



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Understanding how multiple components of a human cell interact together to sustain the basic mechanisms of life remains mostly unknown but makes fast progress through the combination of multiple experimental approaches. Encyclopedia containing the human genome [1] and tissue-based maps of the human proteome [2] enable us to define gene expression profiles and subsequently the protein levels that compose each human cell type. Advanced quantitative methods (quantitative Polymerase Chain Reaction, mass spectrometry) estimate the abundance of mRNA and proteins per cell [3]. Progresses in microscopy allow imaging protein–protein interactions at resolution approaching (macro)molecule level for super resolution fluorescent microscopy and nanometric resolution for cryo-Electron Microscopy (EM) [4]. At nanometric dimensions, specimen preservation is crucial and dehydration or chemical cross-linking should be avoided to prevent structural reorganisation of cellular content. In this respect, eukaryotic cells, including mammalian ones, can be vitrified either by plunge freezing or by high-pressure freezing, to preserve a close-to-native frozen hydrated state, and sectioned below −140 °C for cryo-EM imaging [5]. Abundant proteins forming filaments or large assemblies such as ribosomes or nuclear pores are easily recognised in cryo-electron micrographs due to their characteristic shape [4, 6]. However, less abundant biomolecular complexes or small proteins cannot be recognised unambiguously in electron tomograms and their identification relies on labelling tools. Electron-dense gold nanoparticles (AuNPs) conjugated to antibodies are popular tags for EM immunolabelling since their high electron scattering, round shape, and specific dimension provide sufficient contrast to identify the probe [7]. Colloidal AuNPs with size larger than 5 nm in diameter are readily detected even when the cellular sample is contrasted. They offer a large surface area onto which many proteins adsorb [8]. This ability has been exploited to make immunocolloids of popular usage [7]. However, size restricts penetration of those colloids in gel-like biological samples [9]. It has led to the development of gold nanoparticles with diameters below 2 nm and to modification of the gold-to-antibody conjugation because proteins do not adsorb tightly enough on the surface of gold particles below 2 nm [10]. Phosphine-coated 1.4 nm Nanogold® [11] and 0.8 nm ultrasmall AuNPs [12] have increased labelling efficiency but to the cost of an extra step consisting in increasing the size of the gold particles by silver deposition to reveal their positions in the cell sections. Due to the impermeability of the plasma membrane, labelling of specific intracellular targets is generally performed post-embedding on cell sections at temperatures above 0 °C [13]. This protocol is not consistent with high-resolution cryo-EM because vitrified cellular samples cannot be warmed up after sectioning. Therefore, the gold nanoparticles-antibody conjugates need to be delivered into living cells, prior to the vitrification step, using weakly perturbing transduction methods [14]. Ultra-small 0.8 nm AuNPs conjugated to antibodies directed against RNA polymerase II were shown to be delivered into living HeLa cells using a cationic lipid formulation, and the gold labels were detected within the nucleus by Scanning Electron Microscopy (SEM) after resin embedding and silver enhancement [15]. To develop gold particles of sizes directly visible in vitrified cell sections by cryo-EM, mercaptobenzoic acid (MBA)-coated AuNPs of defined sizes around 2 nm [16, 17] appeared extremely promising [18, 19]. We previously attempted to substitute MBA coating on AuNPs by thionitrobenzoate (TNB) [20] to increase ligand exchange kinetics [21]. During the synthesis of 1.4 nm AuNP with sodium borohydride, a fraction of nitro groups underwent reduction to amines, yielding AuNP with a mixed coordination layer consisting of TABs and TNBs (TAB-, TNB-AuNP) named AuZ. This mixed coverage was seen advantageous, in terms of ligand exchange with incoming thiols on the one hand, and colloidal stability on the other hand. Indeed, ligand exchange onto the AuNPs is rarely quantitative and preferentially occurs with the negatively charged TNBs, leaving zwitterionic TABs on the surface as putative protecting groups against strong associations to proteins [22].
In this study, we linked several commercial monoclonal antibodies (mAbs) to 1.4 nm AuZ gold nanoparticle to produce AuNP-mAb conjugates. First, we verified that these conjugates specifically bind to their intracellular targets using fixed and permeabilised cell lines in a classical gold immunolabelling procedure. We confirmed that shielding the gold surface of the AuNP-mAb conjugate with 2 kDa polyethyleneglycol drastically reduced unspecific adsorption of nanoparticles to extracellular constituents but also to intracellular ones [23, 24]. The AuNP-mAb conjugates were then delivered inside living cells by electroporation [25, 26] and the cells containing cytosolic AuNP-mAb conjugates were cultivated before monitoring the cell distribution of the probes (Figure 1). Our data demonstrate that the 1.4 nm AuNP-mAbs enter the cytoplasm where they circulate and associate to their targets. Accumulation of the probes in living cells was mAb-dependent. The accumulation patterns of the AuNP-mAb conjugates anti-GFP, anti-RPB1 (7G5, anti-RNA polymerase II) and anti-nucleoporins were analogous to the classical immunogold labelling ones indicating that the probes bind in cellulo to their targets without perturbing their function. Cytoplasmic-delivered anti-α-tubulin and anti-β-tubulin conjugates in contrast promoted intracytoplasmic aggregates, indicating that real-time binding of these conjugates perturbs microtubule dynamics.

