


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Nephrolithiasis is extremely common, with estimated prevalence of 10.6% in males and 7.1% in females in the United States of America [1]. In 66.0–93.0% of kidney stones, the main component is calcium oxalate, comprising calcium oxalate monohydrate (COM) in 42.8–74.4% and calcium oxalate dihydrate (COD) in 18.6–23.2% [2, 3]. Up to 90% of calcium oxalate kidney stones are categorized as idiopathic as no causative hereditary or acquired disease can be detected [4]. The exact mechanism of idiopathic calcium oxalate stone formation remains to be elucidated but the majority of these stones appears to form by a process of heterogeneous nucleation on a subepithelial calcium phosphate plaque at the papilla, known as the Randall’s plaque.
The plaque is named after Alexander Randall, who, in the 1930’s, described the presence of a subepithelial interstitial calcium plaque at the renal papilla in 19.6% of 1154 pairs of cadaveric kidneys, as an initiating lesion that precedes kidney stone formation [5, 6]. Randall observed that some of the plaques lost their epithelial lining and hypothesized that they would act as a nidus for subsequent stone formation after their exposure to calyceal urine. Actually, Randall reported small calculi attached to the calcium plaque in 2.3% of patients. Additionally, he found several kidney stones to display a smooth depression, indicative of prior papillary attachment and on some of them, remnants of the calcium plaque.
Randall additionally described a second, less frequently occurring papillary lesion, now known as tubular plugs or Randall’s plugs, which consists of crystal deposition in the lumina of the collecting ducts [6]. Tubular plugging rather than interstitial calcium phosphate plaque formation has been hypothesized as the initiating lesion for brushite stone formation and for kidney stones formed in patients with primary hyperparathyroidism, enteric hyperoxaluria and distal renal tubular acidosis [7, 8, 9]. The subject of tubular plugging is however not the topic of this overview.
