


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Oligo-α-pyridylamido anions have been extensively used as modular polynucleating ligands in coordination chemistry. Their all-syn conformation provides an array of N donors suitable for assembling wire-like structures of interest in molecular electronics [1, 2] and magnetism [3], and known as extended metal atom chains (EMACs) [4, 5]. Depending on the number of linked α-pyridylamido units and on the presence of additional coordinating groups at the ligand’s termini, structurally authenticated EMACs range from tri- to undecanuclear [2]. Among parent amines, widely used are di(pyridin-2-yl)amine (Hdpa) [6, 7, 8, 9] and N2,N6-di(pyridin-2-yl)pyridine-2,6-diamine (H2tpda) [10], whose mono- and dianions act as tri- and pentanucleating ligands, respectively (Figure 1).
The synthesis of oligo-α-pyridylamines and related proligands mostly relies on Pd-catalyzed amination of 2-halopyridines (the so-called Buchwald–Hartwig reaction) [11, 12] which possesses great advantages, like mild reaction conditions and high yields [2, 13, 14, 15, 16, 17, 18, 19, 20]. However, beside requiring an expensive and environment-unfriendly catalyst, it can result in inefficient cross-couplings when N-containing heterocycles are involved [21].
Structures of Hdpa, H2tpda, H3py3tren, and complexes [M1M2Cl(py3tren)] (Mi = Fe2+, Co2+, Mn2+) [22].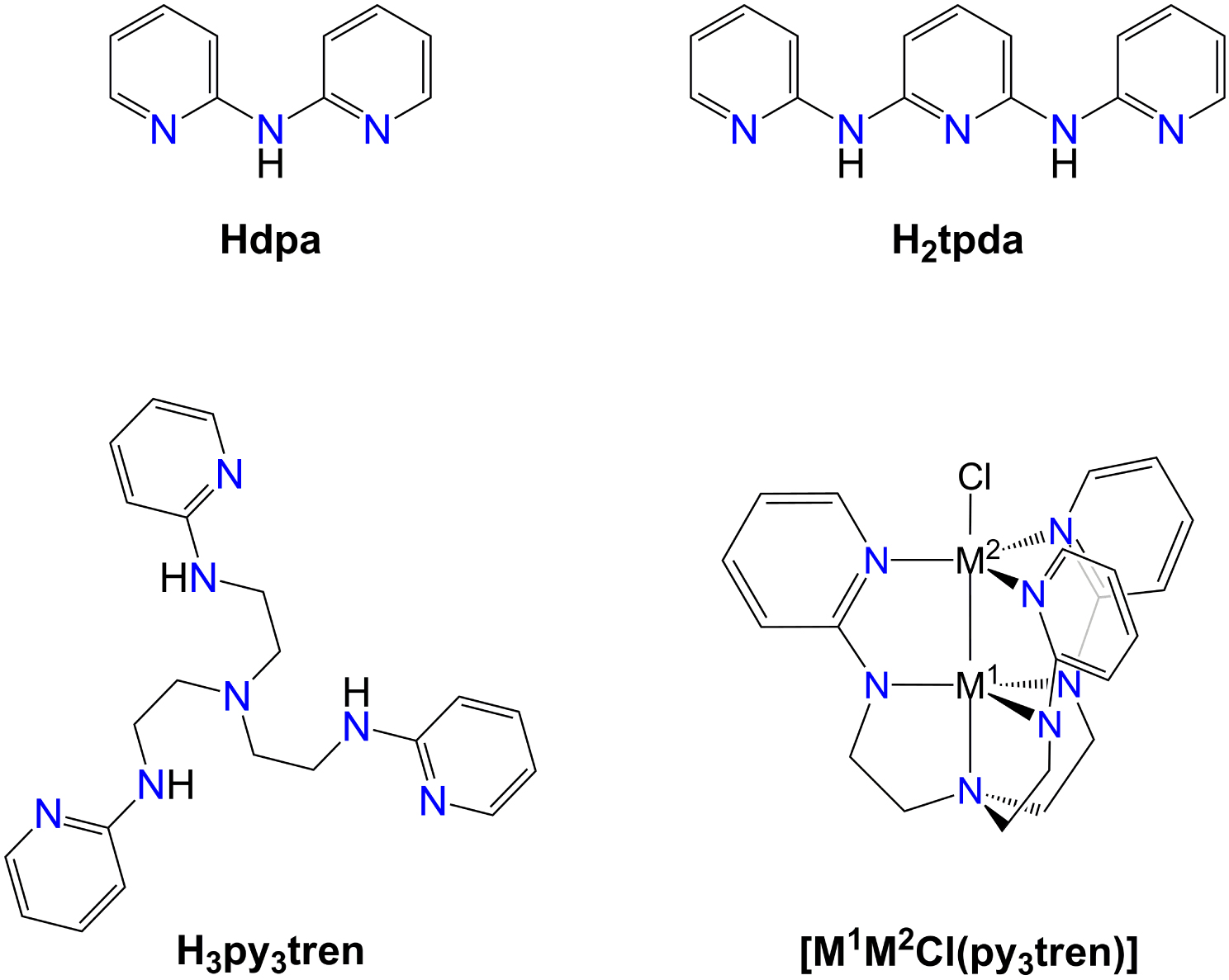
Synthesis and isolated yields of H2tpda (a) [23], HBrdpa (b), and HFdpa (c, d).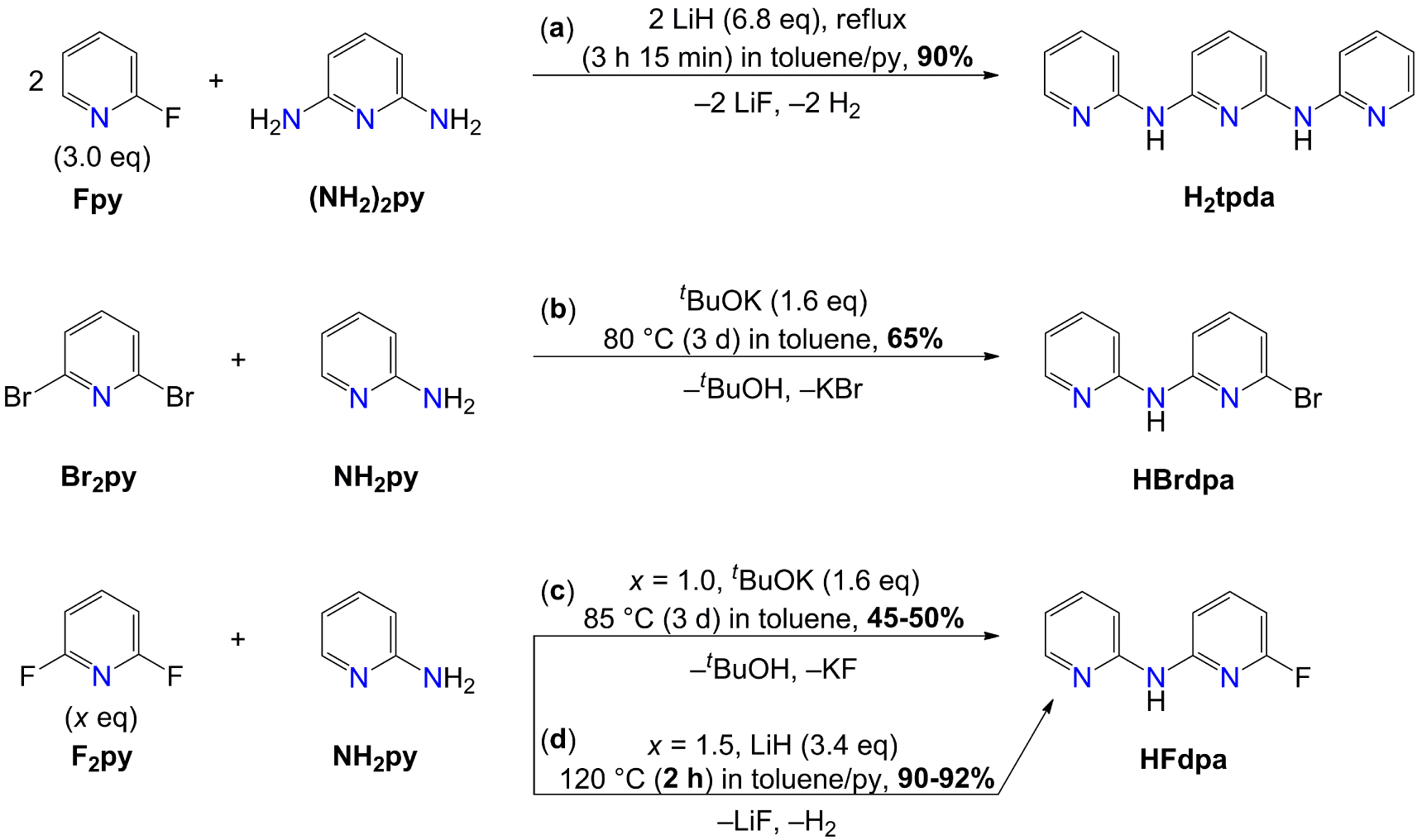
This procedure is far more economical and environment-friendly than Buchwald–Hartwig reaction and can potentially provide access to a variety of oligo-α-pyridylamines and their derivatives.