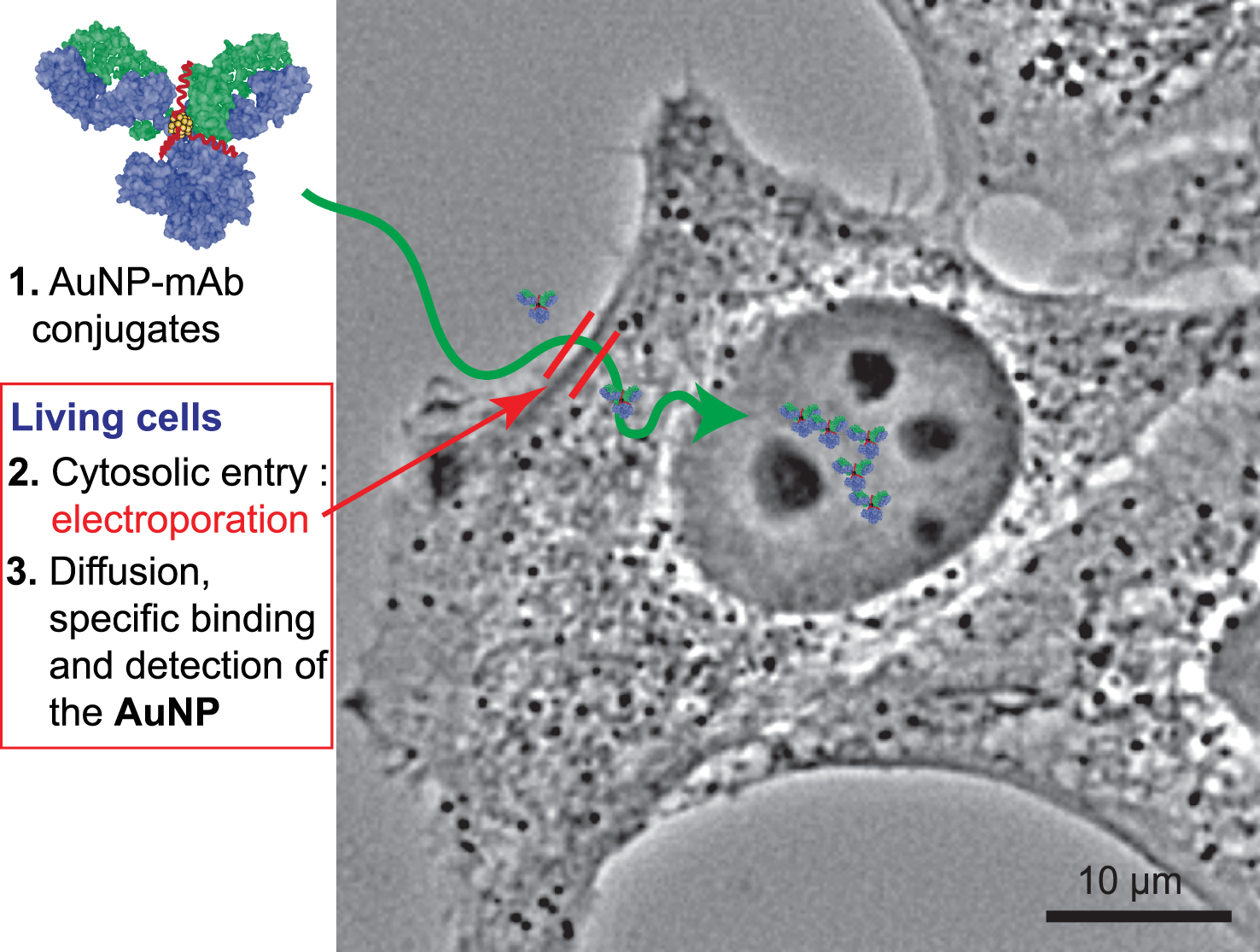
Illustration of gold immunolabelling in living cells. The strategy relies on the synthesis of specific AuNP-mAb conjugates and delivery procedure for the probes to diffuse, even into the cell nucleus, and bind to their specific targets.