Randall’s groundbreaking publications concerning the interstitial calcium plaque were followed by several reports of his contemporaries, who confirmed the presence of papillary plaques in cadaveric kidneys and nephrectomy samples with attached stones in a proportion of these plaques [10, 11, 12, 13].
After a long period with few publications on the matter, research on the subject of Randall’s plaque regained new interest at the end of the 20th century, likely driven by the increased performance of urologic endoscopy, during which Randall’s plaques can be directly visualized at the papillary tip and due to the increased prevalence of Randall’s plaque-associated kidney stone formation. Especially the formation mechanism of Randall’s plaque, which remains incompletely understood, has been addressed in many publications during the last two decades.
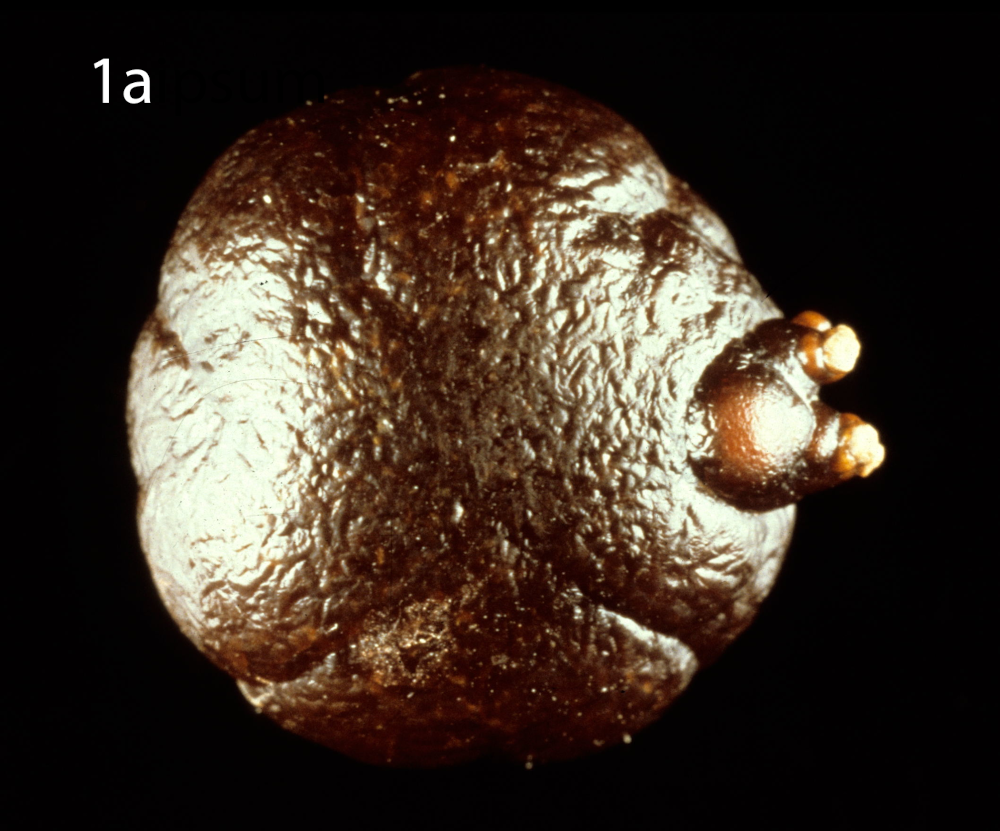

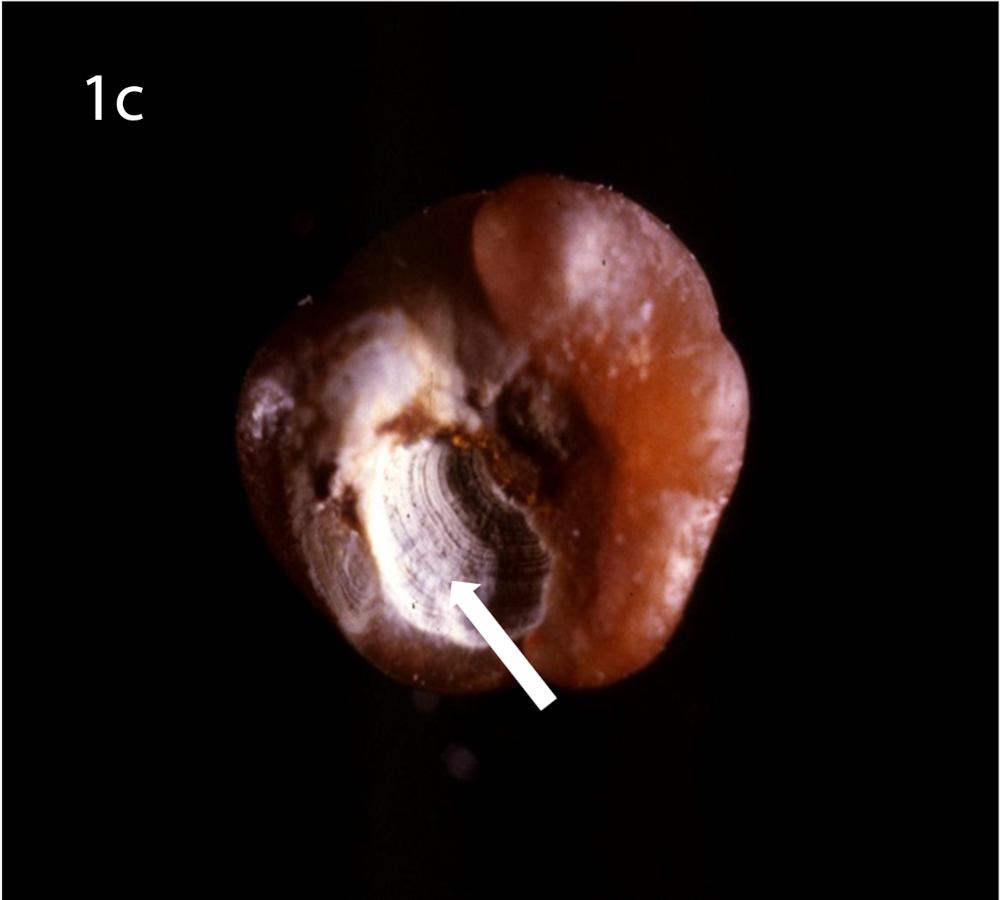
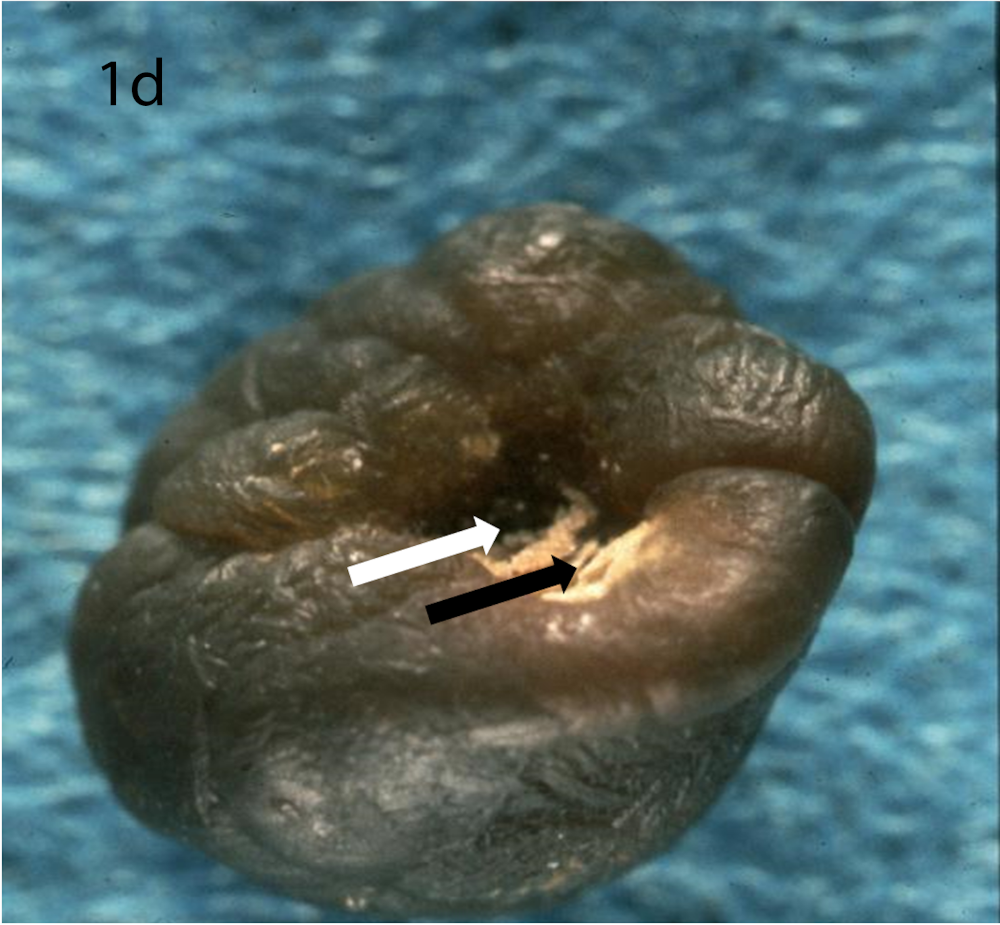
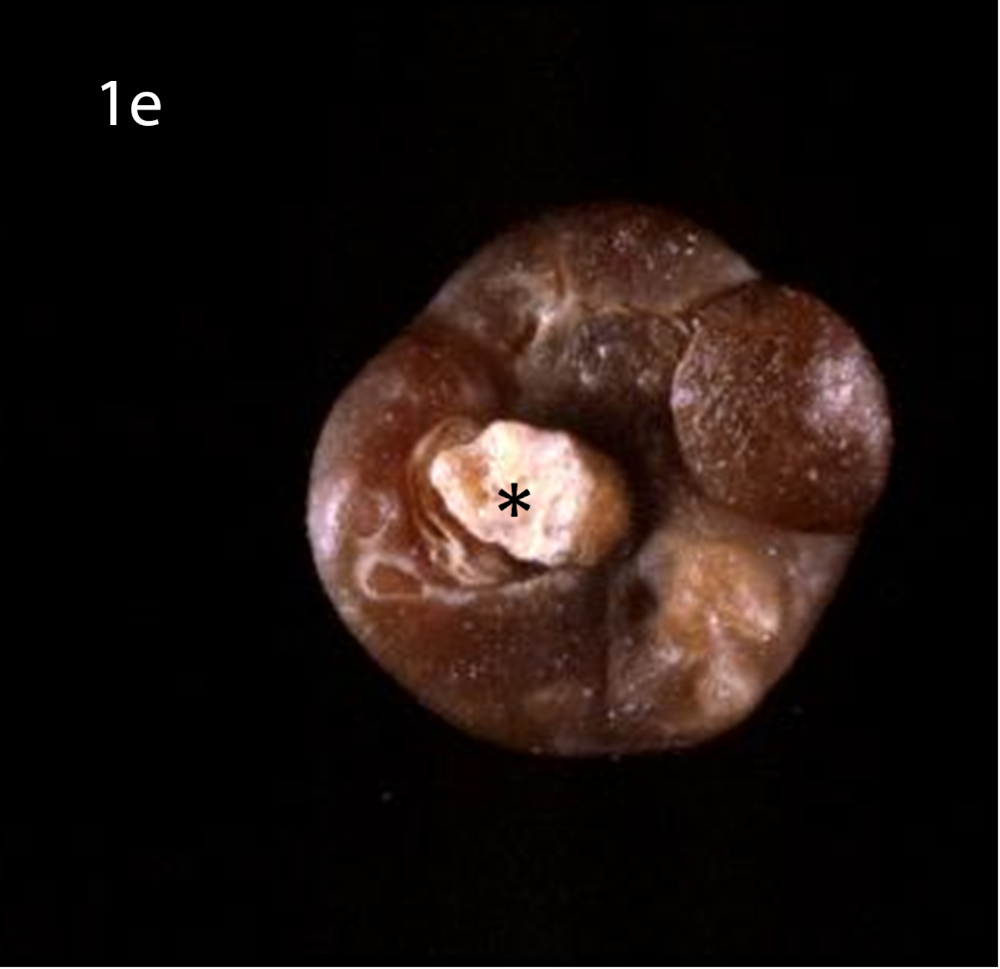
(a) Optical microscopy showing COM stone without papillary umbilication and without Randall’s plaque. (b) Optical microscopy showing sectioned COM stone without papillary umbilication and without Randall’s plaque. (c) Optical microscopy showing COM stone with papillary umbilication. (d) Optical microscopy showing COM stone with papillary umbilication and plaque remnants. (e) Optical microscopy showing COM stone with Randall’s plaque. White arrow: papillary umbilication, black arrow: Randall’s plaque remnants, asterisk: massive Randall’s plaque.
2. Epidemiology
Early publications by Randall [5], Rosenow [10], Vermooten [12] and Haggitt and Pitcock [14] reported the presence of papillary plaques in 8.3–23.0% of unselected autopsy kidney specimens, detected by macroscopic or hand lens examination. The papillary plaques were described as cream-coloured to white lesions, with size ranging from 1–2 mm to a surface occupying the entire papilla. Vermooten [12] described a difference in prevalence between the Caucasian and the native South African population, with respectively 17.2 and 4.3% of subjects displaying Randall’s plaque.
