


 CC-BY 4.0
CC-BY 4.0
1. Introduction
According to the European Commission, the definition of a nanomaterial is the following: “A natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1 nm–100 nm. In specific cases and where warranted by concerns for the environment, health, safety or competitiveness the number size distribution threshold of 50% may be replaced by a threshold between 1% to 50%” [1]. Regarding heterogeneous catalysis, numerous studies have been conducted on nanomaterials serving as catalysts (see for example references [2, 3, 4, 5, 6, 7, 8]). Such catalysts are used [9, 10, 11, 12, 13] for different major chemical processes encompassing Fischer–Tropsch [14, 15, 16, 17, 18, 19, 20], reforming [21, 22, 23] or CO2 methanation [24] as well as for the transformation of bio-renewable substrates and reactions related to the realization of a bio-refinery [25]. Among above cited works, the publications of Dumitriu play a major role [26, 27, 28, 29, 30]. In this contribution dedicated to heterogeneous catalysis, we will present an interplay between the adsorption process of simple molecules and the behaviour of nanometre scale particles [31, 32, 33].
2. Defining a model catalyst
As underlined by Guillen-Hurtado et al. [34], pollution removal in gasoline vehicles is successfully accomplished at high temperature with a Three Way Catalyst (TWC) which catalyses the reactions between oxidizing (O2 and NOx) and reducing (CO and unburned hydrocarbons) species in the exhaust. Most current TWC consist of a ceramic monolithic honeycomb where the different active phases are loaded. These active phases are mainly composed of noble metals, cerium-based oxides and alumina [35]. Several chemical reactions can hence take place on industrial catalysts [36, 37, 38, 39, 40]. Investigations of commercial catalysts [41, 42, 43] is quite difficult even with characterization techniques specific to synchrotron radiation such as X-ray absorption spectroscopy (XAS) [42, 43, 44, 45, 46, 47] able to bring structural information on low ordered materials are used [48].
Following an approach defined and discussed by Boudart: “model catalysts: reductionism for understanding”, a possible first step consists of focusing on the main molecule which is related to the chemical process [49]. In the case of DeNox catalysis (NOx reduction to nitrogen molecule) [50, 51, 52, 53], one may consider the interaction between one molecule i.e. nitrous oxide (NO) and nanometre scale mono metallic particles deposited on one light oxide such alumina. From an experimental point of view, the starting point of our investigation is given by a set of experimental data on the adsorption of NO on nanometre scale clusters of Pt [54, 55, 56] and Rh [57, 58] deposited on different supports. Regarding Pt, Schneider et al. [54] have investigated by XAS [44, 45, 46, 47] a catalyst prepared by impregnation of neutral γ-alumina with a solution of H2PtCl6, 6H2O. These authors noticed that when the catalyst is pre-reduced in situ before the introduction of NO, sintering of particles is considerable above 200 °C. In the study of Loof et al. [55], the catalysts were prepared by coating a monolithic support of cordierite with γ-alumina previously ground in a ball mill together with an aqueous solution of H2PtCl6. They then observed a rapid sintering in NO of nanometre-sized Pt particles by CO temperature-programmed desorption and transmission electron microscopy. Finally, Wang et al. [56] have prepared Pt catalysts by ion exchange of Pt(II) tetraamine hydroxide [Pt(NH3)4(OH)2] onto SiO2 and their results show that nanometre-sized Pt clusters on SiO2 sinter during heating in 1% NO/He at 300 °C. Regarding Rh, Evans et al. [57, 58] have selected a 4 wt% Rh∕γ-Al2O3 sample reduced in situ in a flow microreactor to 573 K under 4% H2∕He. The XAS data obtained from their experiments is consistent with the predominant formation of an oxidic Rh phase. Considering the complete set of results, it was quite surprising to observe that if we consider the adsorption of NO on nanometre scale metallic clusters at temperatures between 300 °C and 500 °C, for Pt a sintering process was measured while for Rh, an oxidation process was clearly pointed out.
