


 CC-BY 4.0
CC-BY 4.0
1. Introduction
With an annual global production exceeding 10 million metric tons, isobutene (2-methyl propene) is a hydrocarbon of great industrial significance. It is mainly used as raw material for producing polyisobutylene (PIB), butyl rubber and fuel additives (isooctane, alcohols-isobutene ethers). PIB is a synthetic rubber that is used to produce lubricants, adhesives, sealants, fuel additives, cling-film, and chewing gum [1, 2]. Methyl tert-butyl ether, ethyl tert-butyl ether and glycerol-isobutene ether are major octane boosters of reformulated gasolines [3, 4]. Due to its high reactivity, isobutene is also involved in catalytic reactions aimed at producing intermediates and chemicals [5, 6]. Thus, the tert-butylation of phenol and isobutene amination are of great importance owing to the usage of products in the manufacture of resins, surface coating, printing inks, antioxidants, drugs, inhibitors, and agrochemicals [7, 8]. Isoprene, which is produced from formaldehyde and isobutylene, is used for isoprene rubber synthesis [5, 9].
Acids, especially solid acids, catalyze most of these reactions. Zeolites, for which the most important property with respect to their use as catalysts is their surface Brönsted and Lewis acidity, are among the best catalysts for the isobutene conversion. As known, zeolites are effective solid catalysts for a variety of heterogeneously catalyzed gas- and liquid-phase processes involved in oil refining [10, 11, 12], petrochemistry [10, 12, 13, 14, 15], and fine chemicals synthesis [16, 17, 18, 19]. The superior catalytic performances of zeolites compared to other catalysts are related to some important properties, namely: (i) particular texture (high internal surface area, uniform pore diameters and pore widths in the order of molecular dimension); (ii) the number and the strength of the acid sites can be adjusted in a wide range; (iii) high thermal and hydrothermal stability; (iv) shape selectivity, which allows for controlling the formation of desired products [10, 11, 15, 20, 21, 22].
The aim of this review is to examine the broad potential of the combination of acidity and shape selectivity offered by zeolites as catalysts in reactions involving isobutene. The following reactions will be taken into account: (i) Prins condensation; (ii) IB dimerization; (iii) ethers synthesis from IB and methanol/ethanol; (iv) ethers synthesis from IB and and glycerol; (v) IB amination; (vi) phenol tert-butylation.
2. Prins condensantion
The Prins condensation is a well-known acid catalyzed reaction consisting of an electrophilic addition of an aldehyde (ketone) to an alkene/alkyne followed by capture of a nucleophile or elimination of an H+ ion [23]. It is a powerful C–O and C–C bond forming method in the synthesis of various organic molecules. Depending on the reagents and reaction conditions, molecules such as dioxanes, saturated and unsaturated alcohols, glycols, acetals, 𝛽-hydroxyacidsand di-olefins can be designed [24 ]. One of the most important industrial applications of Prins reaction is the reaction between isobutene and formaldehyde. This reaction was intensively investigated since 1937 as part of the tehnology used for isoprene/synthetic rubber synthesis. Industrialy, isoprene is produced from formaldehyde and isobutylene in two steps processes (Scheme 1).
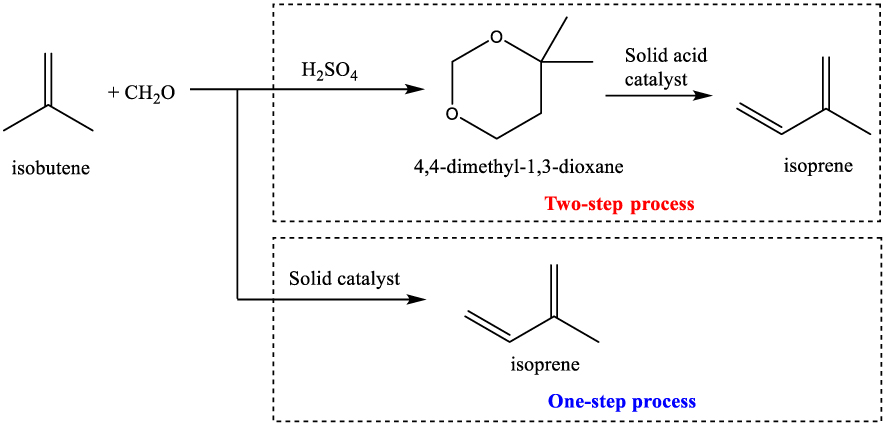
Isoprene synthesis from isobutene and formaldehyde by two-step process and one-step process.
First, sulfuric acid is used as catalyst to obtain initially 4,4-dimethyl-1,3-dioxane (DMD). In the second step, isoprene is produced by cracking DMD in the presence of a heterogeneous catalyst [5]. The low selectivity to isoprene and the use of sulfuric acid in the first stage are the major drawbacks of this technology. To overcome them, research groups have attempted to design one-step processes based on heterogeneous catalysis [9]. Variuos solid materials, including V2O5–P2O5 [25], Nb2O5 [26], Cu/SiO2 [27], SbxOy∕SiO2 [28], AgxSbyOz∕SiO2 [29], heteropoly acids [30, 31], phosphates [32, 33], and sulfates [34] were used as acid catalysts for producing isoprene in gas-phase processes. Unfortunately, most of these catalysts exhibited low isoprene yield, rapid deactivation and difficulty in their regeneration.
Unlike other solid catalysts, zeolites—due to their unique properties (vide supra)—showed very promising potential as catalysts for isoprene synthesis. Venuto and Landis [35] studied for the first time the gas-phase reaction between formaldehyde and isobutene over zeolites. Later, Chang et al. [36] evaluated the performances of H-ZSM-5, H-ZSM-11 and H-ZSM-23 zeolites for Prins reaction carried out in liquid phase. In the 1990s, Dumitriu group revealed that zeolites of various topologies like FAU (faujasites Y), MOR (mordenite), BEA (beta), MFI (ZSM-5) and CLI (clinoptilolite) are effective catalysts for preparing isoprene from formaldehyde and isobutene [37, 38, 39, 40]. More recently, Ivanova and coworkers studied this reaction in the presence of H-ZSM-5, H-Beta, H-Y, Zr-Beta, Sn-Beta, and Nb-Beta catalysts [41, 42]. Yu et al. [43] and Zhu et al. [44] used a series of HZSM-5 catalysts with different Si/Al ratio. Vasiliadou et al. [45] also used H-ZSM-5 catalysts with various Si/Al ratio, but in a liquid phase process. Table 1 summarizes relevant data on the Prins condensation catalyzed by zeolites.
Zeolite catalysts for isoprene synthesis via Prins condensation
| Zeolite catalysts | Reaction temperature (°C) | Main products | References |
|---|---|---|---|
| H-Mordenite | 300 | Isoprene, C1–C4 | [35] |
| H-ZSM-5, H-ZSM-11, H-ZSM-23 | 65–105 (batch) | 3-Methylbutenols, isoprene 3-methylbutanediols | [36] |
| H-ZSM-5, H-M, H-Beta, SAPO-5, Clinoptilolite | 200–400 | C1–C4, isoprene, aromatics | [37] |
| H-ZSM-5, P2O5/H-ZSM-5, SAPO-5, ALPO-5 | 175–400 | C1–C4, isoprene, aromatics | [38] |
| H–Fe-ZSM-5, H–Ga-ZSM-5 | 325–475 | Isoprene, C1–C4, aromatics | [39] |
| HY, USY, H-ZSM-5, H–B-ZSM-5 | 175–390 | Isoprene, C1–C4, aromatics | [40] |
| H-ZSM-5, H-Y, H–Al-Beta, H–Zr-Beta, H–Sn-Beta, H–Nb-Beta | 300 | CO, isoprene | [41] |
| H–Nb-Beta, H–Al-Beta | 300 | CO, isoprene | [42] |
| H-ZSM-5 (Si/Al = 200–800) | 300 | Isoprene | [43 ] |
| H-ZSM-5 (Si/Al = 26–1200) | 240–360 | Isoprene, aromatics | [44 ] |
| H-ZSM-5 (Si/Al = 25–140), H-Beta | 150–180 | 3-methyl-3-buten-1-ol, isoprene, 4,4-dimethyl-1,3-dioxane, 2H-pyran-3,6-dihydro-4-methyl | [45 ] |
Generally, the reaction between formaldehyde and isobutene with zeolite catalysts generate a large distribution of products. Besides isoprene (from Prins condensation), significant amounts of aromatic and light hydrocarbons can be produced [39, 44]. Dumitriu et al. [39] described the reaction pathways occuring on zeolites. Accordingly, Prins condensation starts with the protonation of formaldehyde on a Brönsted acid site generating a carbocation, which further reacts with isobutene to form a tertiary carbocation, and then methylbutenol (Scheme 2). Isoprene results from the dehydration of 2-methylbutene-ol.