Light microscopic examinations of papillary tissue of unselected autopsy kidneys and nephrectomy samples, including kidneys affected by nephrolithiasis, could detect medullary calcifications in all kidneys in the reports of Anderson and McDonald [13] and Haggitt and Pitock [14], while Anderson could only detect calcifications in 12% of autopsy kidneys [11]. It has to be mentioned that in the last study children and infants formed 36% of the study population, with only 5.7% of them displaying Randall’s plaque and frequently examination on only one renal section, not always containing papillary tissue, was performed. Finally, Ruggera et al. [15] reported interstitial calcifications detected by microscopic examination in 42.9% of papillary biopsies performed during ureterorenoscopy in stone-forming patients.
Optical microscopy of eliminated kidney stones grown on Randall’s plaque typically reveals a papillary umbilication, the imprint of the previous papillary attachment, which can be found as an irregular depression, while some stones can display plaque remnants at the umbilication as well [16] (Figure 1). Daudon et al. [16] described papillary umbilication in 19.5% of 45,774 examined calculi. The rates augment to 39.0% taking only spontaneously eliminated stones into account and even to 66.1% considering only spontaneously passed COM stones [4]. Cifuentes Delatte et al. described papillary umbilication in 28.4% of 500 spontaneously passed stones [17] and plaque remnants in 72.4% of 87 umbilicated kidney stones [18]. Letavernier et al. [19] reported 34.1% of 30149 intact calcium oxalate stones to show Randall’s plaque remnants.
Recently, micro-computed tomographic imaging (micro-CT), using 3-dimensional X-ray imaging with microscopic level resolution was introduced by Williams et al. [20], who reported the technique to be able to detect calcium phosphate deposits in papillary tissue samples and in eliminated kidney stones.
Rates of Randall’s plaque detected by endoscopic urological procedures including ureterorenoscopy and percutaneous techniques are much higher compared to the rates detected by optical microscopy as current urological techniques including shockwave, ultrasound and laser frequently destroy kidney stones and Randall’s plaques, resulting in loss of the plaque or papillary umbilication for optical microscopy. Endoscopic urological examination reveals Randall’s plaques as irregular, glossy, whitish lesions surrounding the opening of the collecting ducts [7, 9] (Figure 2). In American studies performing endoscopic urology, Randall’s plaque was detected in 73.7–100.0% of stone-forming patients [9, 21, 22], with 47.8–100.0% of idiopathic calcium stone formers showing evidence of attached stones at the Randall’s plaques [22, 23]. European studies have reported lower rates and detected Randall’s plaque in 56.7% of 289 stone formers [24].
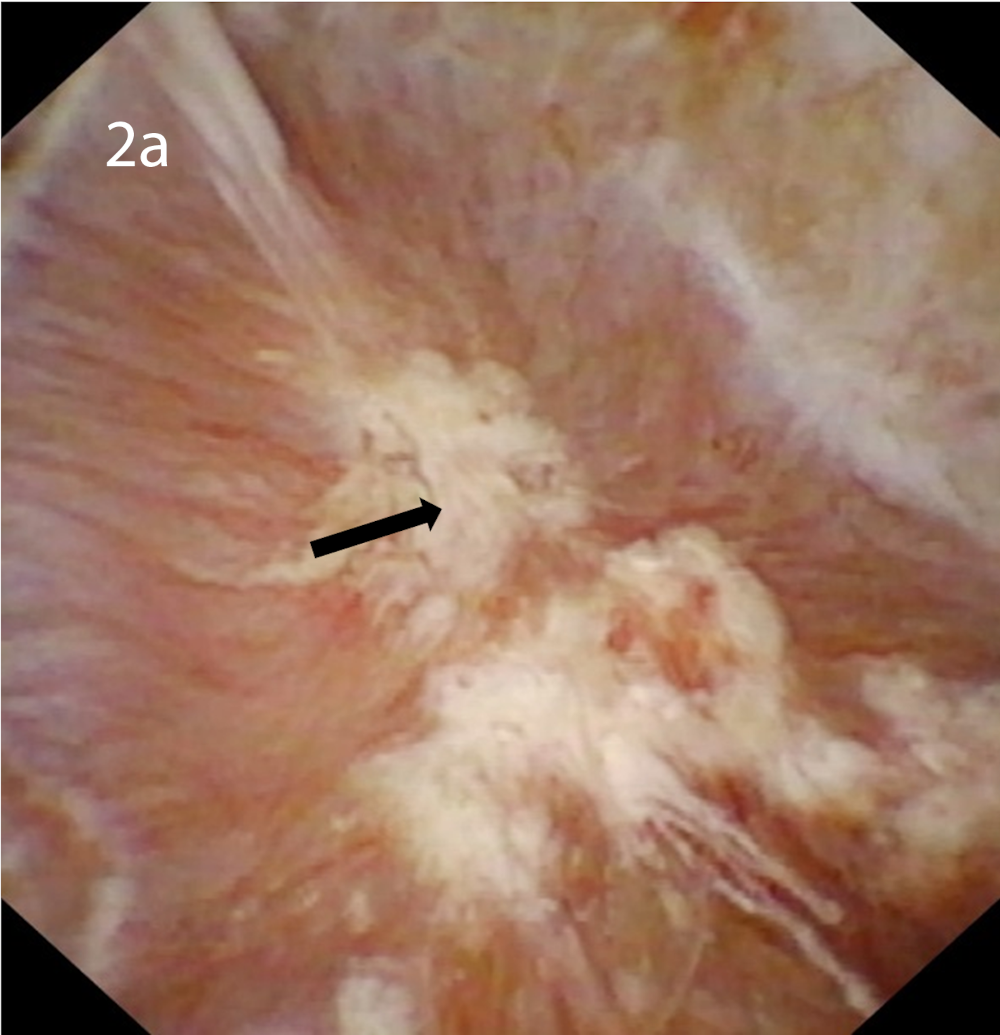
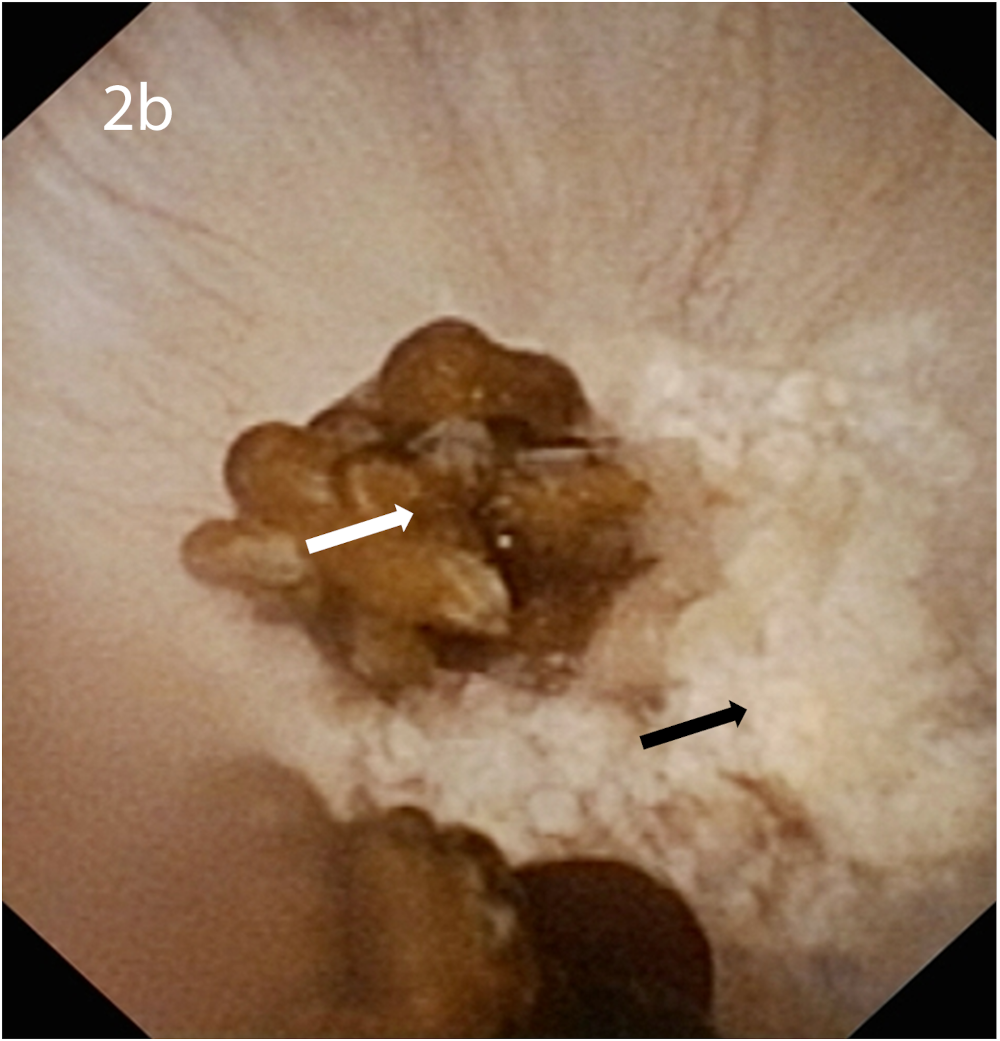
(a) Endoscopic urological view of Randall’s plaques at the papilla, visible as irregular, glossy, whitish lesions. (b) Endoscopic urological view of Randall’s plaques with attached COM stones. Black arrow: Randall’s plaque, white arrow: COM stone.