We herein demonstrate its applicability to the high-yield synthesis of 6-fluoro-N-(pyridin-2-yl)pyridin-2-amine (HFdpa in Scheme 2), a new fluorinated derivative of Hdpa prepared from 2-aminopyridine (NH2py) and 2,6-difluoropyridine (F2py). We then show that HFdpa is an excellent building block for designing more complex proligand architectures, like the tridecadentate tripod H6tren(dpa)3 depicted in Scheme 2. H6tren(dpa)3 is a triply-N-arylated derivative of tris(2-aminoethyl)amine (H6tren, Scheme 2) and contains three Hdpa-like terminations with four different types of nitrogen donors from trialkylamino, alkylarylamino, diarylamino, and pyridyl groups. Simpler, structurally related proligands were used to promote short metal–metal distances in homo- or heterodimetallic complexes, sometimes resulting in unusually high-spin ground states [30, 22]. For instance, H3py3tren in Figure 1 was used to assemble complexes [M1M2Cl(py3tren)] (Mi = Fe2+, Co2+, Mn2+) with M–M distances ranging from 2.287 to 2.531 Å (Figure 1) [22].
Structures of reactants (H6tren, HBrdpa or HFdpa) used in the synthesis of H6tren(dpa)3 (the extra α-pyridylamino units of H6tren(dpa)3 as compared with H3py3tren are highlighted in red). SPa and SPb are potential side products.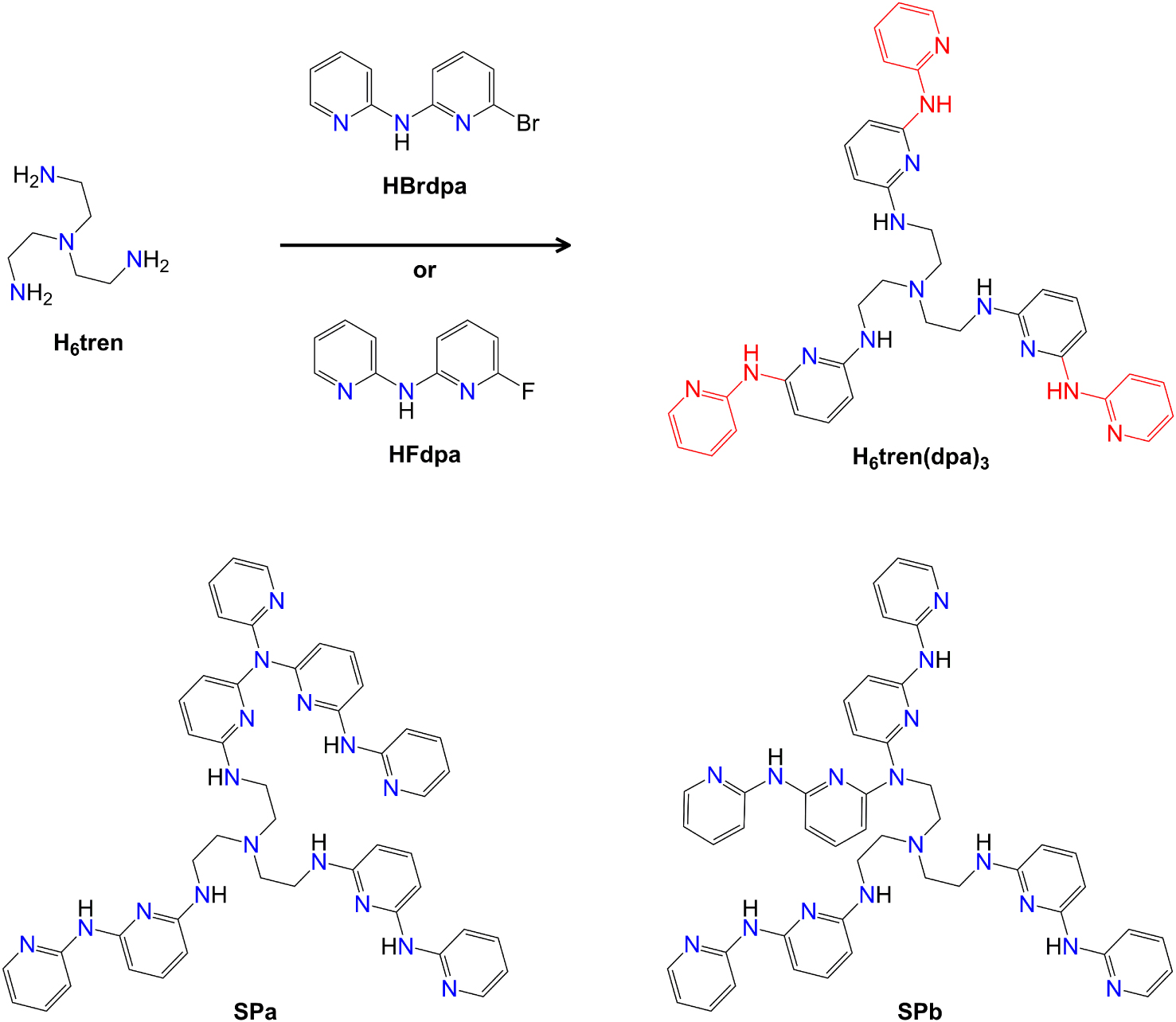
H6tren(dpa)3 contains one more α-pyridylamino unit per branch and can thus be regarded as a higher homologue of H3py3tren. However, its synthesis requires a careful control of reactants and reaction conditions to prevent overarylation of its three additional diarylamino groups.