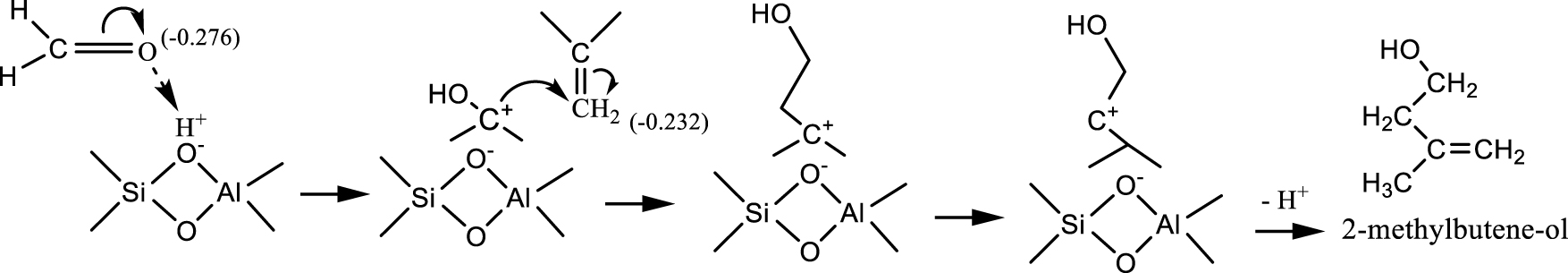
Reaction between formaldehyde and isobutene on Brönsted acid sites (adapted from Ref. [38]).
On the other hand, the protonation of isobutene on the Brönsted acid site can generate a tertiary carbocation (Scheme 3). Note that the formation of the tert-butyl carbocations from isobutene on zeolite surface was confirmed by both theoretical calculations [46, 47] and solid-state NMR spectroscopy [48, 49].
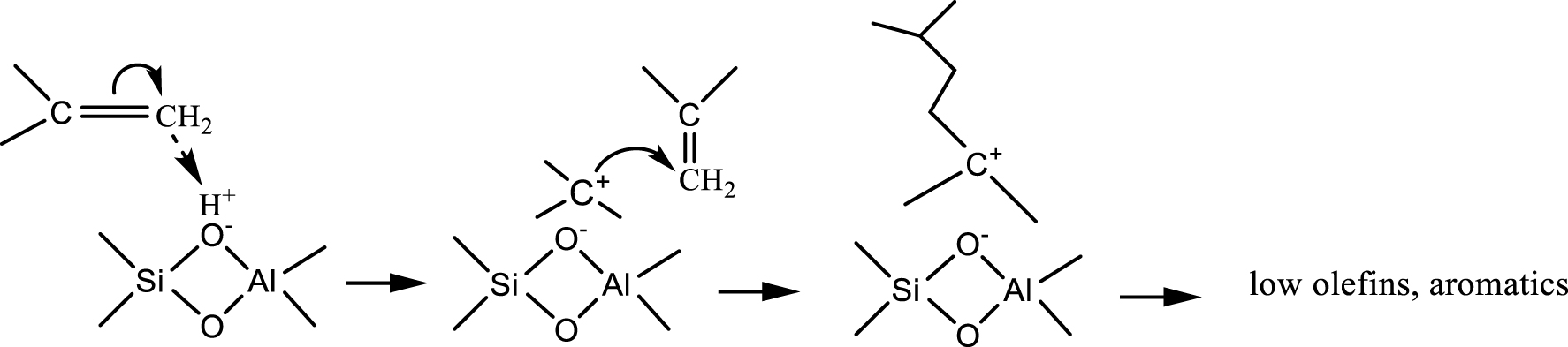
Isobutene dimerization over Brönsted acid sites (adapted from Ref. [38]).
The tert-butyl carbocation reacts with another molecule of isobutene to form an oligomer, and thus various reaction pathways are open. They include processes such as cracking, cyclizations, dehydrogenation of cyclohexanes to aromatics, various isomerizations (cations, olefins, alkylaromatics, etc.). Besides isobutene, isoprene and other olefins resulting from cracking are involved in such processes.
The competition between the reactions showed in Schemes 2 and 3 is strongly dependent on the first stage of the reagent protonation. It depends both on the basic character of reagents (formaldehyde and isobutene) and on the acid strength of the catalytic site. Formaldehyde (charge of − 0.276, Scheme 2) is more basic than isobutylene (charge − 0.232, Scheme 2) and thus it can be more easily protonated, even on weak acid sites [39].
Most of studies carried out over zeolites confirmed this assertion. More exactly, the catalyst activity depended on the strength and the nature of acid sites. The selectivity to isoprene was influenced by the acidity of the catalyst but also by the size/structure of the pores.
In a series of studies, Dumitriu et al. [37, 38, 39, 40] compared zeolites with different topology and acidity. They found that strongly acidic ultrastable Y (USY) and H-ZSM-5 are less selective for isoprene because of numerous side reactions (oligomerization, aromatization, and cracking) which involved isobutene and isoprene. The selectivity to isoprene linearly decreases as the amount of strong acid sites increased [37]. In contrast, the weak Brønsted acid sites were found to be highly efficient for the Prins reaction. Thus, at 250–300 °C, the highest selectivity for isoprene (99–100%) was obtained over H–B-ZSM-5, H–Fe-ZSM-5 and H–Ga-ZSM-5 catalysts. Notable results were also obtained over H–Al-ZSM-5 zeolites with high Si/Al (300) ratio or ZSM-5 zeolite modified by phosphoric acid [37]. The authors considered that in the presence of weak/moderate acid sites the formaldehyde protonation prevailed over isobutene protonation (Schemes 2 and 3), limiting the undesired side reactions.
In a recent study, Yu et al. [43] reported similar conclusions. For a series of H-ZSM-5 zeolites, they found that the Si/Al ratio had a crucial impact on zeolite acidity and catalytic performance in isoprene synthesis. The optimum acid density and acid strength were obtained for a Si/Al ratio of 600. Catalysts with a lower Si/Al ratio have strong acid sites, leading to coke deposits and side reactions.
Wang and Iglesias [50] studied the Prins condensation between isobutanal and isobutene. Over various solid acid catalysts, including H–Al-MCM-41, H-Y, Nb2O5 and H3PW12O40∕SiO2, the selectivity to Prins condensation products was strongly limited by parallel isobutene oligomerization. The authors suggested that the Prins reaction and the oligomerization occur only on Brönsted acid sites. Turnover rates for both reactions were higher on the highly acid catalysts, but the Prins/oligomerization ratio was much higher on the weaker acid catalysts.
Ponomareva et al. [41, 42] studied the single-stage gas-phase synthesis of isoprene in the presence of Al-Beta, Zr-Beta, Sn-Beta, and Nb-Beta catalysts synthesized by isomorphous substitution methods. They found a very good correlation between the Brönsted acid site concentration and the productivity of the catalysts with respect to isoprene (Zr-Beta < Sn-Beta < Nb-Beta < Al-Beta). On the other hand, the amount of carbon monoxide formed as by-product from the decomposition of formaldehyde increased when the number of Lewis acid sites on the catalyst surface increased.
In the studies examined above, the reaction between formaldehyde and isobutene was carried out in the gas phase, at temperatures higher than 250 °C. Under these conditions, the formation of the intermediates was hardly perceptible. To obtain some details on the reaction network, Vasiliadou et al. [45] worked in liquid phase, at 150 °C using H-ZSM-5 zeolites with Si/Al ratio between 25 and 140. Four major molecules were identified in the product mixture: 3-methyl-3-buten-1-ol (I), isoprene (II), 4,4-dimethyl-1,3-dioxane (III), and 2H-pyran-3,6-dihydro-4-methyl (IV) (Scheme 4). Selectivities between 82–90% in Prins condensation products (I and II) were obtained over all catalysts.
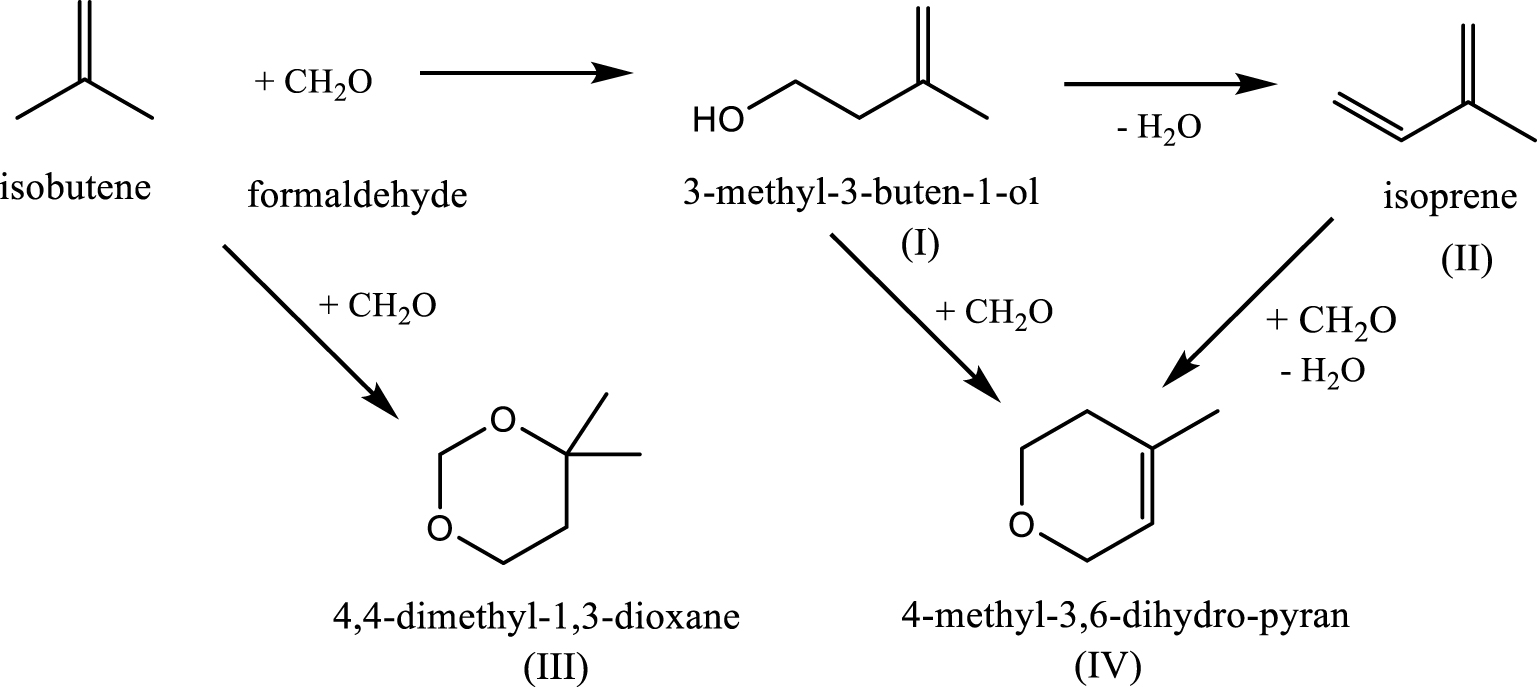
Reaction network of formaldehyde reaction with isobutene in liquid phase (according to Ref. [45]).