A difference in density, determined by computed tomography attenuation values between renal papilla of stone formers and non-stone formers respectively has been reported [25, 26]. In 2013, a study comparing papillary surface plaque coverage determined during percutaneous nephrolithotomy and by means of single- or dual-energy helical computed tomography, reported the absence of correlation between the radiological and endoscopic results and concluded current computed tomography techniques not able to detect Randall’s plaque [27]. Indeed, only huge Randall’s plaque can be visualized with computed tomography. In the future, novel techniques with higher spatial resolution, reduced image noise and improved material differentiation might constitute a good means for the non-invasive detection of Randall’s plaque [28].
Although less prevalent than in stone formers [21, 29] and displaying lower papillary tissue plaque coverage [30], Randall’s plaques are also detected in non-stone-forming patients, supporting the theory of their formation as a precursor lesion for kidney stone formation, with subsequent pathogenic steps required for actual kidney stone formation. Light microscopic examination could detect papillary calcifications in 69.4% of 62 cadaveric kidneys of non-stone formers [31]. Endoscopic urology detected Randall’s plaque in 42.9% of seven non-stone-forming patients in an American study [21], while the rate was 27.7% of 173 non-stone-forming patients in a European study [24]. Finally, incipient papillary Randall’s plaque could be detected in 72.7% of non-stone formers [32].
Endoscopic urological studies reported Randall’s plaque to uniformly affect all or nearly all papilla [9, 21, 22, 24]. Verrier et al. [32] observed that nearly all examined papilla were affected by incipient interstitial calcifications, suggesting that the process of Randall’s plaque formation a minima is extremely frequent.
Randall’s plaque has been described in children [33, 34]. Epidemiological data [19] demonstrate that the proportion of kidney stones formed on Randall’s plaque is maximal in the age category of 20–29 years and that this proportion reduces with age, as opposed to the initial idea that plaque formation and associated kidney stone formation increases with age [12]. Additionally, the age of the patients presenting the largest proportion of Randall’s plaque-associated kidney stone formation has decreased during the last decades [35].
The frequency of Randall’s plaque-associated stone formation is rising. In France, a significant increase in proportion of calcium oxalate stones formed on Randall’s plaque between the early 1980’s and the early 2010’s, in both males and females, but most pronounced in young females was reported [19, 35]. In general, the proportion of Randall’s plaque-associated kidney stone formation increased from 5.7% in the period 1978–1985 to 23.3% during the period 2000–2006 [16, 19]. Additionally, the proportion of papillary tissue covered by Randall’s plaque is correlated with the number of stones formed [30], supporting the theory of Randall’s plaque as an initiating lesion for kidney stone formation and highlighting the plaque’s relevance for recurrent stone formation. The increasing proportion of calcium oxalate stone formation on Randall’s plaque is hypothesized to contribute to the rising frequency of calcium oxalate kidney stone formation in general [36, 37, 38]. Additional epidemiological studies are needed to confirm these findings but are impeded by the frequent loss of papillary umbilication and plaque remnants due to urological fragmentation techniques and by the non-universal use of morphoconstitutional stone analysis [39, 40, 41] comprising optic microscopy and Fourier-transform infrared spectroscopy (FTIR) [42] or X-ray analysis as the gold standard for kidney stone analysis and Randall’s plaque detection.
3. Update on the mechanism of Randall’s plaque formation
3.1. Historical data
Already at the initial description by Randall [5] and later confirmed by other authors [13, 14, 43, 44], it was reported that Randall’s plaque consists of interstitial calcification, with sparing of the tubular lumina, nor were calcifications found in the cytoplasm of tubular cells [14]. Initially, Randall hypothesized that plaque formation begins at the basement membranes of the collecting tubules, initiated by lesions at its epithelium [5]. In the same time period, Vermooten described the localization of the calcifications not only at the collagen fibers of the basement membrane of collecting tubules but also at collagen fibers of the vasa recta and in the interstitium [12, 43]. In 1970, Cooke was the first to report the calcifications to be related to the basement membrane of the thin limbs of the loop of Henle [31]. At the end of the 20th century, Stoller et al. [44] supported the findings of Vermooten [12, 43] and reported the calcifications to be located at the basement membrane of the collecting tubules, the interstitium and the vasa recta.
3.2. Randall’s plaque formation begins in the basement membranes of the thin limbs of Henle’s loop and of the vasa recta
Although the exact mechanism of Randall’s plaque formation as an ectopic calcification remains to be elucidated, it has been demonstrated that Randall’s plaque formation begins in the basement membranes of the thin limbs of the loops of Henle and of the vasa recta at the tip of the renal papilla, with subsequent spreading into the interstitial tissue to the suburothelium [7, 32]. Electron microscopy shows that at incipient Randall’s plaques and papillary tissue adjacent to Randall’s plaques, calcifications can be found around the basement membranes of the thin loop of Henle and of the vasa recta, in close contact with collagen bundles. Calcifications can be found as isolated, small, spherical homogeneous electron-dense deposits but can also be detected as larger deposits with a multilaminated morphology showing radial crystallization [7, 32, 45]. The small spherical deposits can merge to form larger depositions, forming syncytia and losing their identity, spreading in the interstitium, in close contact with collagen bundles and organizing into complete or incomplete rings or cuffs, surrounding the thin loops and the vasa recta. Completely surrounded tubules may show cytoplasm vacuolization or basement membrane detachment as signs of cellular damage. Evan et al. [7], supported by later publications [46], confirmed the absence of crystals in vascular and tubular cells even in larger plaques and reported also the tubular lumina to remain free, except for extensive calcification. The interstitial deposits can extend to and surround the collecting tubules as described by Stoller et al. [44], without affecting the collecting tubular cells, but it is clear that the collecting tubules are not the initial site of Randall’s plaque formation [7, 32].
Scanning electron microscopy of a Randall’s plaque typically shows the presence of calcified tubules and calcified blood vessels [18] (Figure 3). Linnes [9] described in approximately 40% of patients with Randall’s plaque the occurrence of tubular plugs, evidenced by means of ureterorenoscopy. Also Verrier et al. [32] reported tubular plugs consisting of carbonated apatite and amorphous calcium phosphate to be present in 79.6% of papillary tissues with or without incipient Randall’s plaques, but the plugs were not in contact with the interstitial calcifications, suggesting that the formation of incipient Randall’s plaque is not related to these intratubular crystals. The role of tubular plugs and their interaction with interstitial calcium phosphate deposition remains unknown.