2. Results and discussion
Inspirational to our work were literature methods for the synthesis of H3py3tren, which are summarized in Scheme S1 (Electronic Supporting Information). They are based on the prolonged heating of H6tren with 2-bromopyridine (Brpy) [22] or Fpy [31, 32] and excess inorganic base (K2CO3 [22, 31] or Cs2CO3 [32]) under inert atmosphere, so as to promote three consecutive arylations of H6tren primary amino groups. The reported procedures involve the use of DMSO [22] or acetonitrile [32] as solvents, or even solventless conditions [31] which facilitate the work-up step. The highest isolated yield (86%) was obtained using Fpy, K2CO3, and solventless conditions [31], presumably as a consequence of the greater electrophilicity of fluorinated versus brominated pyridines, hence the easier formation of an F-containing σ complex [23].
Extension of the above procedure to the synthesis of H6tren(dpa)3 was tested using two halogenated derivatives of Hdpa as electrophilic reagents: 6-bromo-N-(pyridin-2-yl)pyridin-2-amine (HBrdpa) [33, 34, 35, 36] and HFdpa (Scheme 2). K2CO3 and Cs2CO3 were examined as bases, in both organic solvent-based and solventless conditions.
2.1. HBrdpa and HFdpa
A Pd-catalyzed reaction was reported to give HBrdpa in 86% yield [35], but transition-metal-free procedures were also described. In 2011 Bolliger et al. prepared HBrdpa by reacting 2,6-dibromopyridine (Br2py), NH2py, and KN(SiMe3)2 (1:1.1:1.5 MR; MR = molar ratio) at 100 °C in 1,4-dioxane for 15 min (87% isolated yield) [36]. In 2014 Deng et al. reported that Br2py, NH2py, and tBuOK (1:1:1.6 MR), heated to 80 °C in benzene for three days, also afford the desired product in 65% isolated yield [34]. We tested this last procedure replacing benzene with toluene (Scheme 1b) and obtained an identical yield to that reported in Ref. [34], after purification by silica-gel flash chromatography (FC). The 1H NMR spectrum of HBrdpa is shown in Figure S1.
HFdpa was here synthesized exploiting two different transition-metal-free approaches, which are compared in Scheme 1c,d. The first route (Scheme 1c) is similar to that in Scheme 1b and consists in heating NH2py, F2py, and tBuOK (1:1:1.6 MR) to 85 °C in toluene for three days. Surprisingly, the reaction proved to be significantly less efficient than with Br2py (∼45–50% isolated yield), despite the expectedly higher electrophilicity of fluorinated versus brominated pyridines [22, 31, 32]. The use of a larger excess of tBuOK (2.2 equiv.) did not improve the yield. Therefore, a different approach was attempted (Scheme 1d), whereupon NH2py, F2py, and LiH were heated in toluene/py in a 1:1.5:3.4 MR to exploit C–F bond activation by Li ions [23, 29]. Virtually full conversion of NH2py into HFdpa was observed after only 2 h of reaction time. The work-up step involves removal of excess F2py and NH2py residuals by vacuum treatment and extensive washings with water, respectively, and affords spectroscopically and analytically pure HFdpa in excellent yield (90–92%). The 1H and 13C NMR spectra of HFdpa in CD3CN are shown in Figure 2. The assignment of proton and carbon chemical shifts (𝛿) was based on a detailed 1D and 2D NMR characterization, cross-checking the data from the following NMR experiments: 1H, 13C, 1H–1H Correlated Spectroscopy (COSY, Figure S2), 1H–13C Heteronuclear Single Quantum Coherence (HSQC, Figure S3), and 1H–13C Heteronuclear Multiple-Bond Correlation (HMBC, Figure S4).