The ratio between the products I and II is strongly dependent on the reaction time, the iso-butene/formaldehyde molar ratio and the Si/Al ratio of zeolite. The highest selectivity in isoprene (54.5%) was obtained on H-ZSM-5 (40), after 3 h of reaction, for an iso-butene/formaldehyde molar ratio of three. To develop a reaction mechanism, the authors combined the experimental approach with the DFT method. They suggest that the reaction follows a three-step mechanism: protonation of formaldehyde and electrophilic attack of isobutene, and deprotonation of the resulting carbocation intermediate. The limiting step is the electrophilic addition of isobutene to the formyl group. H-ZSM-5 was identified as the most effective and selective for formation of 3-methyl-3-buten-1-ol. Sequential undesired Prins cyclization and hetero-Diels–Alder reactions are limited by the catalyst pore size.
To conclude, zeolites are able to effectively catalyze the condensation between isobutene and formaldehyde. This reaction only occurs on Brönsted acid sites and it is strongly competed by the isobutene oligomerization. The highest Prins/ oligomerization ratio is obtained on the weaker acid sites. The highest selectivity to isoprene is exhibited by the medium-pore MFI zeolites (pore diameter of 0.55 nm) which prevent the cyclization/ oligomerization reactions. The strong acid sites activate side reactions such as formaldehyde decomposition and oligomerization.
3. Isobutene oligomerization
Isooctane (tri-methyl pentane) is an excellent octane booster compound, with a highly environmental-friendly reputation. Isooctane can be obtained through a simple and low-cost technology, by catalytic dimerization of isobutylene followed by a hydrogenation step (Scheme 5).
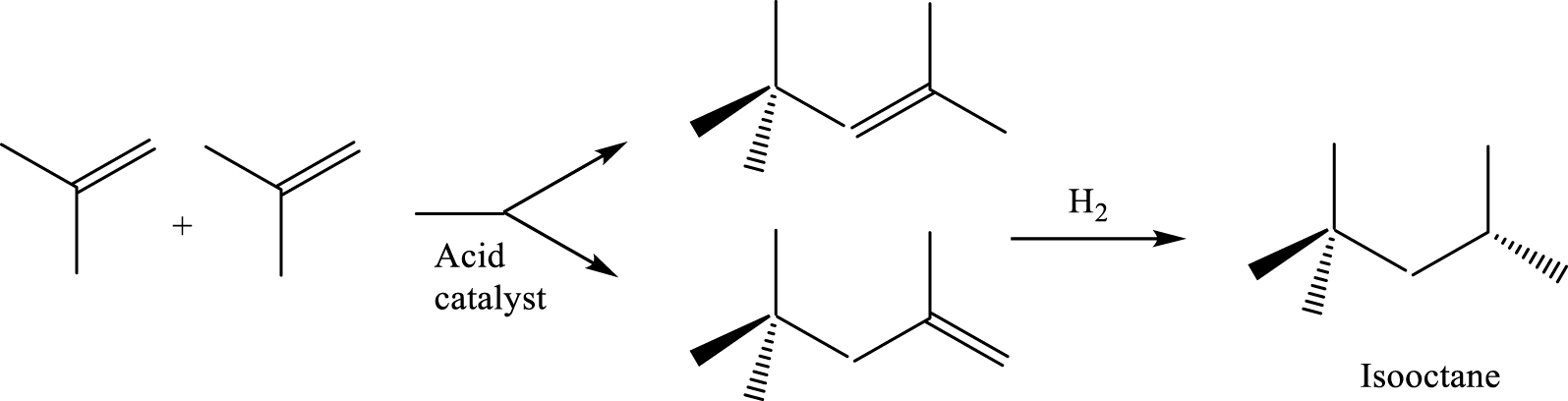
Isooctane formation from isobutene.
Dimerization is highly exothermic (𝛥H = −87.3 kJ/mol) and as a result, important side reactions, leading to iso-C12 and iso-C16 olefins, occur. Various solid acid materials including resins, zeolites, and metal oxides are effective catalysts for the isobutene dimerization [51, 52, 53]. In this section, representative dimerization studies performed on zeolites as catalysts will be examined. The catalytic performances (conversion of isobutene, selectivity towards isooctane, stability of the catalyst) will be assessed in relation to the characteristics of the catalysts (topology, porosity, acidity) and reaction conditions.
In one of the earlier studies, Hauge et al. [54] compared the catalytic behavior of various zeolites and resins for the isobutene dimerization in a plug flow reactor, in liquid phase, at low temperature (30–70 °C). H-Y, H-Beta, H-Mordenite (H-M) and H-ZSM-5 zeolites exhibited high initial activity, but all catalysts rapidly deactivated. In contrast, Amberlist-15 showed a moderate initial activity but a good stability against deactivation. The rapid deactivation of zeolites has been attributed to the formation of high molecular weight oligomers inside the micropores. Yoon et al. [55] have reported similar conclusions in a study focused on the isobutene oligomerization over resins, beta and ferrierite zeolites.
Concerning the selectivity in dimerization processes, besides octenes (dimers), large amounts of trimers (C12) were produced. The ratio between the dimers and trimers depend on the nature of the catalyst, the reaction conditions and the isobutene conversion. Compared to zeolites, the resin catalysts (with large pores) produce more C12 olefins. Typically, the trimers are mainly formed at high temperatures [56, 57] and high conversion [58, 59]. This last aspect is clearly observable in Table 2.
Dependence of selectivity with isobutene conversion*
| IB conversion (%) | 20 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | 100 |
|---|---|---|---|---|---|---|---|---|---|
| Dimer (%) | 92 | 90 | 88 | 85 | 80 | 73 | 62 | 46 | 10 |
| Trimer (%) | 8 | 10 | 12 | 14 | 18 | 23 | 32 | 46 | 77 |
| Tetramer (%) | 0 | 0 | 0 | 1 | 2 | 4 | 6 | 8 | 13 |
*According to Ref. [58]; catalysts: ferrierite, mordenite or ZSM-5.
In the case of zeolites, the topology of the catalyst has a crucial impact on both activity (conversion of isobutene) and selectivity (dimers versus trimers). Yoon et al. [55, 58] evaluated the effect of the zeolite topology in oligomerization of isobutene using beta zeolite (three-dimensional channels, 3D), ferrierite zeolite (two-dimensional channels, 2D), and mordenite zeolite (one-dimensional channels, 1D). The conversion obtained over beta zeolite (99%) was higher than over ferrierite (80%) and much more than over mordenite (20%). Moreover, mordenite has been rapidly deactivated. The amount of trimers produced varied in the order beta (30%) > ferrierite (20%) > mordenite (5%), and this order may be related to the size and the dimensionality of the pores.
The same group studied the effect of zeolite topology (1D versus 3D) using mordenite (1D), beta (3D) and ultra stable Y (USY, 3D) [59]. Significant differences existed between the isobutene conversion profiles as a function of time on stream. On mordenite, the isobutene conversion was rapidly decreased from 90% to 20%, whereas USY and in particular beta exhibited excellent stability for more than 20 h on stream. The selectivity to C12 olefins was very high compared with that over USY and mordenite. In a simple manner, the high stability shown by zeolite beta can be attributed to its 3D porosity. The authors also emphasized the role of the acidity of the catalyst. They tested three beta samples having different Lewis acid sites (LA) to Brönsted acid sites (BA) ratios. The experimental results showed that high LA/BA ratio led to a higher stability against the deactivation and higher trimer selectivity.
Yaocíhuatl et al. [56] have also investigated the effect of the acidity. Zeolites like H-Y, H-Beta and H-M have been modified by impregnation with nickel, using different Ni salts as precursors. The addition of Ni resulted in an enhancement of the acid sites total number and the acid strength. Additionally, the concentration and the strength of the Lewis acid sites increased after zeolite modification. Ni/zeolite catalysts were more active and selective to dimerization reaction than the parent zeolites. The high catalytic activity, selectivity and stability of the modified zeolites in isobutene dimerization was attributed to the specific acidic properties of nickel modified zeolites. The Lewis acid sites are responsible for the selective adsorption of isobutene on the catalyst surface in the proximity of the Brönsted acid sites.
The favorable role played by the presence of Lewis acid sites has been pointed out by Yoon et al. [60] for a series if dealuminated Y zeolites obtained by steaming at different temperature. The catalyst having the highest amount of Lewis acid sites showed the highest stability, but also the highest selectivity to trimers and tetramers. In order to increase the Lewis acidity, USY zeolite has been loaded with AlCl3 [61]. Stable isobutene conversion and high selectivity for trimers and tetramers were attained over the modified zeolite with high ratio of Lewis acid site-to-Brönsted acid site.