(a) Scanning electron microscopy showing a normal papilla. (b) Scanning electron microscopy showing a Randall’s plaque fragment in a kidney stone with calcified tubules and tubular plugs. (c) Scanning electron microscopy showing a Randall’s plaque fragment in a kidney stone with calcified tubules. Black arrow: opening of a collecting duct, white arrow: calcified tubule, orange arrow: calcium phosphate tubular plug.
3.3. In situ calcium phosphate precipitation driven by calcium phosphate supersaturation and the role of hypercalciuria
The mechanism for calcium phosphate deposition at the basement membrane of the thin loops of Henle and of the vasa recta is incompletely elucidated but a process of in situ calcium phosphate precipitation, driven by high local calcium phosphate supersaturation, influenced by high calcium concentration and high pH is assumed. As no intracytoplasmic crystals or calcifications in the tubular epithelial cells are detected [7] and incipient Randall’s plaque are not in contact with tubular plugs [32], a process of intratubular calcium phosphate crystal formation, driven by high calcium phosphate supersaturation in the tubular fluid of Henle’s loop [47] with subsequent paracellular or transcellular transport-like endocytosis [48], resulting in basolateral release of calcium phosphate crystals or increased calcium phosphate excretion is less likely. The hypothesis of in situ interstitial calcium phosphate crystal formation is supported by the description of a medullary concentration gradient for calcium, caused by passive calcium diffusion from the loops of Henle, predominantly at the thick ascending limb, and from the descending vasa recta [49, 50, 51]. It is hypothesized that high calcium delivery out of the proximal tubule, as seen in idiopathic hypercalciuria, where there is reduced proximal tubular calcium reabsorption [52], results in increased passive calcium diffusion at the thick ascending limb of Henle, increasing the medullary calcium concentration. Descending vasa recta tend to wash this outer medullary calcium to the inner medulla but are permeable to calcium, resulting in passive calcium diffusion to the interstitium, increasing the medullary calcium concentration and contributing to the calcium phosphate supersaturation at the papillary tip. Additionally, reduced water reabsorption at the collecting duct due to increased serum calcium levels at the vasa recta, can additionally increase calcium phosphate supersaturation [53]. Finally, it has been hypothesized that there is a high papillary pH in patients with Randall’s plaque [54], further contributing to high papillary calcium phosphate supersaturation. Calcium diffusion from the thin limbs of Henle’s loop is assumed to contribute very little to the high calcium phosphate supersaturation at their basement membranes as no vectorial calcium or phosphate transport occurs at this site and the permeability for calcium and phosphate is very low. The diffusion of calcium from the descending vasa recta close to the thin limbs is assumed to be the major source of calcium phosphate supersaturation at the interstitium in their vicinity. In fact, the basement membranes of the thin limbs of the loop of Henle are thick, composed of collagen and mucopolysaccharides, an electrostatically charged matrix, easily attracting calcium phosphate with progressive calcification. Recently, Evan et al. [55] demonstrated that Randall’s plaque formation begins at the ascending thin limbs of Henle’s loop and not at the descending thin limbs. These findings support the theory of calcium washdown from the interstitial tissue at the thick ascending limb to the interstitial tissue at the papillary tip and the ascending thin limbs by the vasa recta, as the descending vasa recta are preferentially located near the ascending thin limbs of Henle’s loop and as the ascending thin limbs are impermeable to water as opposed to the descending thin limbs [56], impairing dilution of the local high interstitial calcium concentration.
In this “vas washdown” theory, with subsequent in situ calcium phosphate precipitation at the basement membranes of the vasa recta and of the thin limbs of the loop of Henle, calcium concentration of the tubular fluid delivered to the thick ascending limb is an important determinant of Randall’s plaque formation. Many findings support this. First, idiopathic hypercalciuria is the most common metabolic abnormality in idiopathic calcium oxalate stone formers [57]. The coverage of papillary tissue by Randall’s plaque is correlated with calciuria [29, 58]. Randall’s plaque is more pronounced in idiopathic hypercalciuric stone formers compared to other stone formers [8]. The importance of calciuria could also explain the difference in frequency of Randall’s plaque described by Vermooten [10] between Caucasian subjects and the native South African population, who are known to have lower levels of calciuria. Finally, in a study comparing calcium oxalate stone formers related and unrelated to Randall’s plaque, Letavernier et al. [19] described higher serum levels of osteocalcin to be associated with Randall’s plaque formation, and to a lesser extent increased serum calcium levels and phosphate reabsorption rate. As the osteocalcin gene has a vitamin D receptor response element in its promotor region, increased osteocalcin levels are a marker of vitamin D receptor activation. These findings suggest the implication of vitamin D activation in Randall’s plaque formation. Activated vitamin D (1,25-OH2-vitamin D), by means of increased gastro-intestinal calcium absorption, is an important mediator of renal calcium excretion. Letavernier et al. [19] speculated that the increased prevalence of Randall’s plaque-associated kidney stone formation might be partly attributed to the increased use of vitamin D supplements. Although controversial, some publications have reported the relationship between vitamin D supplements and kidney stone formation, especially when combined with calcium supplements [59]. Recently, combined administration of vitamin D and calcium resulted in accelerated calcification in an animal model of Randall’s plaque formation [60]. The role of vitamin D supplementation in the formation of Randall’s plaque in predisposed subjects was again raised by the authors but remains a subject of controversy and debate.
According to this theory, measures that augment proximal tubular calcium reabsorption and hence reduce calcium delivery to the thick ascending limb, like thiazide diuretics and low sodium diet, could reduce Randall’s plaque formation. On the other hand, increased Randall’s plaque formation can be generated by further increase in calcium phosphate supersaturation at the papillary tip, which is typically induced by a Western diet rich in salt and protein, the major dietary determinants of renal calcium excretion. Additionally, high protein intake results in high urinary phosphate excretion and an acid load, leading to basolateral, interstitial bicarbonate excretion in parallel to tubular proton excretion by alpha-intercalated cells of the collecting ducts, additionally raising calcium phosphate supersaturation. Indeed, besides correlation with calciuria and low urinary volume, Kuo et al. [29] described low urinary pH to correlate with Randall’s plaque papillary coverage.
3.4. Crystalline and organic composition of Randall’s plaque
The main crystalline components of Randall’s plaque are carbonated apatite (carbapatite) and amorphous carbonated calcium phosphate [4]. Amorphous carbonated calcium phosphate is more predominantly found at the core of the Randall’s plaque, suggesting that it is the first crystalline phase formed, with subsequent formation of the more stable apatite at the surface [61]. Sodium hydrogen urate can be detected in 3.4% of Randall’s plaque identified during morphoconstitutional kidney stone analysis [4, 62]. Additionally, whitlockite is detected in 1.0% of Randall’s plaques, identified at kidney stone surface [4] and has also been observed in incipient Randall’s plaque in the papilla [32]. Brushite can be detected in 0.34% of Randall’s plaques, identified at kidney stone surface [4]. Finally, minor compounds including octacalcium phosphate, struvite, calcite, uric acid, bobierrite, ammonium urate, potassium urate, opaline silica and porphyrines have been detected as well [4, 17]. The variety of the different crystalline phases detected at the Randall’s plaque and the variable levels of carbonation, ranging between 5 and 35% [16] likely reflect the involvement of different formation mechanisms.