Top: atom-labelled structure of HFdpa and its 1H NMR spectrum in CD3CN (298 K, 600.13 MHz); 𝛿H (ppm) = 1.94 (quintet, residual protons in CD3CN), 2.19 (s, water, OH). Bottom: 13C NMR spectrum of HFdpa in CD3CN (298 K, 150.90 MHz); 𝛿C (ppm) = 1.32 (septet, CD3CN), 118.32 (s, CD3 CN). Processing parameters (TopSpin 4.0.6 [37]): SI = TD, LB = 0.30 and 2.00 Hz for 1H and 13C NMR spectra, respectively. The hidden spectral regions contain no signals, except for: 𝛿H = 5.44 ppm (s, dichloromethane, CH2).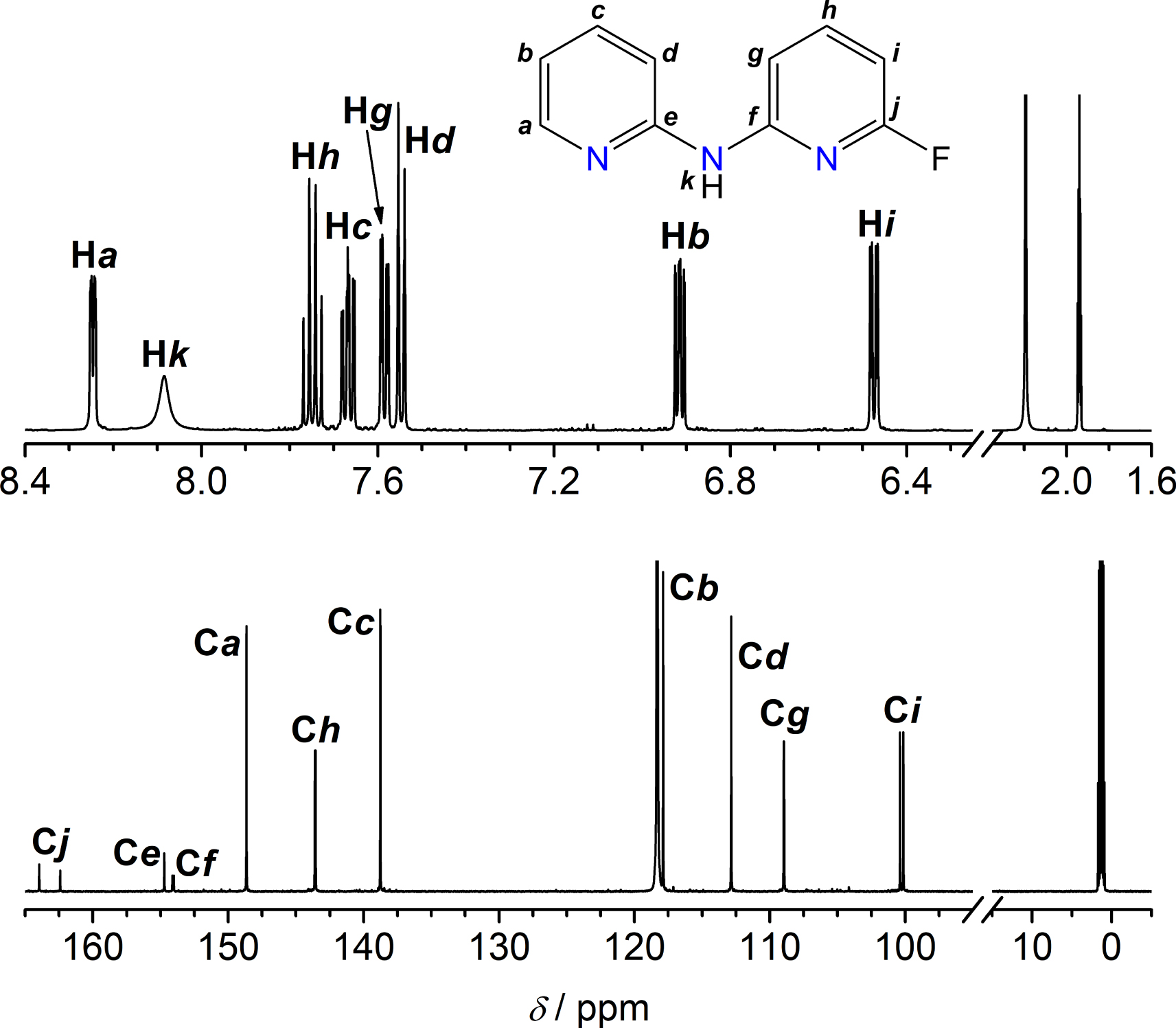
The 1H NMR spectrum contains eight well-resolved signals, with identical integrated intensities. Proton signals from the non-halogenated ring (Ha, Hb, Hc, and Hd) possess the same hyperfine pattern and similar J-couplings as in HBrdpa (see Figure S1). On the other hand, protons of the fluorinated pyridine ring (Hg, Hh, Hi) show additionally split resonances due to the heteronuclear 1H–19F coupling, with J constants consistent with those observed in Fpy [38]. The 13C NMR peaks from Cf, Cg, Ch, Ci, and Cj are also split into doublets by the 13C–19F coupling (only Cj–F and Ci–F splittings are clearly visible at the bottom of Figure 2; all 13C–19F couplings are listed in Experimental Section 4.2.1). Such heteronuclear couplings are diagnostic, and they were crucial for assigning the 𝛿C of these five C atoms. The HMBC spectrum (Figure S4) clearly highlights the presence of 1H–19F and 13Cj–19F couplings. In fact, the (Hg, Cj), (Hh, Cj), and (Hi, Cj) cross-peaks are split along the y axis (F1) due to 13Cj–19F coupling. In addition, the two peaks of each 2D doublet are shifted along the x axis (F2) due to the 1H–19F coupling.
2.2. H6tren(dpa)3
Scheme 3 summarizes all synthetic pathways explored for the preparation of the new tripodal proligand. In all these cases, a slight excess of halo-derivative (3.2 equiv.) was used.