Al-Kinany et al. [57] used a supported phosphoric acid on H-Y zeolite in the oligomerization of isobutene. The addition of phosphor species resulted in an enhancement of Brönsted acid sites total number. The modified zeolite exhibited a very high isobutene conversion and selectivity to isooctane up to 65%. Park et al. [62] have shown that the catalytic performance of zeolites for the isobutene dimerization can be controlled by optimizing both textural and acidic properties. Mesostructured aluminosilicates (MSU) assembled from zeolite beta seeds have been prepared, characterized and compared with commercial beta and USY zeolites. As shown in Table 3, the MSU catalyst was more active and stable in comparison to the commercial zeolites.
Isobutene conversion (%) versus TOS over zeolites and mesostructured catalysts*
| TOS (h) | 1 | 2 | 3 | 5 | 10 | 20 | 30 |
|---|---|---|---|---|---|---|---|
| MSU-Beta | 55 | 48 | 42 | 38 | 36 | 35 | 35 |
| Beta | 48 | 39 | 27 | 19 | 13 | 11 | 11 |
| USY | 43 | 16 | 9 | 6 | 5 | 5 | 5 |
*According to Ref. [62].
The high stability exhibited by the MSU samples has been attributed to their improved textural properties. Larger pores facilitate mass transfer to and from the active sites, whereas in zeolites the reaction is likely controlled by internal diffusion. On the other hand, the authors consider that the acidity promoted by the MSU catalysts also played a crucial role. Compared to beta and USY zeolites, MSU catalysts have a larger amount of Lewis acid sites. A synergetic interaction between Lewis and Brönsted acid sites has been proposed to increase catalytic activity and stability. Selectivities higher than 80% to isooctane have been obtained over all catalysts.
High activity and selectivity to C8 olefins have also obtained by Torres et al. [63] over a Beta-zeolite membrane. This behavior has been attributed to the surface acidity of the membrane and the control of short residence time within the zeolite pores.
Koskinen et al. [64] demonstrated the crucial role played by the solvent in the isobutene dimerization process. ZSM-13 and ZSM-5 zeolites have been used as catalysts, while CO2 and propane was used as solvents. The reaction has been carried out in a continuous stirred tank reactor, at 100 °C and 5.0–9.0 MPa. For both solvents and catalysts, the selectivity to isooctenes has been higher than 75% (at 50% isobutene conversion) and the initial isobutene conversion has been above 80%. However, the catalyst stability has been higher in CO2. Thus, the conversion with CO2 was 56% during 200 h on stream, whereas with propane it was 32% for 120 h on stream.
Tiako Ngandjui and Thyrion [65] studied the kinetics of the isobutene oligomerization on H-mordenite. The linear dependence between the initial rates and the initial reactant concentration suggested a first-order kinetics. The initial rates also showed that the reaction followed a Rideal mechanism. The pathway reaction was found to be a rake type.
To conclude on the isobutene dimerization on zeolites:
- The topology of the catalyst has a crucial impact on both activity (conversion of isobutene) and selectivity (dimers versus trimers). Zeolites with two/three-dimensional channels are more active and stable catalysts than the one-dimensional channel zeolites. However, the former generate more amount of trimers.
- The nature of the acidity also plays an important role in dimerization: a high LA/BA ratio led to a higher activity and stability against the deactivation, but to a lower dimer selectivity.
4. Etherification reaction: MTBE and ETBE synthesis
Methyl tert-butyl ether (MTBE, more than 35 million tons annually) and ethyl tert-butyl ether (ETBE, 4 million tons annually) are major octane boosters of reformulated gasolines [3]. They are produced by etherification reactions between the corresponding alcohol (methanol, ethanol) and isobutene, in the presence of an acid catalyst, according to the following reactions:
MTBE/ETBE formation over various zeolites (selected results)
| Zeolite catalyst | Ether | Temperature (°C) | WHSV (h−1) | IB conversion (%) | Selectivity to MTBE/ETBE (%) | References |
|---|---|---|---|---|---|---|
| ZSM-5 | MTBE | 82 | 3.4 | 30.5 | 100 | [70] |
| ZSM-11 | 25.1 | 99.6 | ||||
| MOR | 14.4 | 58.3 | ||||
| RE-Y | 11.77 | 96.0 | ||||
| Beta | 36.9 | 37.7 | ||||
| ZSM-5 | ETBE | 120 | 3.4 | 6.9 | 100 | [74] |
| ZSM-5 | MTBE | 90 | 3.6 | 20 | 96 | [77] |
| ZSM-5 26.2 | MTBE | 80 | 4.9 | 41 | 100 | [85] |
| Y | MTBE | 75 | 3.25 | 27 | 100 | [86] |
| ZSM-5 | 70 | 26 | 100 | |||
| MOR | 85 | 21 | 100 | |||
| Beta | 60 | 50 | 100 | |||
| Omega | 85 | 25 | 100 | |||
| USY | MTBE | 100 | 14 | 80 | 90 | [87] |
| Beta | 70 | 95 | 85 | |||
| Beta | ETBE | 67 | 2.0 | 22 | 90 | [88] |
| USY | 80 | 11 | 100 | |||
| ZSM-5 | 95 | 5 | 100 | |||
| Omega | 90 | 5 | 100 | |||
| Beta 15 | ETBE | 100 | 1.0 | 85 | 79 | [90] |
| Beta 32 | 80 | 78 | ||||
| Beta 72 | 77 | 80 | ||||
| Beta 124 | 0.5 | 100 | ||||
| ZSM-5 | MTBE | 115 | n.a. | 89 | 100 | [91] |
The first studies on the ability of zeolites to catalyze the reaction between isobutene and lower alcohols were carried out at the end of the 1980s. Chu and Kühl [70] compared two medium-pore ZSM-5 and ZSM-11 zeolites with a commercial A15 catalyst. In liquid phase, under conditions similar to those used in the commercial technology, zeolites gave higher selectivity to MTBE than A15 resin. In addition, the selectivity on zeolites was less sensitivity to methanol/isobutene ratio and to temperature variation. The high selectivity of zeolites has been attributed to their pore size and structure. The authors considered that methanol diffuses faster than isobutene within the zeolite channels. The isobutene molecule encounters an excess of methanol on zeolite surface and reacts to form MTBE with high selectivity. In the same work, Chu and Kühl compared various zeolites, i.e., beta, mordenite, rare-earth-exchanged Y, ferrierite, ZSM-5 and ZSM-11 in the reaction methanol-isobutene carried out in vapor phase. Small-pore ferrierite was inactive for this reaction since isobutene cannot enter the zeolite pores. As in the liquid phase, ZSM-5 and ZSM-11 exhibited high activity and selectivity. In contrast, mordenite and beta showed low selectivity to MTBE, which is in line with the lack of shape selectivity in the diffusion of reagents in these large-pore zeolites.
Tau and Davis [74] carried out a similar comparative study for the ETBE synthesis from ethanol and isobutene over sulfonated resins and ZSM-5 zeolite. In both vapor-phase and liquid-phase tests, the conversion over ZSM-5 was lower compared to that obtained on resin catalysts. In other study, Assabumrungrat et al. [75] found that even though the catalytic activity of beta zeolite was lower than that of resins, the selectivity of ETBE was much higher than that of resins. Le Van Mao et al. [76] showed that triflic acid loaded Y-type zeolite was as active as the sulfonated resins in the (gas phase) synthesis of MTBE. Moreover, the zeolite catalyst produced less by-products and was more thermally stable than the resin-based catalyst.
Chang et al. [77] studied the reaction between methanol and isobutene in vapor-phase (70–110 °C), on H-ZSM-5 zeolite and titanosilicate TS-1. The authors found that the strength of the acid sites and the adsorption strength of TS-1 were weaker than those of H-ZSM-5. As a result, higher selectivity to MTBE was observed, and catalyst deactivation or coke formation did not occur on TS-1 catalyst. The kinetic data fit with the Langmuir–Hinshelwood mechanism, which assumes the reaction between adsorbed methanol molecules with isobutene adsorbed at two different acid sites is the rate-determining step.
In a series of studies published in the 1990s, the Marcelin group investigated the behavior of various parent and modified zeolites as catalysts in the MTBE synthesis [78, 79, 80, 81, 82, 83, 84]. Working at elevated temperatures (up to 175 °C) and low pressures (150 kPa), H-Y and H-ZSM-5 zeolites exhibited higher selectivity to MTBE than the commercially used Amberlyst-15 resin catalyst [78]. H-ZSM-5 was more suitable for high temperature formation of MTBE because of its excellent selectivity towards MTBE and low deactivation behavior. Note that high selectivity to MTBE and deactivation stability for H-ZSM-5 catalysts were also latter reported by Ahmed et al. [85]. To explain the higher selectivity to MTBE observed on zeolites, Kogelbauer et al. [82] studied the adsorption behaviors of different catalysts and their impact on the MTBE synthesis. The experimental tests showed that ca. 2.5 molecules of methanol were adsorbed per acid site on H-ZSM-5 and H-Y zeolites, whereas isobutene formed only a 1:1 adsorption complex. On a commercial resin catalyst equal amount of methanol and isobutene were adsorbed per acid site. The high adsorption of methanol on zeolites was concluded to play a key role in suppressing the formation of by-products due to isobutene oligomerization.