3. Surface science to understand heterogenous catalysis
An elegant way to understand in detail the results of these experiments is given by the numerous investigations which have been dedicated to the adsorption of nitrous oxide on metallic surfaces [59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69]. The literature indicates that the ability of transition metal surfaces to dissociate simple molecules (N2, O2, NO, etc.) decreases across the transition series [59, 63]. More precisely, a review of the literature distinguishes two families of metals (Figure 1) following the adsorption mode which can be either dissociative or molecular [59, 63].
The melting points of the transition metals, and rare earths, versus atomic number. On this diagram we also show a suggested correlation between the propensity for a metal to dissociate NO, and its melting point (according to Brown and King [63]).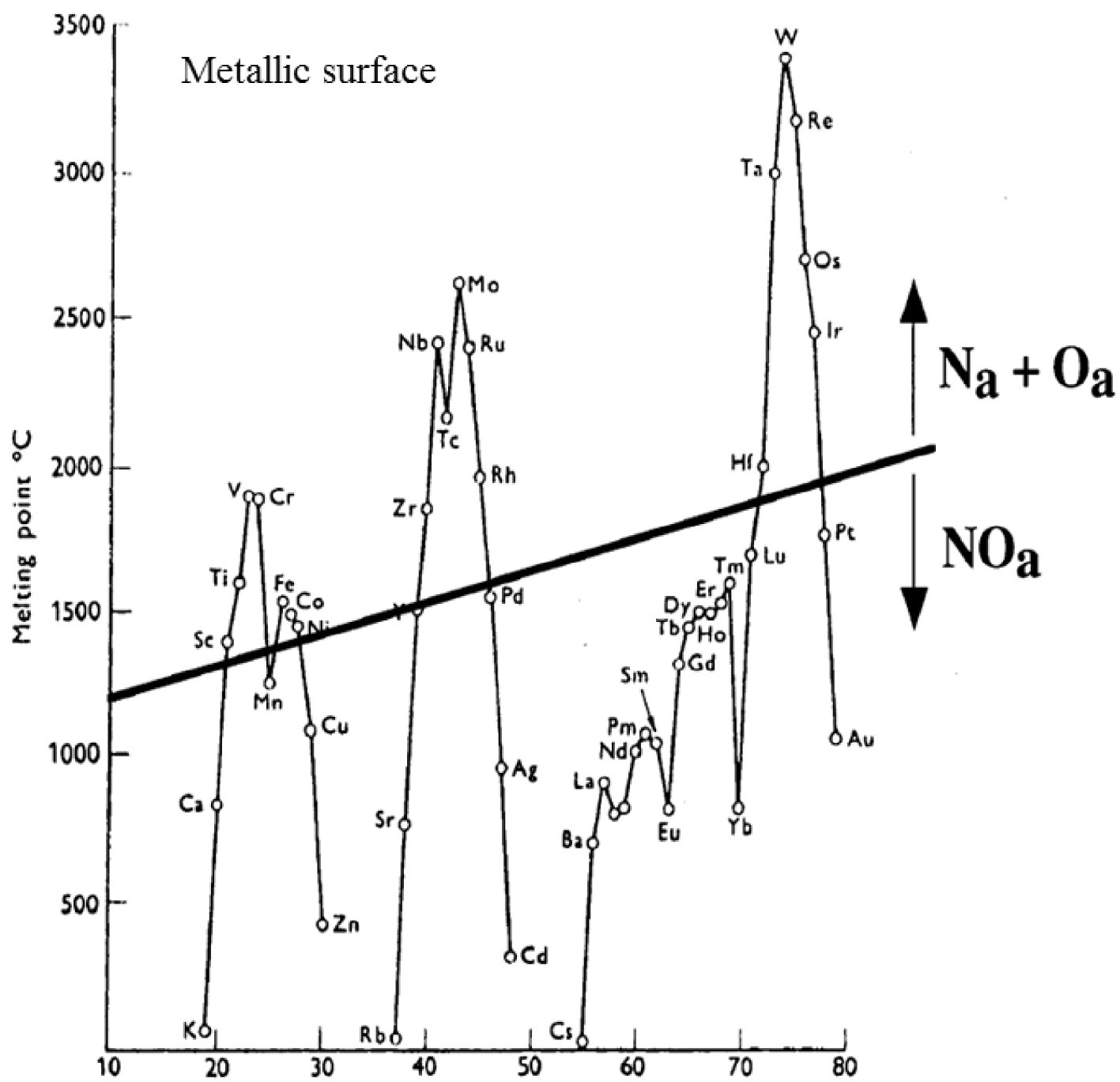
Note that the melting points are a direct measure of the cohesive energies of the elements. The higher the metal cohesive energy, the greater is the propensity for NO dissociation. It is worth to keep in minds that the adsorption process even of small molecules is a complex process. Different NO adsorption geometries are observed, metallic surfaces may undergo reconstructions, and different factors such as the coverage as well as the temperature play a key role [59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69].