Randall’s plaque contains an organic matrix rich in proteins, glycosaminoglycans and lipids, containing collagen fibers and other yet unidentified fibrillary material, membrane vesicles and other cellular degradation products [45, 63]. Already described by Haggitt and Pitcock in 1971 [14], electron microscopy shows that deposits can have a multilaminated morphology [7, 32]. The lamination reflects the alternating layers between mineral phases as shown by the light regions and organic material represented by the electron-dense layers [64, 65]. Evan et al. [65] reported that all mineral deposits, even the smallest, are surrounded by organic material, with larger deposits described to “float in an organic sea”. Already reported in historical papers [12, 43], Khan et al. [45] confirmed the mineral spherical deposits to be in close association with collagen fibers and described the presence of membrane vesicles. The authors hypothesized that in the process of aggregation and merging of the initial spherical mineral deposits to form larger deposits, collagen and membranous degradation products interact with the enlarging calcification, getting calcified as well. Very recently, Gay et al. [63] demonstrated at higher magnification that, although plaques are located in collagen-rich regions, there is a gap between the collagen fibers and microcalcifications in the incipient calcium plaque, assuming no close interaction with collagen in the early steps of Randall’s plaque formation. The authors confirmed the findings of Khan et al. [45] and reported the presence of vesicles containing calcium phosphate crystals. A predominant macromolecule detected at Randall’s plaque is osteopontin [66], which is located at the interface of the apatite crystal and the surrounding organic matrix [65]. The exact role of this crystallization modulator [67, 68], which is also implicated in pathological vessel calcification [69] and bone mineralization, is unknown. Other macromolecules detected at Randall’s plaque organic matrix include osteocalcin, bone sialoprotein and inter-alpha-trypsin inhibitor heavy chain 3 [70].
Very recently, Winfree et al. [71] described, in a study using microscopic fluorescence, an autofluorescence signal unique to Randall’s plaque, present on calcium oxalate stones and in papilla and different from the signal detected from the attached stone. As autofluorescence originates from proteins and metabolites, these findings suggest the presence of a unique, yet unidentified proteinaceous or metabolic deposition in the organic components of Randall’s plaque, which is absent in stone matrix and urine. Such results may be in line with the presence of some specific metalloproteins in the Randall’s plaque. Actually, high levels of zinc [72] have been identified at Randall’s plaque, that may be related to inflammatory processes and to the presence of metalloproteinases.
3.5. Alternative theories on Randall’s plaque formation
3.5.1. The vascular theory of Randall’s plaque formation
The vascular theory of Randall’s plaque formation, postulated by Stoller and colleagues [73, 74], hypothesizes that injury to the vasa recta located at the renal papilla, could lead to an atherosclerosis-like reaction resulting in vessel wall calcification, through the implication of oxidative stress and production of reactive oxygen species (ROS). Erosion of the vessel wall calcification into the surrounding interstitium could subsequently lead to Randall’s plaque formation. Vasa recta at the renal papillary tip are hypothesized to be especially prone to injury due to several reasons: the changing blood flow from laminar to turbulent, the hypoxic and hyperosmolar environment and the decreased vascular resistance and flow velocity in the ascending vessels. The theory is supported by the association between cardiovascular disease [75], aortic and coronary artery calcification [76], cardiovascular risk factors like hypertension [77], metabolic syndrome, obesity [78] and diabetes mellitus [79] and kidney stone disease. Additionally, similarities between vascular calcification and Randall’s plaque formation are reported, comprising the involvement of collagen and membrane vesicles and peripheral growth of the calcification [45], the implication of inflammation and oxidative stress [80], involvement of the same macromolecules regulating urine crystallization and vascular calcification like osteopontin, similarities between the different types of calcifications involved at incipient Randall’s plaque and cardiovascular calcification [63] and the detection of lipid droplets in interstitial cells of the medulla [81]. Additionally, the protective effect of statins for the development of kidney stones was described, suggesting the involvement of a common pathogenic step [82]. So far however, no report has described the presence of plaques or calcifications in the vasa recta, which disproves this theory.
3.5.2. Theory involving progression of proximal intratubular calcifications
Recently, several publications [83, 84, 85] provide arguments to support a novel version of a hypothesis postulated in 1954 by Carr [86] in which the distal interstitial Randall’s plaque is hypothesized to be formed secondary to intratubular crystal deposition in the proximal upper medulla and using the “medullo-papillary complex” as a functional unit, consisting of a papilla with the corresponding medulla, displaying specific pressure and chemical gradients. Micro-CT and electron microscopy of nephrectomy samples showed the presence of proximal intratubular calcifications in all kidneys, even in those without Randall’s plaques. The amount of proximal intratubular crystal deposition correlated with the presence of Randall’s plaque interstitial distal mineralization. The theory postulates that proximal intratubular crystal deposition, caused by high calcium phosphate supersaturation in short nephrons that display low fluid velocity, precedes distal interstitial calcification due to changes in fluid dynamics secondary to obstructed tubular lumina in the proximal medulla.
3.5.3. The involvement of cellular osteogenic differentiation
Another theory on Randall’s plaque formation hypothesizes the involvement of osteogenic differentiation of cells like pericytes, fibroblasts, vasa recta endothelial cells and/or tubular epithelial cells. First, osteopontin, the main protein identified in Randall’s plaque [65], is also a known marker of osteogenesis. Additionally, increased expression of the osteogenic markers Runt-related transcription factor 2 (Runx2), osteocalcin and osteopontin has been reported in Randall’s plaque papillary tissue compared to normal papillary tissue [87]. Mezzabotta et al. [88], performing experiments on papillary cells from a medullary sponge kidney (MSK) patient, reported spontaneous calcium phosphate deposition and osteogenic differentiation with expression of osteogenic markers by cells resembling pericytes or stromal stem cells. The authors hypothesized that the papilla might be a perivascular niche where pericytes, involved in other ectopic calcification processes and present around the vasa recta can undergo osteogenic differentiation leading to Randall’s plaque formation. These findings were however contradicted by a study of Evan et al., who could not detect interstitial mineral deposition in areas positive for Runx2 and osteoblast transcription factor (osterix) gene expression in papillary biopsies of MSK patients [89]. Additionally, no Runx2 or osterix gene expression was observed at Randall’s plaque in idiopathic calcium stone formers. Zhu et al. [87] showed that under osteogenic conditions also renal interstitial fibroblasts can display an osteogenic phenotype, forming calcium phosphate nodules similar to Randall’s plaque calcium phosphate deposition, with increased expression of osteogenic markers, hypothesizing the implication of osteogenic differentiation of fibroblasts in the formation of Randall’s plaque. Finally, Priante et al. [90] reported the osteogenic differentiation of human renal proximal tubular cells with expression of osteogenic markers and leading to deposition of calcium phosphate crystals in osteogenic conditions. Also Khan et al. [91, 92] hypothesize that cellular osteogenic differentiation is implicated in Randall’s plaque formation and postulate that in conditions of stress, like exposure to calcium oxalate crystals, hyperoxaluria, hypercalciuria or hypocitraturia, renal epithelial cells or vasa recta endothelial cells undergo osteogenic differentiation, associated with increased expression of osteogenic markers, reduced expression of crystallization inhibitors like fetuin and matrix Gla protein and the release of membrane vesicles.



(a–c) Scanning Electron Microscopy showing the interface of Randall’s plaque and COM crystals at different magnification. (a) Black arrow: COM stone, white arrow: Randall’s plaque. (b) White arrow: carbonated apatite spherical deposits at Randall’s plaque, orange arrow: COM crystals. (c) Black arrow: carbonated apatite spherical deposits at Randall’s plaque, orange arrow: COM crystals.
4. Mechanism of stone formation
The disruption of the papillary epithelium with subsequent exposure of the plaque to the calyceal urine [45, 93] occurs through an unknown mechanism, although the involvement of matrix metalloproteinases and sheer force due to the growth of the plaque has been suggested [94].
Although Randall initially hypothesized Randall’s plaque to act as a nidus for the formation of different types of stones including calcium oxalate, calcium phosphate and uric acid [6], it is now known that the bulk of stones formed on Randall’s plaque are COM stones [17, 46]. Analysed by FTIR, 89.5% of 10462 Randall’s plaque-associated kidney stones were made of COM pure or mixed with COD [62]. Alternatively, 88% of calcium oxalate stone formers have endoscopic evidence of Randall’s plaque [21] and 66.1% of spontaneously passed COM stones display papillary umbilication [4]. Randall’s plaque was most common in calcium oxalate stone formers compared to stone formers of other stone types [58]. Additionally, the crystalline phase in direct contact with the Randall’s plaque is almost exclusively COM [16] (Figure 4). Other crystalline phases may be present, like COD, hydroxyapatite and uric acid but these are not in direct contact with the Randall’s plaque [35, 93]. Calcium oxalate crystal nucleation is driven by calcium oxalate supersaturation, influenced by urinary volume, urinary calcium and oxalate concentration and concentration of crystallization inhibitors like citrate.