To understand the influence of the acid strength of zeolites upon gas-phase formation of MTBE, the authors tested a series of dealuminate H-Y zeolites [79, 80]. The strong increase of the TOF in MTBE as the concentration of the acid sites was reduced was attributed to the interaction between extra-lattice Al and Brönsted acid sites. For the same purpose, partially ion-exchanged H-Y zeolites with cations like Li, Na and Rb were tested in process [81]. No significant effect upon initial rates for the formation of MTBE was observed. In contrast, deactivation due to the formation of olefin oligomers had a strong impact on the steady-state activities. Finally, the authors modified the parent zeolites, i.e., H-Y, H-M or H-ZSM-5 by adding triflic acid (TFA) [83]. In the case of H-Y zeolite, an increase in activity for MTBE formation was observed only for levels of TFA up to 3 wt%. Further incorporation of TFA resulted in a blockage of the active sites, leading to a lower activity. A more severe case of site blockage was observed for the TFA-modified HZSM-5 and H-M. In these cases, the experimental evidence showed no increase in catalytic activity. A similar beneficial effect on the reaction rate was observed when H-Y, H-M or H-ZSM-5 zeolites have been modified by ion-exchange with ammonium fluoride [84].
The Poncelet group has investigated the behavior of zeolites having different topology and Si/Al ratios in the MTBE or ETBE synthesis [73, 86, 87, 88]. Amberlyst-15 has been used as reference catalyst. When the reaction between methanol and isobutene has been carried out in gas phase over different dealuminated acid zeolites, the following sequence in activity was observed: Beta = Amberlyst-15 > H-Y > H-Omega > H-ZSM-15 > H-M > SAPOs. Dealumination has a beneficial effect on the reaction, the highest conversions being reached for bulk Si/Al ratios between 13 and 35, according to the type of zeolite. Over all zeolites, the yield of MTBE increased as reaction temperature increased, reaching a maximum at 60–70 °C. For the best catalyst (H-Beta), the authors extended their investigation to the liquid-phase reaction [87]. Zeolite H-Beta (Si/Al ratios of 12.2 and 36) exhibited a higher activity than Amberlyst-15 at temperatures between 40 and 100 °C and similar MTBE selectivity up to about 90% conversion of isobutene. At higher conversion, the resin is slightly more selective than the H-Beta, because the oligomerization of isobutene is more pronounced over zeolite. MTBE yields of 85–90% have been reached with both catalysts. H-beta kept its high activity during a period of more than 50 h on stream. The catalytic activity of H-beta catalyst has been related to the external specific surface area, and to the concentration of bridging hydroxyls and silanol groups in the pores.
Collignon and Poncelet [88] have realized a similar comparative study of different solid catalysts (sulfonated resins and zeolites) for the ETBE synthesis. Vapor phase reaction between ethanol and isobutene has been carried out over USY, Beta, and ZSM-5 zeolites with different Si/Al ratios, using Amberlyst-15 as a reference catalyst. Beta exhibited the highest external surface area and showed higher activity than the other zeolites: Beta zeolite > USY > Mordenite > Omega > ZSM-5 was observed. Selectivity up to 100% has been obtained over both Beta zeolites and Amberlyst-15 below 55 °C. On zeolite, the reaction occurs on bridging AlOHSi acid sites, with higher yields of ETBE for those with high content of acid sites (AlOHSi) and low SiOH/AlOHSi ratios. Extra-framework Al species have a detrimental influence on the reaction.
More recently, Vlansenko et al. [89, 90] explored the behavior of beta zeolites with different Si/Al ratio for the ETBE synthesis carried out in a continuous flow reactor, in both liquid and gas phase. A good correlation between the rate of ETBE synthesis and the concentration of weak acid sites has been observed. The authors concluded that the active sites of H-Beta are Brönsted hydroxyls representing internal silanol groups associated with octahedrally coordinated aluminum in the second coordination sphere.
According to the studies reviewed in this section, beta seems to be the best zeolite catalyst for the etherification between isobutene and methanol/ ethanol. To better understand the behavior of this zeolite, Hunger et al. [92] studied the adsorption of isobutene and methanol on a zeolite beta by in situ MAS NMR spectroscopy, under continuously-flow conditions. The experimental data showed that silanol groups, located at the external surface of the zeolite particles are responsible for the formation of adsorbate alkoxy complexes between the reagents. These complexes, assigned to species with carbenium-like properties, may be important for the reactivity of acidic zeolites in MTBE synthesis.
Kogelbauer et al. [93] investigated the coadsorption of methanol and isobutene on HY zeolite using the IR spectroscopy. The study revealed that isobutene, once adsorbed, quickly oligomerizes, even at low temperature. When the methanol has been adsorbed before isobutene MTBE is selectively formed. In other words, methanol effectively inhibits the adsorption and subsequent oligomerization of isobutene on the acid sites of the zeolite.
5. Glycerol-isobutene etherification
Nowaday, glycerol is produced in large amount (about 4 million tons per year) as by-product in the biodiesel industry. Unfortunately, the glycerol surplus has still a limited range of industrial applications. In order to enhance the sustainability of the biodiesel industries, it is mandatory to develop new ways of transforming glycerol. Among the promising ways able to convert glycerol into value-added products is the production of glycerol-based fuel additives [4, 94, 95]. For example, the tert-butyl ethers of glycerol (TBG) synthesized via etherification reactions between glycerol and isobutene can be used as octane-boosters for gasoline.
The reaction between glycerol and isobutene is a Brönsted acid catalyzed reaction and produces a large game of mono-, di- and tri-ethers (Scheme 6) [96]. By-products like isobutene oligomers and tert-butyl alcohol can be also formed. Note that only di-TBG (DTBG) and tri-TBG (TTBG) have the properties required by the fuel additives.

Reaction pathways for the etherification of glycerol with isobutene (adapted from Ref. [96]).
Various solid catalysts including resins, sulfonated carbons, heteropolyacids and microporous/mesoporous aluminosilicates have been explored in this process. For a recent review on heterogeneous catalysis in the ethers synthesis, see Refs. [96] and [97]. The most important results obtained in the presence of zeolite catalysts will be examine below.
Table 5 summarizes representative zeolite-based catalysts investigated in the reaction between isobutene and glycerol.
IB-Glycerol etherification over zeolites (selected results)
| Zeolite | Temperature (°C) | Pression (bar) | IB/G | Reaction time (h) | Glycerol conversion (%) | Selectivity to GTBE | References |
|---|---|---|---|---|---|---|---|
| Nd-Beta | 70 | 15 | 3 | 2 | 93 | 75 | [98] |
| Y | 80 | Auto | 4 | 5 | 82 | 57 | [99] |
| Beta | 75 | 10 | 4 | 48 | 100 | 84 | [100] |
| Hierarchical Beta | 75 | Auto | 4 | 24 | 77 | 35 | [101] |
| ZSM-5 | 120 | Auto | 4 | 0.25 | 59 | 22 | [102] |
| Beta | 90 | Auto | 4 | 10 | 57 | 29 | [103] |
| USY | 90 | Auto | 4 | 4 | 75 | 21 | [104] |
| Beta | 90 | 1 | 4 | 8 | 97.7 | 95 | [105] |
| 90 | 4 | 10 (TOS) | 95 | 96 | |||
| Beta Y | 60 | Auto | 4 | 1 | 84 | [106] | |
| Y | 8 | 88.7 | |||||
| Beta | 90 | Auto | 4 | 4 | 96.7 | [107] | |
| MOR | 4 | 66.1 | |||||
| USY | 4 | 100 | |||||
| USY | 75 | Auto | 3 | 6 | 100 | [108] | |
| Beta | 100 | ||||||
| Beta | 60 | Auto | 4 | 8 | 100 | [109] | |
| 90 | 18 | ||||||
| Y | 60 | 94.8 | |||||
| 90 | 100 |
Because the crucial factors determining the catalytic activity and the nature of the products formed in the etherification process are the Brönsted acidity and the porosity/texture of zeolites, most studies have focused on the behavior of the catalyst with respect to these properties. Most often, zeolites are compared to commercial catalysts, especially to resins. Klepáčová et al. [109] studied the solventless etherification of glycerol with isobutene (or tert-butyl alcohol) on resins and two large pore H-Y and H-Beta zeolites. At 60 °C, glycerol conversion up to 100% and selectivity to TBG up to 92% were obtained over resins. Good yield to DTBGs were also obtained over zeolites (65.8% and 80.6% for H-Y and H-Beta), but these catalysts suffered rapid deactivation due to their small size of their pores. When the etherification reaction was carried out in the presence of solvents, the best productivity in DTBGs was obtained over HY zeolite [106]. However, even with a solvent, the catalyst easily deactivated.
Ozbay et al. [110] compared different commercial catalysts i.e., Amberlyst-15, Amberlyst-36, Amberlyst-35, Amberlyst-16, Relite EXC8D, Lewatit K2629, H-beta, H-M and Nafion SAC-13. Amberlyst-15, which has the highest Brönsted acidity, gave the highest glycerol conversion at 90–100 °C. However, this material is unstable at temperatures higher than 110 °C. On the other hand, the experimental results also proved the importance of diffusion resistance on the reagents conversion: the penetration of glycerol to the active acid sites is limited, especially in zeolites, which have smaller pore diameters.
Miranda et al. [103] studied the role of the acidity (nature, concentration, strength) and textural properties in the TBG synthesis for various acid catalysts, such as Amberlyst-15, silica, alumina, silica alumina and four types of zeolites, i.e., FAU, MOR, BEA and MFI. The experimental data showed that the glycerol etherification is not only a function of the amount of Brönsted acid sites, but that it further proceeds via a product shape selectivity mechanism. Thus, the reaction rate strongly depended on the accessibility in the pores of the catalyst (Table 6). The authors stated that the etherification of glycerol by tert-butyl alcohol occurs through a successive reaction sequence and follows an Eley–Rideal type mechanism.