Synthetic routes to H6tren(dpa)3, starting from HBrdpa (a, b, c, d, d′) or from HFdpa (e, f). DMAP is 4-(dimethylamino)pyridine. Isolated yields and MRs of H6tren(dpa)3 and SPa were determined by 1H NMR spectroscopy after purification. All reactions were carried out under N2 atmosphere.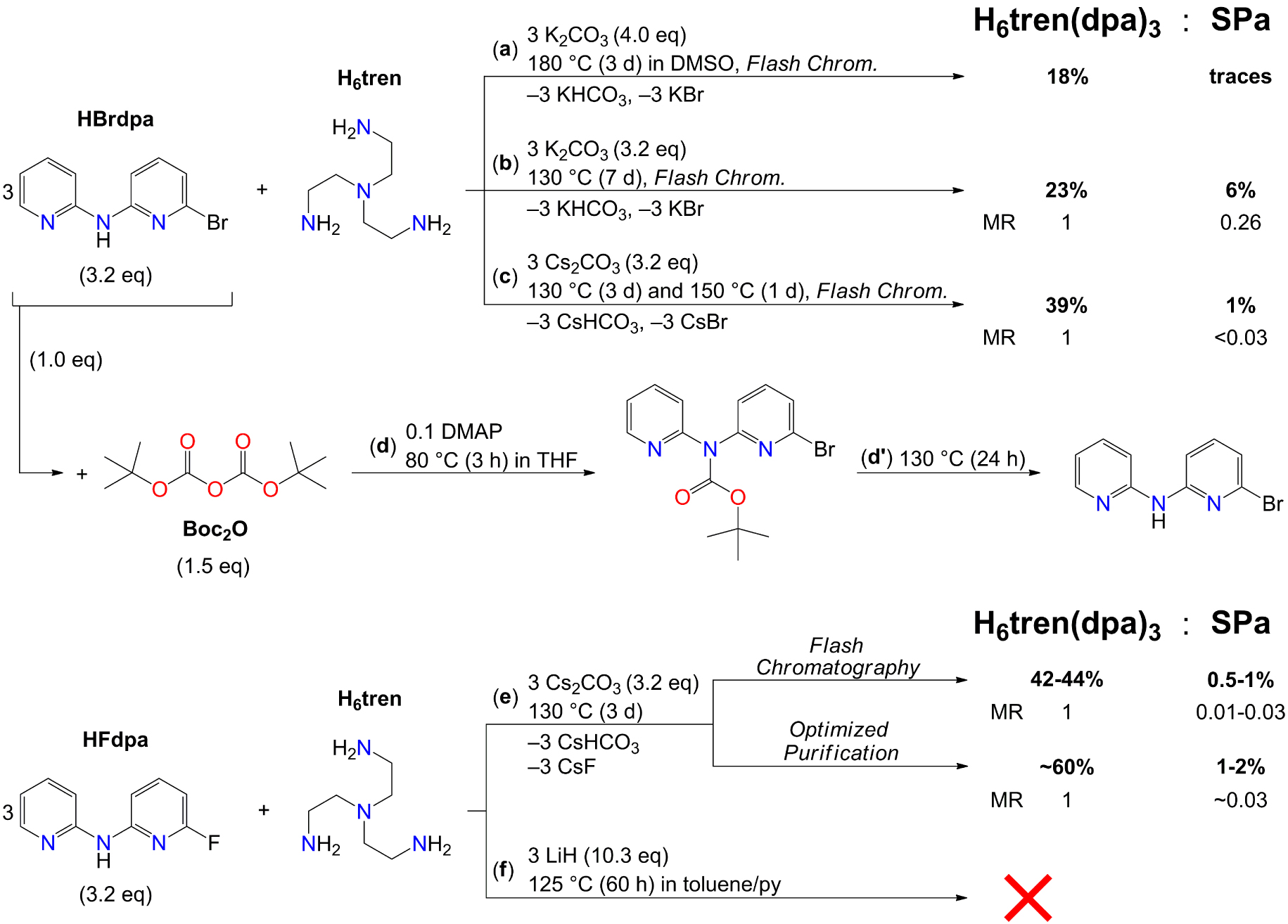
H6tren(dpa)3 proved to be synthetically accessible in substantial yields under similar, but not identical, conditions to those used to prepare H3py3tren [22, 31, 32], as shown in Scheme 3a–c,e. Products were isolated by adding CH2Cl2 to the reaction mixture, washing the solution with saturated aqueous NaHCO3 and then with water, drying it over MgSO4, and evaporating the solvent under reduced pressure. The dark orange solid so obtained was finally purified by gradient FC with Et2O:EtOH (or by alternative methods) to remove unreacted HFdpa and intermediate products, such as bipodal H6tren(dpa)2 and monopodal H6tren(dpa).
Prolonged heating of H6tren, HBrdpa, and K2CO3 (4.0 equiv.) at 180 °C in DMSO, following the procedure in Ref. [22] (Scheme 3a), gave high conversion (98%) but with extensive charring of the reaction mixture and very poor isolated yield (∼18%). Charring was much reduced working in solventless conditions and at lower temperature (130 °C), where K2CO3 (3.2 equiv.) promoted significant conversion (74%) [31]. Notice that HBrdpa (and similarly HFdpa) have rather low melting points and are in a molten status at this temperature. After work-up and purification by gradient FC, the product was isolated as a yellow oil in 23% yield (Scheme 3b). However, NMR spectroscopy and ESI-MS spectrometry showed the presence of one side product in 0.26:1 MR with H6tren(dpa)3, namely SPa in Scheme 2 (details on 1H NMR and ESI-MS detection of SPa can be found in the Electronic Supporting Information). SPa co-elutes with the main product during FC purification and is produced by further nucleophilic attack of dipyridylamino nitrogens on HBrdpa (overarylation). The other possible overarylation by-product SPb in Scheme 2 was not detected in this work.
As a possible countermeasure to avoid overarylation, the diarylamino group of HBrdpa was protected by tert-butyloxycarbonyl (Boc). Boc is probably the most common protecting group for primary and secondary amines, as it withstands basic and nucleophilic attacks [39]. Protection of HBrdpa was carried out quantitatively following a literature protocol (Scheme 3d) [40]. Unfortunately, the protecting group was cleaved at 130 °C over long times, likely due to water traces in the starting substrate (Scheme 3d′). In fact, Boc-amines are cleaved in the presence of water already at 100 °C [41, 42]. Since the formation of H6tren(dpa)3 does not proceed significantly below 130 °C, this strategy was abandoned.
Replacing K2CO3 with Cs2CO3 was found critically important to improve regioselectivity. Cesium bases such as CsOH, CsHCO3, Cs2CO3, and CsF, are indeed known to promote mono-N-alkylation of primary amines, suppressing overalkylation [43, 44]. In fact, Cs2CO3 (3.2 equiv.) under solventless conditions (Scheme 3c) gave a lower conversion than K2CO3 (52% versus 74%) but with a much improved regioselectivity and a higher isolated yield (39%).
As a final improvement of the procedure, use of HFdpa as an electrophilic reagent (Scheme 3e) gave a significantly higher conversion (77%), with an almost complete suppression of overarylation and a slightly higher isolated yield (∼42–44%). The composition of the reaction mixture probed by 1H NMR before purification (Table 1) indicates that the higher conversion attained using HFdpa is accompanied by a more favorable MR between H6tren(dpa)3 and the side products (H6tren(dpa)2 and SPa), confirming an enhanced regioselectivity. Notice also that a 63% conversion to H6tren(dpa)3 implies at least ∼85% efficiency for each N-arylation step. The higher purity of the product from Scheme 3e is also reflected by its physical state: a lightweight yellow solid, as contrasted with the oils obtained under the conditions of Scheme 3a–c. Unfortunately, despite many attempts we were unable to grow X-ray-quality crystals of the compound.