Cohesion energy of the nanometre scale metallic cluster for different morphologies versus the number of atoms inside the cluster (from Refs. [70, 71]).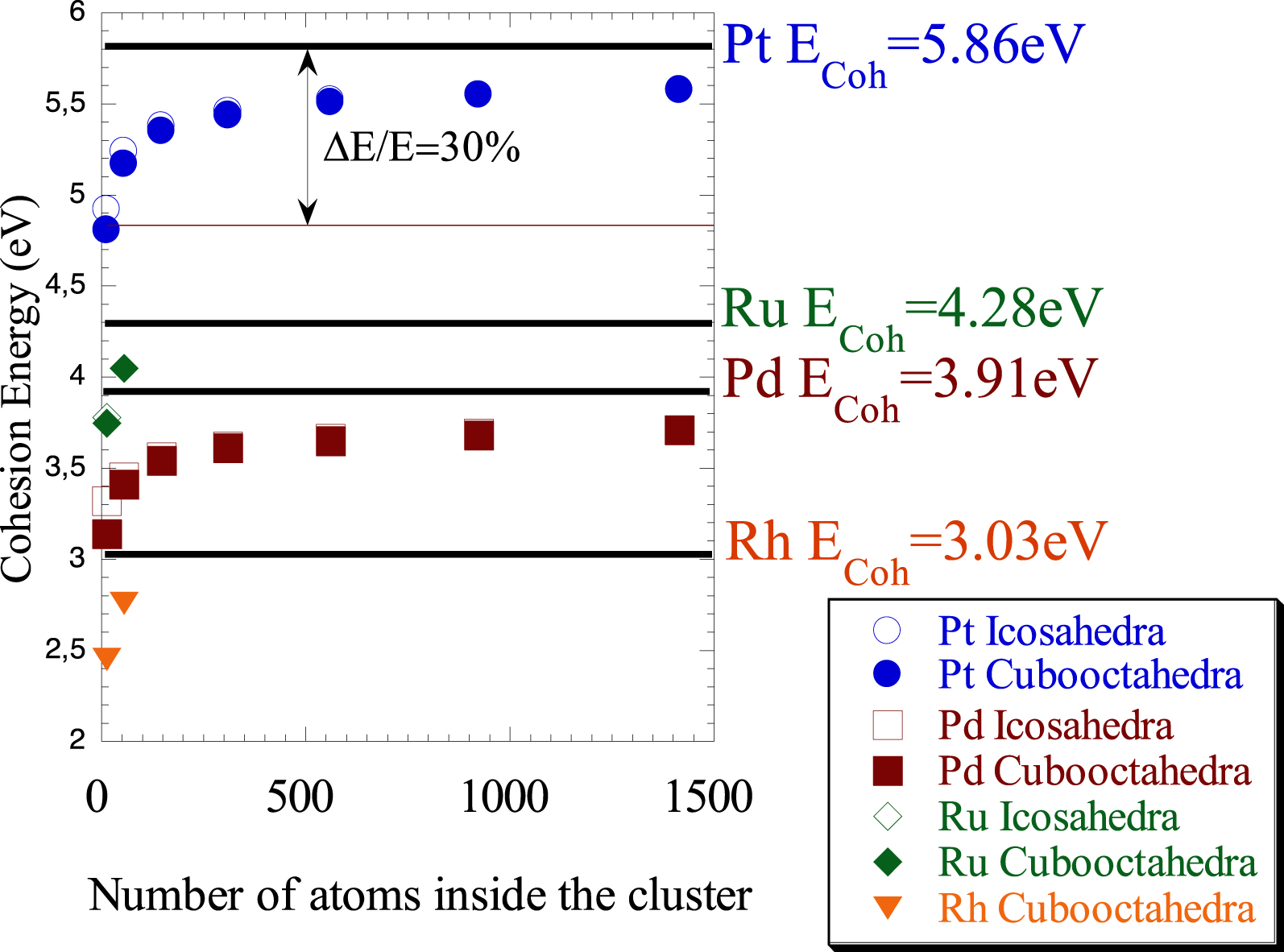
4. From surface science to nanoscience
To move from surface science to nanoscience, one simple question arises. How does the Brown and King diagram changes when we consider nanometre scale metallic cluster (instead of metallic surface)? The answer is given in different investigations of the electronic properties of nanometre scale metallic particles [70, 71]. As it can be seen on Figure 2, a significant decrease of the cohesive energy, around 30%, is observed independently of the nature of the metal and the morphology of the cluster.
The projection of this experimental finding in nanoscience (Figure 3) leads to the conclusion that both families of metals and their associated NO adsorption mode, which can be either dissociative or molecular (Figure 1), can be used for nanoscale metal particles. In that case, using experimental data obtained on different metals [31, 32, 33], one may establish a relationship between the adsorption mode of NO and the behaviour of nanometre scale metallic particle. More precisely, an oxidation process of the metallic cluster corresponds to a dissociation mode of NO while a sintering process is related to a molecular adsorption (Figure 3). From a catalytic point of view, the line which separates the two groups defines an ideal catalyst (no sintering and no disruption), its structural characteristics being stable.