Glycerol conversion as a function of reaction time over zeolites
| TOS (h) | 0.5 | 1 | 2 | 3 | 5 | 7 | 10 |
|---|---|---|---|---|---|---|---|
| Beta | 18 | 29 | 41 | 44 | 51 | 52 | 53 |
| Y | 2 | 7 | 10 | 16 | 20 | 27 | 31 |
| ZSM-5 | 1 | 3 | 5 | 7 | 9 | 10 | 11 |
| Mordenite | 5 | 6 | 6 | 7 | 7 | 8 | 9 |
*According to Ref. [103].
In a recent detailed study, Bozkurt et al. [108] compared the performance of more than 70 solid acid catalysts. Unmodified and modified ion exchange resins, zeolites, silica, and heteropolyacids were tested under similar conditions. In general, the acid strength of zeolites increased by modification, affecting the product selectivity in etherification. For example, the desired glycerol tertiary butyl ether (DTBG and TTBG) selectivity improved from 66 to 85 wt% by hydrothermal steam treatment of zeolite H-Y and from 75 to 80 wt% with partial La+-exchange of zeolite H-Beta, at high glycerol conversions. Turan et al. [111] obtained similar results in a study focusing on comparing the performance of resin catalysts with zeolites. Differences in product selectivity over Amberlyst 35 and zeolite beta have been attributed to the much lower concentration of acid sites in zeolite beta.
González et al. [101, 112] studied the role of Brönsted acidity and zeolite porosity for the etherification of glycerol with tert-butanol. For this purpose, three commercial Na-zeolites (mordenite, beta and ZSM-5) have been modified by protonation, dealumination, desilication-protonation, lanthanum-exchange and fluorination. These modifications generated only moderate effect for ZSM-5 and mordenite. In contrast, the introduction of fluorine in the beta zeolite framework generated higher amounts of stronger acid sites, which were able to transform glycerol until the glycerol TTBG. Thus, fluorinated beta yielded the best conversion (75%) and selectivity to TBGs (37%) with the formation of glycerol triether in low amounts. These values were comparable to those obtained at the same reaction conditions with an Amberlyst-15, an acid catalyst traditionally used for this reaction.
In order to improve the catalytic performance in etherification, the acidity and the texture of the zeolites have been modified in various ways. Estevez et al. [113] dealuminated ZSM-5 and Y zeolites by acid treatment with HCl and functionalized them with sulfonic acid groups. These solids, exhibiting higher acidity, gave good yields to TBG (13%). González et al. [114, 115] incorporated sulfonic groups into beta, ZSM-5 and mordenite zeolite. In all cases, the presence of sulfonic acid sites improved the catalytic activity of the zeolites studied. Zhao et al. [98] studied the etherification reaction over rare earth (La3+, Ce3+, Nd3+, and Eu3+) modified H-Beta zeolites. At 70 °C, Nd3+-beta exhibited the best catalytic result (67% of yield to TBGs). The highest activity attained on this catalyst is related to the highest acidity that it also exhibited. Xiao et al. [99] observed an improvement in the catalytic activity of H-Y zeolite washed with citric acid and nitric acid, in comparison with the unmodified H-Y. This behavior was associated with the modification of zeolite texture. Simone et al. [102] synthesized nanostructured MFI-type zeolites consisting in a three-dimensional disordered network with 2.5 nm thickness supporting each other. This catalyst, which exhibited a high proportion of the acid sites on the external surface (enabling a favorable accessibility of the reactants to these sites) was more active that the traditional MFI zeolite in the etherification reaction. Viswanadham et al. [107] obtained better results (95% of glycerol conversion and selectivity to TBGs of 99%) on a nano-beta zeolite in a continuous flow reactor, due to the presence of inter-crystalline mesopores, which were absent in the typical beta zeolite. Saxena et al. [105] showed that the desilicated beta zeolite with enhanced (meso)porosity exhibited 98% glycerol conversion with 99% selectivity to DTBG and TTBG). The modified zeolite catalyst also exhibited stability in catalytic performance with the reaction time.
Briefly, the selectivity to the desired ethers increases with increasing Brönsted acidity, improving the textural properties (increased surface area, pore size, pore volume and additional mesoporosity), and improving the accessibility of Brönsted acid sites. The selectivity in the etherification process can be also affected by the reaction conditions, such as temperature, reagent mole ratio, and reaction time. For instance, a higher temperature is associated with more di-isobutene production [109] as isobutene dimerization rate is more temperature sensitive [88].
6. Isobutene amination
Tert-butylamine is an important intermediate for producing pharmaceuticals, pesticides, rubber additive, and water treatment chemicals. Most of the methods proposed for its synthesis use strong acids/alkaline and highly toxic substances, which produce large amounts of waste [116]. Compared to these unfriendly methods, the production of tert-butylamine by direct amination of isobutene over solid acids (1) is a notable example for the replacement of a stoichiometric reaction by environmentally benign heterogeneous catalysis [117, 118].
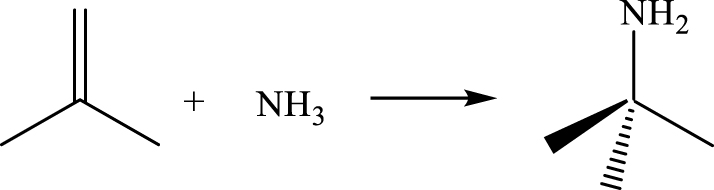
Concerning the amination catalysts, both already in use and under evaluation, it is interesting to note that zeolites are widely investigated [120, 121, 122, 123, 124, 125, 126, 127, 128, 129]. Deeba et al. [124] first reported direct amination of isobutene over zeolites. The authors found that the activity exhibited by various zeolites, including H-Y, RE-Y, Na-Y and H-M, correlates with the number of their strong acid sites. The necessity of acid catalysis was demonstrated by the negligible activity of the nonacidic Na-Y zeolite. The carbenium ion (generated by the protonation of isobutene by either a Brönsted acid or an adsorbed ammonium ion) was suggested as key intermediate for the reaction.
Mizuno et al. [122] studied the isobutene amination over H-ZSM-5, H-M, H-Y, H–FER, H–OFF zeolites having a wide range of Si/Al ratios and over other solid acid and base catalysts. The authors concluded that the amination reaction occurs only on the Brönsted acid sites. Moreover, the number and the strength of these sites were considered the critical factors controlling the reaction. H-ZSM-5 with a SiO2∕Al2O3 of 81 was the most active catalyst among the solid tested. At 200 °C and 0.1 MPa, the amination was zero order with respect to NH3 partial pressure and positive order with respect to isobutene partial pressure. The same group has already previously reported the excellent behavior of zeolite ZSM-5 as a catalyst for the amination of isobutene [121].
Lequitte et al. [125] investigated the reaction between isobutene and ammonia at 250–450 °C and 1.0–6.0 MPa over a series of acidic zeolites with BEA, MFI and FAU topology. The reaction rates increased with the Si/Al ratio of the catalyst and no influence of the zeolite structure was observed. The catalytic activity expressed per proton was higher on MFI than on BEA or FAU zeolites. The large pore BEA and FAU zeolites showed a higher resistance to deactivation with the strongly basic polyalkylamines formed in process. A Langmuir–Hinshelwood mechanism involving adsorbed species has been proposed. A high product yield (above 85 wt%) in the direct amination of isobutene to tert-butylamine on zeolite beta has also been reported by Zhao et al. [126].
In a more recent work, Gao et al. [127] have studied the relationship between the zeolite structures and acidities and the catalytic performance for the direct amination of isobutylene to tert-butylamine. Thus, zeolites with suitable pore diameter (>0.5 nm) or with large side pockets/cups in the outside surface (Beta, EU-1, ZSM-11, ZSM-5, MCM-49, MCM-22) and a certain number of mid-strong acid sites of the zeolites exhibited good amination performance. The equilibrium conversion was >46.4% and the tert-butylamine selectivity was >99.0%. In contrast, ZSM-35 (narrow pore diameter), ZSM-23 (narrow pore diameter), SAPO-11 (weak acidity) and mordenite (too high strong acidity) zeolites exhibited low isobutylene amination activity. A linear relationship between the amination activity and the amount of Brönsted acid sites with mid-strong strength has been observed for ZSM-5 zeolites with different Si/Al ratios. Zhang et al. [128] have also revealed the favorable effect of the amount of the Brönsted acid sites for ZSM-11 zeolite catalyst.
To better understand the mechanism of isobutene amination and to identify key structural properties responsible for catalytic behavior, the Bell group realized a combined experimental and theoretical study [129]. The authors suggest that the active sites are the Brönsted acidic protons located within the zeolite pores. The small-pore zeolites with one-dimensional channels (i.e. FER) are inactive because tert-BuNH2 blocks the pore mounts. In the case of the medium/large-pore zeolites (MFI, FAU, MOR), the intrinsic reaction rate and activation energy are dependent on zeolite topology. Additionally, kinetic measurements and FTIR spectroscopy revealed a strong competition for adsorption on Brönsted acid sites between isobutene and tert-butylammonium ions. The reaction mechanism proposed are illustrated in Scheme 7 (k2 is the rate constant for carbenium formation and k3 is the rate constant for nucleophilic attack by NH3 to form a tert-butylammonium ion).