Composition (mol%) of the reaction mixture and conversion (mol%) of HXdpaa
| Reaction | HXdpa | H6tren(dpa)3 | H6tren(dpa)2b | SPa | Conversion |
|---|---|---|---|---|---|
| c (X = Br) | 48 | 34.5 | 15.5 | 2 | 52 |
| e (X = F) | 23 | 63 | 12.5 | 1.5 | 77 |
aFrom 1H NMR spectroscopy before purification. bBipodal intermediate.
Considering its excellent performance for the synthesis of HFdpa, the LiH/toluene/py system was finally tested. However, prolonged heating of H6tren, HFdpa, and LiH (10.3 equiv.) at 125 °C in toluene/py failed to produce H6tren(dpa)3 (Scheme 3f), suggesting that these conditions are probably effective only for the N-arylation of aromatic amines.
1H and 13C NMR spectra of H6tren(dpa)3 are presented in Figure 3. For the assignment of proton and carbon chemical shifts, 1D spectra were flanked by the following 2D NMR experiments: 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC (Figures S5, S6, and S7–S8, respectively). The 1H NMR spectrum presents nine well-resolved signals with 𝛿 ranging from 5.21 to 8.15 ppm and identical integrated intensities. Seven of these signals (Ha, Hb, Hc, Hd, Hg, Hh, and Hi) correspond to the aromatic H atoms, while the two remaining resonances at 𝛿 = 5.21 and 7.77 ppm arise from the amino protons Hn and Hk, respectively. Interestingly, Hn is not a singlet as usually occurs for amino protons, but is instead a triplet due to the 1H–1H correlation with the closest aliphatic protons Hl (Figure S5). The high-field region shows one pseudo-quartet at 3.37 ppm and one triplet at 2.76 ppm, which have identical areas but are twice as intense as each aromatic resonance. They correspond to the aliphatic H atoms Hl and Hm, respectively. In H6tren, these two signals are found at 𝛿 = 2.40 and 2.61 ppm in CD3CN, showing that they are both shifted downfield upon arylation. The multi-branch nature of H6tren(dpa)3 is also confirmed by the 1H–13C HMBC spectrum which exhibits a long-range 1H–13C correlation (3 J) between Hm and Cm of a different branch of the same molecule (Figure S8).
Top and middle: atom-labelled structure of H6tren(dpa)3 and its 1H NMR spectrum in CD3CN (298 K, 600.13 MHz); 𝛿H (ppm) = 0.89 (t, 3 J = 7, n-hexane, CH3), 1.28 (m, n-hexane, CH2), 1.94 (quintet, residual protons in CD3CN), 2.15 (s, water, OH), 5.44 (s, dichloromethane, CH2). Bottom: 13C NMR spectrum of H6tren(dpa)3 in CD3CN (298 K, 150.90 MHz); 𝛿C (ppm) = 1.32 (septet, CD3CN), 118.31 (s, CD3 CN). Processing parameters (TopSpin 4.0.6 [37]): SI = TD, LB = 0.30 and 2.00 Hz for 1H and 13C NMR spectra, respectively. The hidden region of the 13C spectrum contains no signal.
H6tren(dpa)3 contains thirteen amino nitrogens and its chromatographic band tails and broadens considerably during column chromatography, which may lead to high product losses. To bypass FC, an alternative purification procedure was developed. After the work-up, the crude product was dissolved in CH2Cl2 and passed through a very short silica gel plug. This treatment is sufficient to remove the intermediate species, such as underarylated by-products H6tren(dpa)2 and H6tren(dpa), which contain free −NH2 moieties and are strongly retained on silica gel (basic aluminum oxide gives incomplete separation). After complete removal of the solvent, the solid residue was repeatedly triturated with Et2O and n-hexane to remove all unreacted HFdpa (see Section 4.2.3 for details). Since H6tren(dpa)3 is only slightly soluble in Et2O and insoluble in n-hexane, this method leads to minimal product losses. In fact, the isolated yield increases to ∼60%, indicating virtually complete recovery (see Table 1). Rewardingly, the spectroscopic and analytical purities evaluated by 1H and 13C NMR, and by elemental analysis, respectively, are not worse than for the chromatographed material. Finally, it is important to note that solvent residuals are invariably retained by the purified product, even after prolonged vacuum treatment. In particular, the product purified by FC contains 0.44–0.66 mol of EtOH per formula unit along with Et2O traces. The alternative purification procedure described in Section 4.2.3 instead leaves 0.35–0.40 mol of Et2O per formula unit plus traces of n-hexane.
3. Conclusions
In this work, the synthesis of oligo-α-pyridylamines and complex architectures based thereon was shown to proceed smoothly via fluoroarenes. The new fluorinated intermediate HFdpa was prepared in excellent isolated yield (90–92%) from F2py, NH2py, and LiH in toluene/py, following a transition-metal-free route. HFdpa is an excellent building block for the introduction of Hdpa-like moieties in organic structures. As an example, the new tridecadentate proligand H6tren(dpa)3 was designed, synthesized, and isolated in good yield (∼60%) via a triple N-arylation of H6tren with HFdpa and Cs2CO3 in solventless conditions. HFdpa turned out to be more reactive than the corresponding brominated derivative, while Cs2CO3 was preferable over K2CO3 as a base, as it considerably improves the regioselectivity of the reaction by eliminating overarylation almost completely.
We are now investigating the coordination chemistry of H6tren(dpa)3 and, in particular, its ability to promote the assembly of transition-metal ions into EMAC structures.
4. Experimental section
4.1. Materials and methods
All chemicals were of reagent grade and used as received, unless otherwise noted. Pyridine was distilled over KOH (115–116 °C) and stored over KOH pellets prior to use. Compound HBrdpa was synthesized by slight modification of a known procedure [34] (details can be found in the Electronic Supporting Information). H6tren was purified by distillation over CaH2 (10% w/w) under reduced pressure (137 °C, 17 mmHg).
