordination layers, the magnetic behavior of the two compounds is very similar. At room temperature, the χMT values are 6.40 (1) and 6.64 cm3 mol−1 K (2), which are close to those expected for the sum of the corresponding noninteracting isotropic metallic ions (6.25 cm3 mol−1 K for g = 2.00 for both CrIII and MnII). Upon cooling, the χMT products for 1 and 2 increase continuously. This evolution indicates that the exchange interactions between the two metallic CrIII and MnII ions are dominated by ferromagnetic interactions. To quantify the average interaction, the inverse susceptibility (Figs. 4a and S4a) is fitted using a Curie–Weiss law in the 40–300 K window and yields Curie–Weiss temperatures of 9.9 K for 1 and 9.0 K for 2. Upon further cooling, both compounds enter in a ferromagnetic phase with a Curie temperature 6.0 K for 1 and 5.8 K for 2 (Figs. 4b and S4b). These results match with those found for previous 2D [MnCr] oxalate-based compounds, the slight modifications being due to slight changes in the metrics of the coordination spheres of each metallic ion that influence the exchange interaction between the two metal ions.
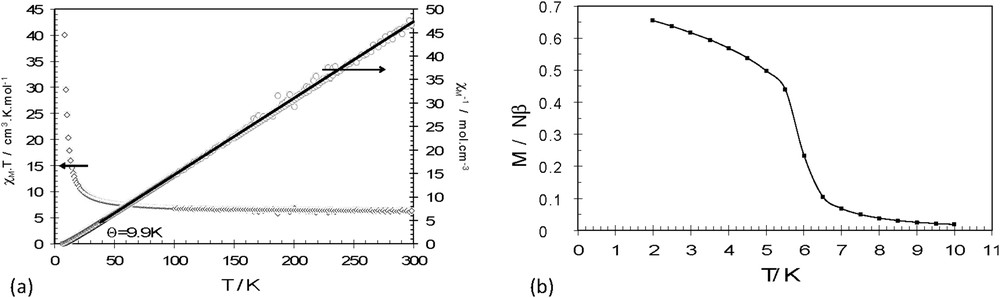
Thermal variation of χMT and χM−1 (a) and field-cooled magnetization (b) for 1.
The space groups of both compounds are compatible with ferroelectricity [25], so that this property has been tested. Because of leakage currents, the ferroelectricity measurements performed using the same procedure as for C[MnCr(C2O4)3(CH3CH2OH)] (C+ = 1-(hydroxyethyl)-4-(N,N-dimethylamino)pyridinium) [15b] were not conclusive.
4 Conclusion
The presented results confirm the ability of amidinium-based cations to template the formation of 2D oxalate-bridged bimetallic [MnCr] networks. Two striking features appear when comparing the templating activity of the mono(amidinium) monocations with the one of the previously used bis(amidinium) dications [20]. First, the absence of H-donating ability of the dialkyl-substituted mono(amidinium) monocation limits the intermolecular interaction between the cation and the coordination network and leads solely to nondirectional interactions. The absence of competition between coordination and hydrogen bonds led to flattened 2D coordination layers instead of the corrugation previously observed. Most importantly, the asymmetry brought about by the absence of the second amidinium moiety has favored the crystallization of the compounds in noncentrosymmetric space groups and, in the case of 2, led to a spontaneous resolution process. Accordingly, such template cations open new possibilities toward new multifunctional oxalate-based compounds.
Acknowledgments
This work was supported by the “Centre national de la recherche scientifique” (CNRS), “Université de Strasbourg” (UdS), “Université Joseph-Fourier” (UJF), and the “Agence nationale de la recherche” (ANR) within the framework of the ANR-08-JCJC-0113-01 project, in particular through a postdoctoral grant to C.M.
Vous devez vous connecter pour continuer.
S'authentifier