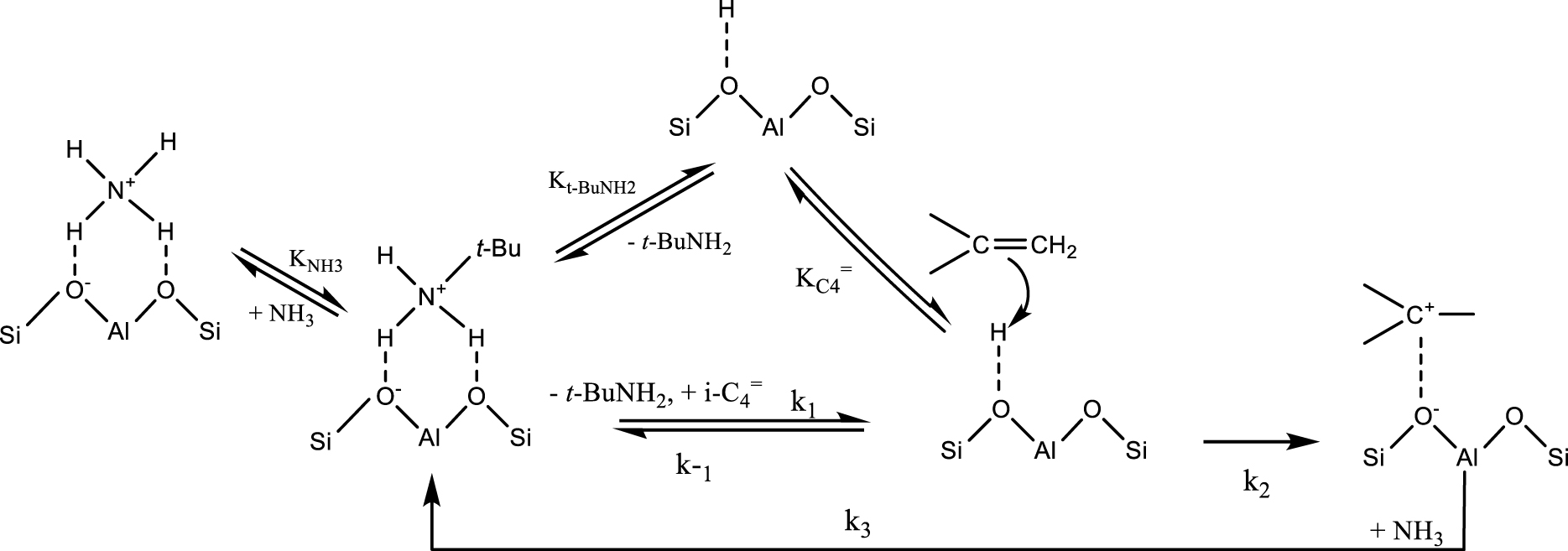
Mechanism of t-BuNH2 formation over MFI zeolite (adapted from Ref. [129]); k−1 represents the constant for t-BuNH2 desorption, k1 represents the constant for isobutene adsorption.
DFT simulations showed that at very low partial pressures, tert-butylamine desorption is rate limiting. At sufficiently high tert-butylamine partial pressures (>0.03 kPa), protonation of isobutene to the corresponding carbenium ion limits the rate of amination. Based on these results, a pseudo steady-state rate expression has been proposed:
7. Phenol tert-butylation
The catalytic tert-butylation of phenol with isobutene/tert-butyl alcohol is of great fundamental and industrial importance owing to the usage of alkylated products in the manufacture of resins, surface coating, printing inks, antioxidants, drugs, inhibitors, and agrochemicals [134]. This is a typical Friedel–Crafts alkylation catalyzed by both Lewis and Brönsted acids, in homogeneous or heterogeneous catalytic processes [135, 136, 137]. The development of solid acid catalysts is of major importance in the context of efficiency, safer and cleaner technologies. The use of solid catalysts, including ion-exchange resins [137], heteropoly acids [138], clays [139], mesoporous materials [140] and zeolites/zeo-types materials have been extensively investigated during the recent decades. For a review on this subject, see Ref. [136]. Due to their unique structure, thermal stability and reusability, zeolites and mesoporous aluminosilicates are the most promising heterogeneous acid catalysts. In line with the topic of this review, the reactions between phenol and isobutene/tert-butanol catalyzed by zeolites will be examined in this section.
The tert-butylation of phenol is a complex process. As shown in Scheme 8, various products, including tert-butyl phenyl ether (TBPE), ortho- and para-tert-butylphenol (2-, 4-TBP), 2,4-di-tert-butylphenol (2,4-DTBP), 2,6-di-tert-butylphenol (2,6-DTBP) and 2,4,6-tri-tert-butylphenol (2,4,6-TTBP) can be formed.
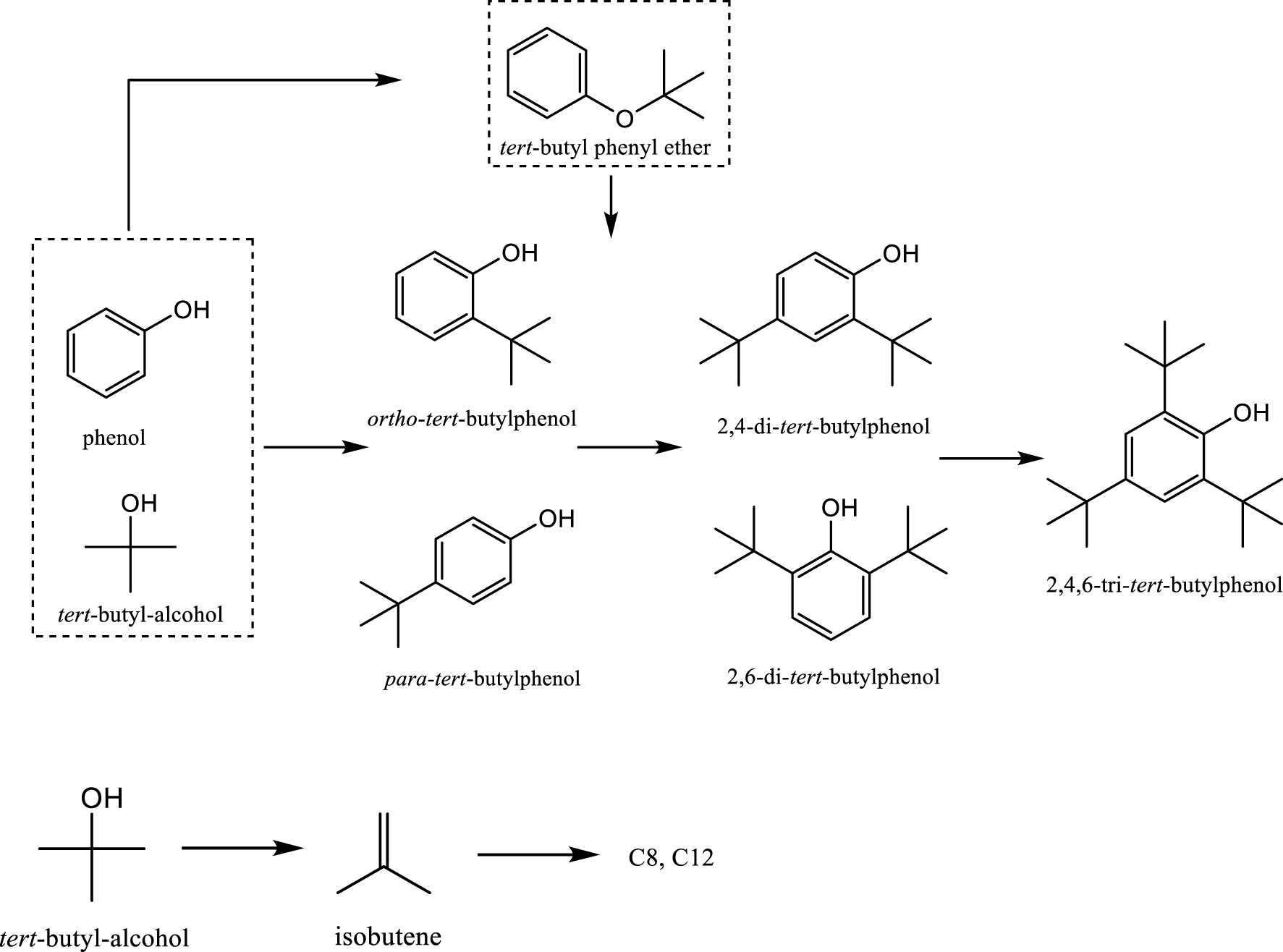
Reaction pathway for the tert-butylation of phenol with tert-butyl-alcohol over zeolites.
As will be shown below, the product selectivity can efficiently be tuned by choosing zeolites with suitable acidic and textural properties, as well as the reaction temperature and reaction time.
7.1. Effect of zeolite topology
Dumitriu and Hulea [141] studied the tert-butylation of phenol in liquid phase, at 70 °C, in the presence of H-form dealuminated zeolites with FAU, BEA and MOR topology. The three-dimensional interconnecting pore system of BEA and FAU zeolites showed higher catalytic activities than the monodimensional system of pores of the MOR zeolites. On the other hand, for each type of zeolite, the phenol conversion was higher when the Si/Al ratio was higher. As concerns the evolution in time of the product selectivity, as shown in Figure 1, in the early stages of the reaction the main product is TBPE and its concentration reaches a maximum after 1 h of reaction. After that, a decrease in the yield of TBPE and an increase in the yield of TBP are observed.