Cohesion energy of 3d, 4d and 5d nanometre scale metallic clusters. Here, all the metallic clusters of these metals have the same size and the temperature is the same.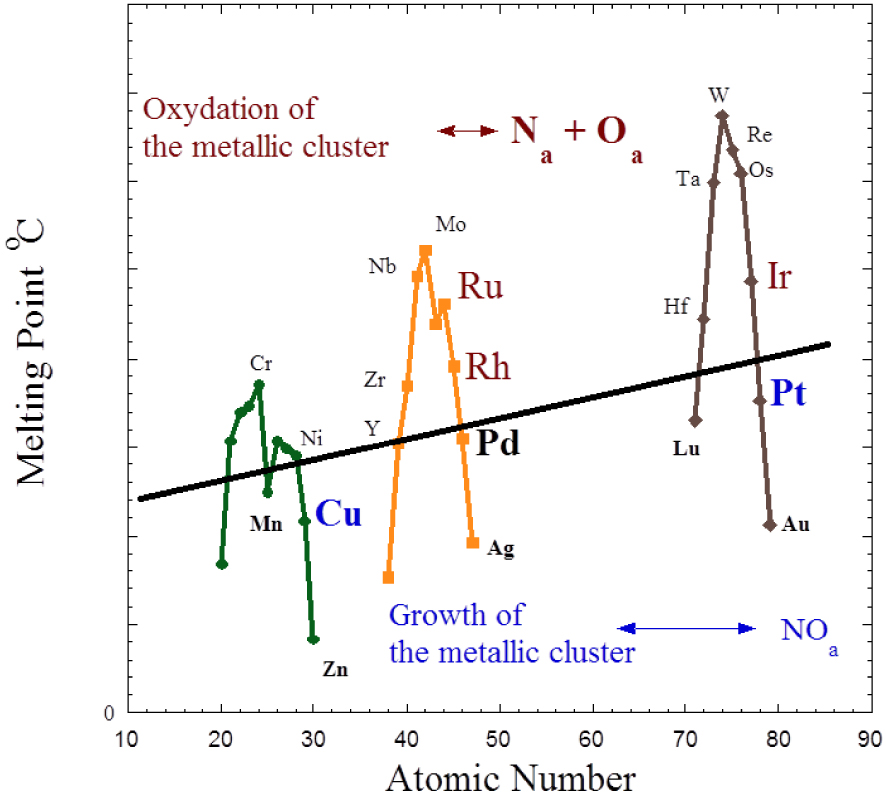
5. Crucial contribution from density functional theory
Density functional theory (DFT) [72, 73, 74] constitutes an approach which has led to several significant breakthroughs on the adsorption of small molecules on metallic surface [75, 76, 77] as well as on nanometer scale metallic clusters [78, 79, 80]. The important role of DFT in catalysis and materials science is today undeniable [73, 74]. Today one can say that DFT is a reliable tool to model nanometer scale metallic clusters and other catalytic systems.
Different 13 atom-sized transition metal (Pt, Ir, Ru, Rh, Cu, Pd and Ni) clusters have been selected to elucidate the fundamental adsorptive properties and interactions of NO on nanometre scale monometallic catalysts [81]. The complete set of simulations is in line with the initial model based on experimental data. Indeed, the dissociation of NO on Ru, Ir, Rh, and Ni, which lead to the formation of an oxide, was recovered. At the opposite, the DFT calculations indicate no NO dissociation for Pt, Cu, and Pd.
Similarly, using density functional theory, Takagi et al. [82, 83] have calculated the activation energies of NO dissociation reactions on several M clusters (M = Cu, Fe, Co or Ni) containing 55 atoms. These authors reported that Fe55, Co55 and Ni55 are reactive toward NO dissociation in agreement with experimental findings, whereby the reactivity followed following order Fe55 > Co55 > Ni55. Also, NO dissociation was most favoured on small Rh clusters [84, 85]. It seems thus that such relationship between the adsorption mode of NO and the behaviour of the nanometre scale metallic cluster is valid.