Thin-layer chromatography (TLC) was performed on aluminum oxide cards (Fluka) or silica gel 60 plates (Merck), and spots were visualized by UV irradiation at 254 nm. Elemental analysis was performed using a ThermoFisher Scientific Flash 2000 analyzer. The electronic spectrum in THF solution was recorded up to 2000 nm on a Jasco V-570 double beam UV–Vis–NIR spectrometer, using a quartz cuvette (optical path length l = 0.5 cm). ESI-MS measurements were conducted on a 6310A Ion Trap LC-MS(n) instrument (Agilent Technologies) by direct infusion of CH2Cl2 solutions, in positive ion mode. The 1D and 2D NMR spectra (1H, 13C, 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC) were recorded at 298 K in CD3CN, on AVANCE III HD (600.13 and 150.90 MHz for 1H and 13C, respectively) and AVANCE400 (400.13 MHz for 1H) FT-NMR spectrometers from Bruker Biospin. The chemical shifts are expressed in ppm downfield from Me4Si as external standard, by setting the residual 1H (13C) signal of CD3CN at 1.94 ppm (1.32 ppm, CD3) [45]. Coupling constants are in Hz. Spectrum analysis was carried out with TopSpin 4.0.6 software [37]. IR spectra were measured in ATR mode on a JASCO 4700 FT-IR spectrometer between 400 and 4000 cm−1 with 2 cm−1 resolution. The following abbreviations are used in reporting NMR data: br = broad singlet, dd = doublet of doublets, ddd = doublet of doublets of doublets, t = triplet, pt = pseudo-triplet, dpt = doublet of pseudo-triplets, q = quartet, pq = pseudo-quartet, m = multiplet; IR data: s = strong, m = medium, and w = weak.
4.2. Synthesis
4.2.1. HFdpa
NH2py (1.3643 g, 14.496 mmol) and LiH (0.3940 g, 49.57 mmol) were introduced in a round bottom flask under N2 atmosphere, and were stirred together for five minutes. Then, F2py (2.5050 g, 21.767 mmol) and pyridine (7.5 mL) were added to give a yellow suspension, which was slowly heated to ∼90 °C, until a lively gas evolution and a rapid color change to orange were observed. Anhydrous toluene (20 mL) was then added to slow down the reaction. When the gas evolution subsided, the temperature was carefully raised to 120 °C and the mixture was heated for two hours, to give a dark red suspension. The reaction was followed by TLC on silica gel plates (one drop of reaction mixture in 0.5 mL of CH2Cl2; eluent petroleum ether: Et2O, 1:1 v/v; Rf (NH2py) = 0.02; Rf (HFdpa) = 0.26; F2py is undetectable) and 1H NMR spectroscopy in CD3CN. The reaction mixture was allowed to cool to room temperature, and then the solvent was removed under vacuum to give a brown solid. The residue was cooled in an ice bath and the excess of LiH was carefully quenched with small pieces of ice. When effervescence ceased, water (30 mL) was carefully added, and the suspension was stirred overnight at room temperature. The mixture was filtered on a fritted glass funnel to give a light-brown solid, which was washed with water (3 × 20 mL). Then, the solid was repeatedly extracted with CH2Cl2 (3 × 20 mL) to give a dark red solution. The solvent was removed under vacuum and the solid obtained was redissolved in CH2Cl2 (15 mL). The solution was then filtered over a very short silica gel plug (∼2.0 g of SiO2, ∼2 cm thickness) placed over a thin layer of celite. After the filtration, additional CH2Cl2 (3 × 7 mL) was passed through the silica gel plug. The solvent was evaporated and the residue well-dried under vacuum to give the product as a light-brown solid, which required no further purification (2.5316 g, 13.381 mmol, 92.3% isolated yield).
Mp 105.4–107.5 °C. Anal. Calcd for HFdpa: C, 63.49; H, 4.26; N, 22.21%. Found: C, 63.40; H, 4.34; N, 22.01%. 1H NMR (CD3CN, 298 K, 600.13 MHz): 𝛿H (ppm) = 8.25 (1H, ddd, 3 J(a,b) = 4.9, 4 J(a,c) = 1.9, 5 J(a,d) = 0.8, Ha), 8.08 (1H, br, Hk), 7.75 (1H, pq, 3 J(h,g) ∼3 J(h,i) ∼ 8.0, 4 J(h,F) = 8.7, Hh), 7.67 (1H, ddd, 3 J(c,d) = 8.4, 3 J(c,b) = 7.2, 4 J(c,a) = 1.9, Hc), 7.58 (1H, ddd, 3 J(g,h) = 8.0, 5 J(g,F) = 2.4, 4 J(g,i) = 0.4, Hg), 7.55 (1H, dpt, 3 J(d,c) = 8.4, 4 J(d,b) ∼5 J(d,a) ∼ 0.9, Hd), 6.92 (1H, ddd, 3 J(b,c) = 7.2, 3 J(b,a) = 4.9, 4 J(b,d) = 1.0, Hb), 6.47 (1H, ddd, 3 J(i,h) = 7.8, 3 J(i,F) = 2.5, 4 J(i,g) = 0.4, Hi). 13C NMR (CD3CN, 298 K, 150.90 MHz): 𝛿C (ppm) = 163.2 (1C, d, 1 J(j,F) = 234.6, Cj), 154.7 (1C, s, Ce), 154.1 (1C, d, 3 J(f,F) = 16.2, Cf), 148.7 (1C, s, Ca), 143.6 (1C, d, 3 J(h,F) = 8.2, Ch), 138.8 (1C, s, Cc), 117.9 (1C, s, Cb), 112.9 (1C, s, Cd), 109.0 (1C, d, 4 J(g,F) = 4.2, Cg), 100.3 (1C, d, 2 J(i,F) = 36.6, Ci). IR (ATR): (cm−1) = 3269 (w), 3218 (w), 3186 (w), 3101 (w), 3025 (w), 1659 (w), 1628 (m), 1609 (m), 1599 (m), 1591 (m), 1569 (m), 1538 (m), 1527 (m), 1476 (m), 1466 (m), 1437 (s), 1416 (s), 1358 (m), 1298 (m), 1279 (m), 1264 (m), 1245 (m), 1221 (m), 1215 (m), 1150 (m), 1144 (m), 1101 (w), 1072 (w), 1055 (w), 1025 (m), 1011 (w), 997 (w), 985 (w), 867 (w), 861 (w), 847 (w), 769 (s), 752 (s), 723 (m), 720 (m), 671 (w), 659 (w), 625 (m), 601 (m), 553 (m), 515 (m), 462 (w).