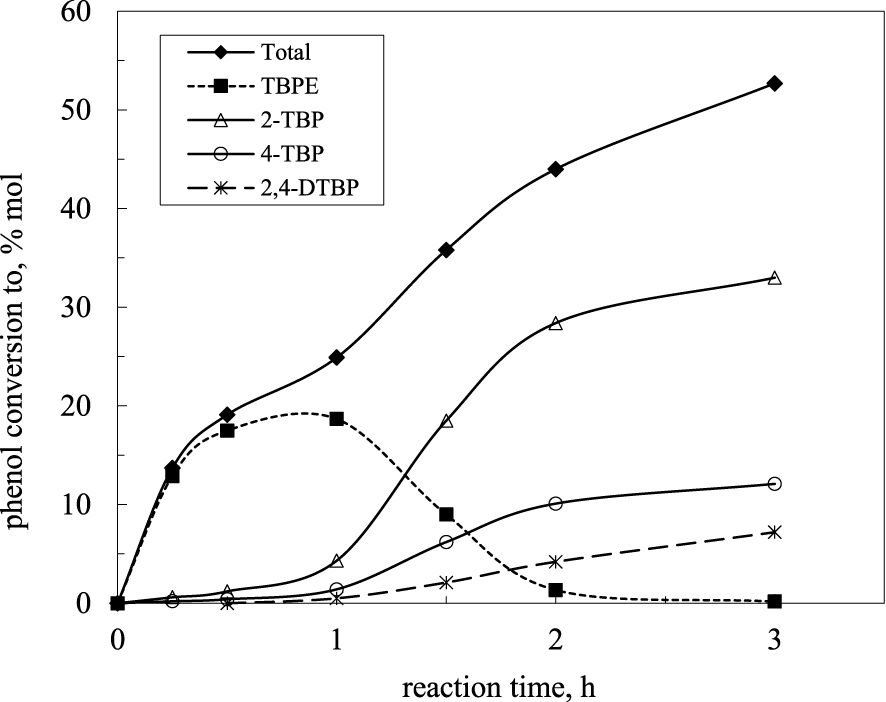
Phenol tert-butylation over USY catalyst: evolution of product selectivity [141].
Mono and di-C-alkylated phenols become the main products after 2 h of catalytic reaction. Such an evolution of the O- and C-alkylated products were observed for all zeolites tested in this study, but also in the presence of other zeolites, such as dealuminated MCM-68 [142] MCM-22, MCM-36 and ITQ [143]. Corma et al. [144] showed that over Y zeolite, at low temperature (30 °C), the major product obtained at this temperature was TBPE.
For the butylation reaction carried out in liquid phase, the distribution of the C-alkylated products strongly depended on the geometric constrictions of zeolite [141, 145]. Thus, the para isomer (4-TBP) was obtained over Beta zeolites, whereas the ortho isomer (2-TBP) was the main product over Y zeolites. A 1:1 ration between 4-TBP and 2-TBP was obtained in the presence of MOR zeolites. The formation of bulky di-tert-butyl phenols occurred mainly in the large cavities of Y zeolites.
When the reaction between phenol and TBA was performed in vapor phase, higher phenol conversion was obtained and the main products was 4-TBP, regardless the zeolite topology [146, 147, 148, 149, 150, 151]. Amount of 2,4-DTBP was also regularly obtained. In contrast, the TPBE was obtained only in small amounts.
Due to their small pores, ZSM-5 zeolite exhibited low activity for tert-butylation of phenol [152]. To increase the accessibility of reagents to the surface of the zeolite, some research groups have prepared zeolites with hierarchical porosity [153, 154, 155, 156, 157, 158]. Xu et al. [153] prepared a hierarchical ZSM-5 zeolite using a dual templates method. With this catalyst, the conversion of phenol was 81.2%, while it was only 16.4% on the conventional ZSM-5. Over hierarchical zeolite, the selectivity to 4-TBP and 2,4-DTBP was 52.2% and 40.6%, respectively. The high phenol conversion and 2,4-DTBP selectivity were attributed to the presence of the hierarchical porosity in this material. Xu et al. [154] have stated similar conclusions for a series of hierarchical ZSM-zeolites used as catalysts in the gas phase reaction between phenol and TBA.
Hsu et al. [155] prepared nanocrystals (40–50 nm) of ZSM-5 zeolite using a polymer as a template. After a thermal treatment, the interparticle spaces between the nanocrystals created mesopores and macropores with a pore volume of 1.12 cc/g and an external surface area of 478 m2/g. Tested in liquid phase tert-butylation of phenol with TBA, nanocrystalline zeolite ZSM-5 gave very high conversion of phenol (95%) and the main alkylated product was 2,4-DTBP. This particular behavior is due to the great accessibility of the reagents/products at the surface of the zeolite.
Chen et al. [156] prepared hierarchical ZSM-5 zeolite aggregates possessing open/accessible mesopores. The reaction between phenol and TBA has been used as probe reaction for this catalyst. Phenol conversion more than 30% was observed and the major products were 4-TBP and 2,4-DTBP.
7.2. Effect of zeolite acidity on catalytic selectivity
As shown above, the product distribution is strongly dependent on the catalyst porosity. However, some experimental studies have shown that there is also an effect of the acidity of the catalyst on the selectivity [159, 160, 161]. It has been shown that 4-TBP is produced mainly on moderate strength acid sites, whereas 2-TBP is obtained over weak acid sites. A strong acid sites also enhanced the selectivity to 2,4-DTBP. Based on their experimental results, Dumitriu and Hulea [141] stated that in the early stages of the reaction the O-alkylation occurred at a high rate on the acid sites, regardless of their acid strength.
7.3. Mechanism
Alkylation of phenol with both olefins and alcohols is an electrophilic aromatic substitution. It is widely accepted that over solid acid catalysts, with olefins as alkylating agents, the electrophile is a carbenium ion formed on a Brönsted acid site. In the case of alkylating phenol with alcohol, the electrophile can be the protonated alcohol (an alkoxonium species) or a carbenium ion derived from alcohol dehydration [162]. The electrophile may attack the phenolic OH or the aromatic ring, giving O-alkylation or C-alkylation products, respectively. It has also been suggested that C-alkylation products could be formed through intramolecular rearrangement of the kinetically favored O-alkylation product [163]. In the case of the butylation of phenol with TBA, this hypothesis was contradicted by the results obtained over different zeolite catalysts [141].
For tert-butylation of phenol with TBA over zeolites, two reaction routes have been suggested: the stepwise and the concerted mechanisms [164]. In the stepwise mechanism, in the first step the TBA molecule adsorbs on an acid site to form a tert-butyl carbenium ion by dehydration. This ion can either deprotonate to form isobutene, or react with co-adsorbed phenol, to form the tert-butylation product. In the concerted mechanism, the TBA molecule adsorbs on an acid site through an Oalcohol–Hzeolite hydrogen bond. In this state, TBA reacts with co-adsorbed phenol, to form the tert-butylation product.
Computational results revealed that tert-butylation of phenol preferentially occurs through a concerted path involving co-adsorption and reaction of tert-butyl alcohol and phenol without prior dehydration of tert-butyl alcohol, rather than through a stepwise path via dehydration to form a carbenium ion as the first step followed by tert-butyl cation attack on the 2- and 4-position on phenol [164].
However, Zhao et al. [162] have shown that the alkylation of phenol with cyclohexanol to decalin over Beta zeolite follows a stepwise mechanism. Using in situ 13C MAS NMR spectroscopy, they found that the alkylation occurs primarily via electrophilic attack of a cyclohexyl cation, not an alkoxonium ion, on the phenolic OH or π electrons in the aromatic ring. Cyclohexanol needs to be almost completely dehydrated before the rate of alkylation is measurable.
8. Conclusions and outlook
The processes examined in this review are based on acid catalyzed reactions involving highly reactive molecules. These reactions can therefore occur even at low temperatures, favoring the use of resins as main solid acid catalysts. As commercial catalysts, resins are inexpensive and well-performing catalysts. On the other hand, the reagents and/or the products are easily involved in side reactions, generating various by-products, which are responsible for the low selectivity of the process and the deactivation of the catalysts. In this context, the use of zeolites, as alternative catalysts for resins, provides notable advantages. They have an adjustable acidity and thus certain reactions can be favored or avoided. They limit the formation of large species, which cannot be accommodated in pores. Compared to resins, they are more thermally stable.
Zeolites with various topology have been tested in the reactions examined in this review. Among them, Y and especially beta zeolite, with large 3D pores seem to be the best catalysts in these processes. The large pores these zeolites are very beneficial for the free diffusion of the bulky species within the pore system, resulting in a lower deactivation rate and higher activity. ZSM-5 zeolite also has an exceptional ability to catalyze these reactions.
The research results analyzed in this review show that sustainable catalytic routes for converting isobutene into fuel additives and chemicals are realistic. Although promising progress has been obtained for each way, further research is still needed in order to produce the knowledge necessary to design the “ideal” catalysts and large-scale processes. Meanwhile, for shape selectivity purposes, medium pore zeolites will probably continue to play the most important role. However, I believe there may be new opportunities for zeolites with more than one type of pore in the same structure, which will offer new possibilities for isobutene transformation. Additionally, the encouraging results discussed above could create the basis for new research projects in which oil-based isobutene will be replaced by bio-isobutene. Indeed, it is important to note that in 2019 Global Bioenergies announced that runs using wheat straw were successfully performed in the Leuna demo plant, leading to the production of cellulosic isobutene. [https://fr.style.yahoo.com/global-bioenergies-first-production-isobutene-163800780.html?]
Conflicts of interest
Authors have no conflict of interest to declare.
Vous devez vous connecter pour continuer.
S'authentifier