5.1. Is this simple model valid for similar molecules?
A simple question arrives. Can we expect to use such relationship for other molecules? One more time, we can start from surface science. When a diatomic molecule such as NO approaches a metal surface [86], it may encounter three kinds of potential wells corresponding respectively to a physisorbed state (far from the surface), a molecular chemisorbed state and finally to a dissociative chemisorption state. Dissociative chemisorption is the most stable situation if
This is a general tendency and the evolution of atomic adsorption energy versus the number of electrons in the d band is almost the same for the elements N, O, and also C. We can note that the frontier between nondissociative and dissociative adsorption can be given roughly by the dissociation energy of the molecule of interest. For example, regarding the 3d transition elements, the frontier is between Fe and Co for N2 and between Ni and Cu for NO, a tendency in line with the different values of the dissociation energy of these two molecules (N2 = 9.76 eV, NO = 6.50 eV). Regarding the adsorption of O2 on nanometre scale metallic clusters, in situ XAS experiments shows that even Pt clusters are not stable versus the adsorption of O2 [87]. At this dimension, it seems that there are no “noble” metals, an observation in line with the low value of the dissociation energy of this molecule (O2 = 5.12 eV).
5.2. Is it possible to use this simple model to predict some simple chemical reactions on nanometre scale monometallic cluster?