At this point, it is worth to recall some physicochemical properties of biological apatites to assess the interface between the Randall’s plaque and COM crystallites. Biological apatites which belong to physiological or pathological calcifications share physicochemical characteristics [95, 96, 97]. The morphology of biological apatite nanocrystals can be described as a thin platelet morphology elongated towards the c-axis with crystal size of less than 50–100 nm length and a few nanometers of thickness [98]. As discussed in detail in several papers [99, 100, 101], it seems that apatite nanocrystals display an apatitic core and a more or less structured surface hydrated layer including non-apatitic domains. As discussed previously, on this amorphous surface of biological apatite, proteins are present, with Tamm–Horsfall protein (THP) and osteopontin identified at the interface. Such structural characteristics are thus not compatible with an epitaxy between the Randall’s plaque and COM crystals [102, 103]. As illustrated by Figure 4c, large randomly oriented COM crystals are trapped on a phase of carbapatite crystals embedded in proteins acting as a “glue” [104]. This mechanism explains why patients without risk to generate kidney stones (the size of COM crystals is compatible with natural expulsion) become at risk due to the presence of the Randall’s plaque.
Other mechanisms of calcium oxalate stone formation on Randall’s plaque have been proposed. First, the efficient nucleation of calcium oxalate by apatite has been demonstrated in vitro [102, 103]. Evan et al., in a study on biopsy specimens comprising papillary stones with their renal attachment [93], described the plaque, exposed to calyceal urine, to be covered by a “ribbon” composed of alternating layers of organic material and mineral phases, which were identified by FTIR as amorphous phosphate. Eventually, progressing through the alternating layers of the interface, the authors reported that calcium oxalate crystals get mixed with apatite and progressively become the dominant mineral phase, resulting in calcium oxalate stone formation. Also Khan et al. reported Randall’s plaque crystal deposits to be present under a fibrous layer [45] and hypothesized that stone growth occurs by organic matrix-associated nucleation of calcium oxalate or by the transformation of the outer layer of calcium phosphate crystals into calcium oxalate crystals [91]. It has been suggested that in case of low diuresis or high acid load, proton secretion in the distal nephron results in low urinary pH leading to dissolution of calcium phosphate crystals, giving rise to increased calcium concentration and increasing calcium oxalate supersaturation, resulting in replacement of the external calcium phosphate crystals at the plaque by calcium oxalate crystals by a process of crystal dissolution and recrystallization [105, 106, 107]. Sethmann et al. [46] however described in a study examining papillary stones the presence of an initial layer of COM crystals at the surface of the Randall’s plaque, subsequently covered by a “crust” containing calcium phosphate, suggesting no macromolecular interface at COM crystal precipitation.
5. Recent developments
Recently, research on Randall’s plaque is not just merely focusing on factors influencing urinary lithogenicity or supersaturation but also recognizes factors aiding crystal nucleation or crystal germination, as the first step of Randall’s plaque formation [108], like membrane vesicles, which can be linked to inflammation.
5.1. The role of membrane vesicles in crystal nucleation
The presence of membrane vesicles at Randall’s plaque was first described by Khan et al. [45] and later confirmed by Verrier et al. [32]. Recently, in their nanoscale analysis of incipient Randall’s plaque, the same research group [63] confirmed these observations as they described the presence of different types of calcifications. Next to the earlier described microcalcifications, situated at collagen-rich areas, which can either be homogeneous, rounded, electron-dense, small spheres with an approximate diameter of 50 nm or display a larger, multilaminated morphology with 0.5–2 μm diameter [45], they observed various types of nanocalcifications: solitary mineral particles with an average diameter of 100 nm, but also vesicles with a nitrogen-rich “membrane”. These membrane vesicles contain nanocrystals with a diffraction pattern that suggests the presence of crystalline apatite and can be found solitary or in clusters, giving rise to larger depositions and subsequently form microcalcifications. Membrane vesicles or exosomes, consisting of phospholipids can be liberated by all cell types as a response to certain physiological and pathological stimuli and have been suggested to be implicated in other physiological and pathological calcification processes including bone formation [109] and vascular calcification [110]. In vitro, it has been demonstrated that phospholipids induce apatite precipitation [111]. In animal models, experimentally induced calcium oxalate crystals are always accompanied by membrane fragments [112]. Although it remains unclear from which cells these membrane vesicles are released, their presence at the incipient Randall’s plaque, their nanocrystal content and the analogy with other calcification processes lead the authors to hypothesize membrane vesicles to be implicated in the very first steps of Randall’s plaque formation by aiding the heterogeneous nucleation of calcium phosphate.
In addition, Gay et al. [63] described a variety of compositions of different mineral particles. All mineral particles were mainly composed of calcium phosphate but more than half of them additionally had calcium carbonate located at their centre, while other mineral particles do not contain calcium carbonate but display high amounts of organic compounds. The authors suggested that both carbonate and organic compounds act as driving factors for nucleation of the respective mineral particles.
5.2. The role of inflammation, oxidative stress and immunity
Although Evan et al. [7] explicitly reported the absence of cellular injury, inflammation and interstitial fibrosis, recently, the involvement of the immune system, inflammation, oxidative stress, apoptosis and kidney injury in the pathogenesis of Randall’s plaque and in the formation of calcium stones in general, has received much attention [113, 114, 115].
First, certain biomolecules detected at Randall’s plaque, including THP, osteopontin and inter-alpha-trypsin inhibitor are also implicated in the immune/inflammatory system. Additionally, the high zinc content of Randall’s plaque described by Carpentier et al. [72] points towards an inflammatory mechanism involved in Randall’s plaque formation, as zinc has been demonstrated to be implicated in inflammation [116, 117]. Further, the presence of sodium hydrogen urate, although rare, in Randall’s plaque, was accompanied by various cells, suggesting an inflammatory process [62]. It is known that not only sodium hydrogen urate, but also many other crystals, are involved in inflammation by triggering the nod-like receptor protein 3 (NLRP3) inflammasome pathway and other pathways leading to IL-1 production and innate immune cell recruitment [118]. Additionally, Taguchi et al. [66] described the upregulation of gene expression pathways associated with inflammation, oxidative stress and kidney injury and the increased expression of proinflammatory cytokines, immune cells and cellular apoptosis in Randall’s plaque tissue compared to non-Randall’s plaque papillary tissue of calcium oxalate stone formers. Sun et al. [119] reported increased gene expression of inflammatory cytokines in papillary tissue of stone formers compared to controls, with the upregulation of CCL-2, CCL-7, CCR-2 and CSF1 suggesting the involvement of monocyte activation. Microarray analysis of genes expressed in renal papillary tissue of stone formers demonstrates an upregulation of genes associated with an M1 inflammatory macrophage phenotype and downregulation of genes linked to an M2 anti-inflammatory macrophage phenotype [120]. In fact, two macrophage phenotypes have been described with M2 anti-inflammatory macrophages being associated with crystal phagocytosis, suppression of stone formation and suppression of inflammatory damage, while M1 inflammatory macrophages would be associated with stone formation [121].