4.2.2. H6tren(dpa)3
Under N2 atmosphere, HFdpa (2.3244 g, 12.286 mmol) and Cs2CO3 (4.0033 g, 12.287 mmol) were introduced into a pear-shaped Schlenk flask. H6tren (0.5492 g, 3.755 mmol) was then added with a syringe, and the mixture was carefully heated to 130 °C with stirring (between 85–100 °C the mixture progressively turns into a dark brown suspension as HFdpa melts). The temperature was kept constant for three days during which the mixture progressively hardened, blocking the magnetic stirring after 24–30 h. The reaction was monitored by TLC on aluminum oxide cards (one drop of reaction mixture in 0.5 mL of CH2Cl2; eluent Et2O:EtOH, 10:0.4 v/v; Rf(H6tren) = 0.00; Rf(H6tren(dpa)3) = 0.20; Rf(HXdpa) = 0.70 for X = F and 0.68 for X = Br) and 1H NMR spectroscopy in CD3CN. The hard light-brown solid so obtained was allowed to cool to room temperature and dissolved in CH2Cl2 (25 mL) with the help of an ultrasonic bath. The dark red solution was washed with a saturated aqueous solution of NaHCO3 (3 × 5 mL) and subsequently with water (3 × 5 mL). The organic phase was then dried over MgSO4 (1 h), filtered over a fritted glass funnel (porosity G3) and evaporated under reduced pressure. The solid residue was then purified by gradient FC (for ∼2.5 g of crude material: basic Al2O3; eluent Et2O:EtOH, from 1:0 to 0.8:0.2 v/v; tgradient = 10 min, flow rate = 10 mL/min, column diameter = 25 mm, column length = 130 mm), to give H6tren(dpa)3⋅xEtOH (x = 0.44–0.66) as a lightweight yellow powder (isolated yield = 41.6–43.5%).
Anal. Calcd for H6tren(dpa)3⋅0.66EtOH: C, 65.51; H, 6.33; N, 26.61%. Found: C, 65.24; H, 6.26; N, 26.99%. UV–Vis–NIR (THF, b = 0.5 cm, 7.32 × 10−5 M): 𝜆max(𝜀) = 239 nm (4.96 × 104 M−1⋅cm−1), 272 nm (5.74 × 104 M−1⋅cm−1), 332 nm (5.89 × 104 M−1⋅cm−1). 1H NMR (CD3CN, 298 K, 600.13 MHz): 𝛿H (ppm) = 8.15 (3H, ddd, 3 J(a,b) = 4.9, 4 J(a,c) = 1.9, 5 J(a,d) = 0.9, Ha), 7.77 (3H, br, Hk), 7.73 (3H, dpt, 3 J(d,c) = 8.4, 4 J(d,b) ∼5 J(d,a) ∼ 0.9, Hd), 7.53 (3H, ddd, 3 J(c,d) = 8.5, 3 J(c,b) = 7.1, 4 J(c,a) = 1.9, Hc), 7.25 (3H, pt, 3 J(h,g) ∼3 J(h,i) ∼ 7.9, Hh), 6.76 (3H, ddd, 3 J(b,c) = 7.2, 3 J(b,a) = 4.9, 4 J(b,d) = 1.0, Hb), 6.60 (3H, dd, 3 J(g,h) = 7.9, 4 J(g,i) = 0.5, Hg), 5.93 (3H, dd, 3 J(i,h) = 8.0, 4 J(i,g) = 0.6, Hi), 5.21 (3H, t, 3 J(n,l) = 5.6, Hn), 3.37 (6H, pq, 3 J(l,m) ∼3 J(l,n) ∼ 6.0, Hl), 2.76 (6H, t, 3 J(m,l) = 6.2, Hm); EtOH (0.66 mol per mole of H6tren(dpa)3) 𝛿H (ppm) = 3.54 (2H, q, 3 J = 7.0, CH2), 2.46 (1H, br, OH), 1.12 (3H, t, 3 J = 7.0, CH3). 13C NMR (CD3CN, 298 K, 150.90 MHz): 𝛿C (ppm) = 159.1 (3C, s, Cj), 155.6 (3C, s, Ce), 154.2 (3C, s, Cf), 148.6 (3C, s, Ca), 139.7 (3C, s, Ch), 138.4 (3C, s, Cc), 116.7 (3C, s, Cb), 112.5 (3C, s, Cd), 100.4 (3C, s, Ci), 99.7 (3C, s, Cg), 54.5 (3C, s, Cm), 40.8 (3C, s, Cl); EtOH (0.66 mol per mole of H6tren(dpa)3) 𝛿C (ppm) = 57.96 (1C, s, CH2), 18.76 (1C, s, CH3). IR (ATR): (cm−1) = 3391 (w), 3258 (w), 3191 (w), 3033 (w), 2952 (w), 2849 (w), 1599 (m), 1568 (m), 1526 (w), 1502 (w), 1467 (m), 1446 (m), 1427 (s), 1358 (w), 1307 (m), 1247 (w), 1223 (w), 1146 (m), 1100 (w), 1049 (w), 986 (w), 768 (s), 721 (m), 615 (w), 599 (w), 510 (w).
4.2.3. Alternative purification of H6tren(dpa)3
The reaction was carried out as described in Section 4.2.2, using HFdpa (2.9992 g, 15.853 mmol), Cs2CO3 (5.1647 g, 15.851 mmol), and H6tren (0.7264 g, 4.967 mmol). To avoid purification by chromatography, the solid obtained after work-up was dissolved in CH2Cl2 (7–8 mL) and filtered over a very short silica gel plug (∼2.5 g of SiO2, ∼2 cm thickness) placed over a layer of celite. After the filtration, additional CH2Cl2 (7 mL) was passed through the SiO2. The solvent was evaporated under vacuum to give a sticky light-brown solid, which was first triturated by stirring in Et2O (25 mL) for 24 h. The solid was then triturated in n-hexane (15 mL) for 2 h, with alternate stirring (20 min) and sonication (10 min). As a further purification step, the solid was again triturated with Et2O (∼20 mL overnight with stirring and then 3 × 6 mL washings) and subsequently with n-hexane (∼20 mL for 2 h with stirring and then 3 × 6 mL washings). After each step the solvent was removed with a narrow bore pipette. Finally, the solid was well dried in vacuum to afford H6tren(dpa)3⋅0.40Et2O as a yellow powder (2.02 g, 2.96 mmol, 59.5% isolated yield). Anal. Calcd for H6tren(dpa)3⋅0.40Et2O: C, 66.08; H, 6.34; N, 26.64%. Found: C, 66.05; H, 6.52; N, 26.43%. The spectroscopic (NMR, UV–Vis–NIR, and IR) properties are identical to those recorded on chromatographed samples, except for the presence of the peaks of Et2O instead of those of EtOH in the 1H NMR spectrum (CD3CN, 298 K, 400.13 MHz): Et2O (0.40 mol per mole of H6tren(dpa)3) 𝛿H (ppm) = 3.42 (4H, q, 3 J = 7.0, CH2), 1.12 (6H, t, 3 J = 7.0, CH3).
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are grateful to A. Mucci (Department of Chemical and Geological Sciences, University of Modena and Reggio Emilia) for stimulating discussion on NMR spectra.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.223 or from the author.