Another point which can be discussed is related to simple chemical reaction on nanometre scale metallic cluster. Again, we can start from the fact that NO reactions on surfaces can also exhibit oscillatory behaviour, particularly in the case of reactions on metals such as Rh and Pt. As underlined by Brown and King [63], oscillatory reactions on metallic surfaces such as the reactions of NO and CO, NO and H2 and NO and NH3, have been reviewed by Imbihl [88], by Nieuwenhuys and co-workers [89], and by Gruyters and King [90].
At least we can use this simple model to predict the behaviour of the metal nanoparticles when a mixture of NO + O2 is considered. For Rh, on which NO molecules undergo dissociative adsorption with the formation of metal–oxygen bond formation, it is possible that the presence of oxygen will not change significantly this simple scheme. Such behaviour is expected for other metals such as Ru or Rh. In contrast, for Pt the situation is more complex because the temperature plays a significant role.
Regarding experiments, Schneider et al. [87] have concentrated their investigation, on the one hand, on the influence of NO, C3H6 in the presence or not of an excess of oxygen and, on the other hand, on the influence of the total mixture. The most striking result is the growth of the platinum particles under NO. Moreover, when the catalyst is submitted either to a mixture of NO + O2 or C3H6 + O2, the particles are not fully oxidised. With the total mixture, the particles both grow and are oxidised. Note that the relative concentrations of the two gases, as well as the temperature, are probably significant parameters.
To a first approximation, we can conclude that for metals above the straight line, namely Rh, Ru or Ir, it is necessary to add to a NO + O2 mixture, a reducing agent in order to re-establish the metallic state of the atoms. This is not the case for metals below the line (Pt,Cu). In this case, we can probably adjust the relative concentration of the two gases NO and O2 to keep a metallic state of the Pt (or Cu) atoms.
Following this approach, we can consider also another simple reaction namely NO + H2. For metals above the straight line, namely Rh, Ru or Ir, we may observe oscillatory behaviours between oxidation of the metallic clusters induced by NO dissociative adsorption and reduction of the metallic clusters induced by H2 dissociative adsorption. This is not the case for metals below the line (Pt,Cu) for which we will observe a growth of the metallic cluster.
5.3. Is it possible to use this simple model to predict some simple chemical reactions on nanometre scale bimetallic clusters?
Is it possible to consider bimetallic catalysts? This purely energetic model leads to a complete rejection of some bimetallic systems [31, 32, 33, 91]. For example, if we consider a RhRu bimetallic cluster (or a RhIr as well as RuIr bimetallic), the NO adsorption process leads probably to the formation of a metal oxide i.e. the dissociation of NO will stop. On the other hand, if we consider a PtCu system, it is probable that the NO adsorption will lead to large clusters.
A guideline for the choice of the bimetallic system is to add to platinum a second metal like Rh or Ru. If we consider the CeO2 support, the PtPd bimetallic seems to be acceptable while the PtPd system supported on alumina has to be rejected.
5.4. Some perspectives
It is possible that other major industrial processes in which nanometer scale clusters are involved namely hydrogen evolution reaction [92] or lithium–oxygen batteries [93] can take advantage of this simple model. For example, it is well known that the electron population of CO increases steadily from Ni to Ti [94, 95, 96]. Although interesting and systematic results have been gathered and are partly explained, this adsorption process still remains a challenge [96].
Also, nanometre scale metallic clusters constitute materials of interest in the case of lithium–oxygen batteries. Recently, Lu et al. [93] report the deposition of subnanometre silver clusters of exact size and number of atoms on passivated carbon to study the discharge process in lithium–oxygen cells. Note that DFT is an efficient technique to investigate the effects of LiO and Li2O adsorptions on small bimetallic nanoparticles [97].
6. Conclusion
In this small review dedicated to DeNox catalysis, we combined experimental data and recent DFT results to show the efficiency of such approach which seems to be already adopted by several research teams. Work is in progress.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgment
FDP and FT wishes to acknowledge the VUB for support, among other through a Strategic Research Program awarded to their group.