Animal and in vitro experiments have demonstrated that intratubular calcium oxalate crystals, formed in conditions of hyperoxaluria, induce an inflammatory response, that implicates ROS production, increased NLRP3 inflammasome activation and expression of inflammation-related genes and increased synthesis of molecules implicated in the inflammatory cascade including osteopontin, matrix Gla protein, fetuin and monocyte chemoattractant protein-1 (MCP-1) [122, 123]. The inflammatory response leads to monocytes migration, surrounding intratubular and interstitial calcium oxalate crystals [112], followed by in situ monocytes differentiation towards macrophages. Depending on the local cytokine milieu and influenced by other mediators, a preferential differentiation towards an inflammatory macrophage phenotype is induced, resulting in tissue damage through the production of ROS and bioactive lipids like prostanoids and leukotrienes versus differentiation towards an anti-inflammatory phenotype, associated with crystal phagocytosis and prevention of kidney injury. M2 anti-inflammatory macrophages have been shown to clear calcium oxalate crystals by processes of clathrin-mediated endocytosis and phagocytosis, followed by release of inflammatory cytokines by the macrophages, leading to the additional recruitment of macrophages, neutrophils and dendritic cells. Patel et al. [124] hypothesized that an imbalance between oxidative and antioxidative forces with increased ROS production, induced by excessive calcium oxalate crystal deposition, may result in mitochondrial damage of the monocytes, leading to impaired crystal elimination capacities and perhaps resulting in macrophage polarization towards an inflammatory phenotype and further enhancing tissue inflammation. Additionally, a possible role of the NLRP3 inflammasome [125] and the androgen receptor [126] in the differential polarization towards an inflammatory macrophage phenotype and tissue damage caused by calcium oxalate crystals has been suggested. Alternatively, the implication of Sirtuin 3 [127], a mitochondrial enzyme that decreases the production of ROS, in the preferential anti-inflammatory polarization of macrophages has been proposed. Finally, while short period exposure to calcium oxalate crystals induces an anti-inflammatory macrophage phenotype, prolonged exposure results in switch to a proinflammatory phenotype [128].
Although most animal and in vitro studies have focused on calcium oxalate crystals, some in vitro studies have reported similar reactions of renal epithelial cells after exposure to calcium phosphate crystals including cellular injury, increased production of ROS, cellular injury, upregulation of inflammatory mediators, production of MCP-1 and increased apoptotic activity [123, 129, 130].
Of note, most of the animal models based upon urinary calcium oxalate crystallization induce crystalline nephropathies (intratubular crystal precipitation) rather than kidney stone formation. Whether calcium oxalate crystals forming stones in urinary cavities induce inflammatory processes has not been proven in humans.
It is hypothesized that repeated and continuous exposure of renal epithelial cells to conditions like hyperoxaluria, hypercalciuria, calcium oxalate or calcium phosphate crystals induces oxidative stress and an inflammatory cascade including polarization towards an inflammatory macrophage phenotype and possibly the production of membrane vesicles as a facilitator of calcium phosphate nucleation. The ensuing continuous inflammation can lead to papillary damage, collagen deposition and calcification and ultimately Randall’s plaque formation. An alternative hypothesis suggests the implication of inflammation only in the process of urothelial rupture after interstitial calcium phosphate deposition, resulting in exposure of the plaque to the calyceal urine and subsequent aggregation of calcium oxalate crystals [130].
Despite these findings, patients exhibiting Randall’s plaque seem not prone to develop chronic kidney disease, although exact data on this subject are lacking.
5.3. Development of an animal model and the role of inorganic pyrophosphate
Advances in the knowledge of the formation mechanism of Randall’s plaque have partly been hampered by the lack of an appropriate animal model of Randall’s plaque formation. In the past, hyperoxaluric animal models [112, 131, 132] developed intratubular calcium oxalate crystal deposits with subsequent stone formation while animal models of hypercalciuria [133, 134] developed intratubular calcium phosphate deposits with subsequent stone formation. Npt2-/- mice, knockout of NPT2 (SLC34A1), a Na+-dependent phosphate transport protein 2A expressed on the luminal surface of the proximal tubule, produce both interstitial and intratubular deposits [135], just like THP-deficient mice [136, 137]. Osteopontin-deficient animals did not consistently show interstitial calcifications [137, 138]. Finally, Na+/H+ exchanger regulatory factor-1 (NHERF-1) knockout mice develop interstitial calcifications that are however not located at the basement membrane of the thin loops of Henle [139]. In conclusion, no model of interstitial calcification deposition as seen in Randall’s plaque was developed until recently.
Pseudoxanthoma elasticum is a hereditary disorder characterized by ectopic calcification of elastic fibers in skin, retina and peripheral arteries. The disease is caused by mutations in the ABCC6 gene, which encodes an ATP-binding cassette transporter, implicated in extracellular release of ATP, which due to its catabolism by ectonuclotide pyrophosphatase phosphodiesterase-1 (ENPP1) is the major source of inorganic pyrophosphate [140]. Inorganic pyrophosphate is a long known mineralization inhibitor and acts through inhibition of calcium phosphate crystallization and precipitation [141]. Low serum levels of inorganic pyrophosphate have been detected in patients with pseudoxanthoma elasticum [140]. Up to 39.8% of pseudoxanthoma elasticum patients have a history of nephrolithiasis [142, 143], with morphoconstitutional stone analysis and results from computed tomography in a few patients suggesting a Randall’s plaque-driven stone formation. Rare cases reported even nephrocalcinosis [144, 145]. Abcc6-/- mice have lower inorganic pyrophosphate urinary excretion and serum levels compared to controls and spontaneously develop interstitial calcifications at the tip of renal papilla with ageing [143]. The demonstrated interstitial calcifications are similar to the calcifications of Randall’s plaque, namely located at the basement membrane of the loops of Henle and the vasa recta specifically at the tip of the renal papilla, consisting of spherulites with alternating concentric layers of calcium phosphate and organic compounds and containing apatite and amorphous calcium phosphate [143]. Supplementation with inorganic pyrophosphate results in increased serum inorganic pyrophosphate levels and reduced tissue calcifications [146, 147, 148], while combined administration of vitamin D and calcium accelerated interstitial calcifications in Abcc6-/- mice [60]. The Abcc6-/- mice animal model of Randall’s plaque formation hence suggests the role of inorganic pyrophosphate in the formation of Randall’s plaque, while the presence of merely hyperoxaluria, hypercalciuria or deficiency in macromolecules is insufficient to constitute a reliable model of Randall’s plaque formation. In fact, not all studies were able to find a difference in urinary calcium excretion between calcium oxalate stone formers with and without Randall’s plaque respectively [19, 149] and urinary calcium excretion in Abcc6-/- was not different compared to wild type mice, suggesting that hypercalciuria is insufficient for the development of Randall’s plaque and that additional deficiency of inhibitors like inorganic pyrophosphate is mandatory. Low urinary levels of inorganic pyrophosphate have been reported in stone formers [150, 151, 152, 153], but currently it is unknown if patients with Randall’s plaque display lower urinary levels of inorganic pyrophosphate compared to patients with stone formation unrelated to Randall’s plaque. Measurement of inorganic pyrophosphate levels has been difficult in the past due to absence of reliable methods and the very low concentrations of inorganic pyrophosphate. As it has been demonstrated that oral pyrophosphate is partly absorbed [154] and increases serum levels, inorganic pyrophosphate supplementation can possibly constitute a treatment option for the prevention of Randall’s plaque formation in the future.
6. Conclusion
The recent development of an animal model of Randall’s plaque formation will likely accelerate research on Randall’s plaque and associated kidney stone formation and will hopefully bring clarity to the exact formation mechanism of both Randall’s plaque and associated calcium oxalate kidney stones. Additionally, the identification of inorganic pyrophosphate as a probable important determinant for the development of Randall’s plaque leads to not only additional, so far unresolved, questions and problems like the development of reliable inorganic pyrophosphate measurement methods but also to some hopeful prospects for the future, as inorganic pyrophosphate supplementation might constitute a treatment option in patients with Randall’s plaque-associated kidney stone formation.
Vous devez vous connecter pour continuer.
S'authentifier