2. Results and discussion
2.1. Synthesis of gold nanoparticle-antibody conjugates
The 1.4 nm AuZ used in this study has been previously described and comprehensively characterised [20]. To conjugate the mouse monoclonal antibodies to AuZ, we took advantage of the disulfide bonds present in the hinge region. These disulfide bonds are easily reduced to sulfhydryl groups using Tris(2-carboxyethyl)phosphine (TCEP) [28], thus creating a conjugation site for the 150 kDa mAb onto TAB-, TNB-AuNP at about 7 nm of the recognised antigen paratope (Figure 2) [29]. The various reaction intermediates can be monitored using a non-reducing SDS-PAGE experiment as illustrated by the conjugation of AuZ to the 7G5 mAb (Figure 2B). The 7G5 mAb targets the 52 times-repeated YSPTSPS sequence at the C-terminal domain of the largest subunit (RPB1) of mammalian RNA polymerase II (Pol II). TCEP at a concentration of 1 mM fully cleaved the disulfide bond holding the heavy chains together as seen in the SDS-PAGE analysis (lane 7G5-SH). The mAb with an initial apparent molecular weight (MW) of 150 kDa was completely transformed into a 75 kDa protein corresponding to a single copy of the heavy and light chains. The reduced mAb was then reacted with a 1.5 molar excess of AuZ to yield a new component with an apparent MW slightly above 150 kDa (lane 7G5Au). Excess AuZ and the first released TNBs were removed by size exclusion chromatography. The purified 7G5Au was then reacted with an excess of thiolated 2 kDa polyethyleneglycol (PEG) to chemically neutralise the remaining reacting TNBs present on the gold particle surface [30] and to shield the gold surface from unspecific binding [8, 23]. A material with a highly retarded electrophoretic mobility was observed (lane 7G5AuPEG), indicative of the shielding ability of PEG.

Synthesis and characterisation of the AuNP-antibody conjugate. (A) Disulfide bonds of the mouse monoclonal antibody targeting RPB1 of RNA polII (clone 7G5) are reduced at the hinge area with TCEP. The sulfhydryl groups at the hinge can then react with AuZ by thiolate exchange. After clearing the conjugate from excess AuZ, the remaining reacting thiolates on the AuNP surface are exchanged with thiolated 2 kDa polyethyleneglycol (PEG). (B) Non-reducing SDS-PAGE analysis of the reaction intermediates. Illustration of the antibody derived from protein data bank entry id 1IGY [27]. (C) Gallery of two-dimensional class averages from negatively stained 7G5AuPEG particles. (D) Representative class averages representing 2-D views of unlabelled (top) or gold-conjugated (bottom) IgG molecules. The bar represents 15 nm in C and D. Masquer
Synthesis and characterisation of the AuNP-antibody conjugate. (A) Disulfide bonds of the mouse monoclonal antibody targeting RPB1 of RNA polII (clone 7G5) are reduced at the hinge area with TCEP. The sulfhydryl groups at the hinge can then react with ... Lire la suite
To assess the molecular organisation of the probe, the purified antibody-gold particle was visualised by transmission EM after negative staining with 2% uranyl acetate. Single particle analysis was performed to observe the position of the AuNP relative to the antibody, to determine the proportion of Au-bound IgG molecules and to assess the integrity of the AuNP-bound antibody. The negatively stained protein moieties create a stain exclusion volume which diffuses less electrons than the surrounding medium. This property was used to select 80431 molecular images from 1125 micrographs that were aligned and clustered into 100 classes using Xmipp [31] (Figure 2C). After rejection of false positives, the remaining 36671 images were clustered into 100 classes. About 88% of the uranyl acetate-excluding materials were found associated with an electron-dense particle whose size is consistent with the input AuNP. The other images were found in classes representing characteristic views of non-conjugated IgG molecules such as 15 nm long, Y-shaped molecules (Figure 2D, top). The classes showing a dark electron-dense gold nanoparticle were heterogeneous and showed several extensions about 5 nm in size which could each correspond to Fab or Fc fragments. The 3-branched classes resemble the Y-shaped IgG molecule while the other classes are likely to represent two IgG molecules bound to the same AuNP particle with different rotational registers (Figure 2D, bottom). Strikingly, the electron-dense gold particle was always located at the centre of the multibranched structures, indicating that the AuNP associates with the hinge region of the IgG which exposes several thiolates following mild reduction step [32]. When the AuNP is bound to a single IgG molecule, the latter adopts a classical Y shape, suggesting that conjugation does not affect the molecular organisation of the antibody.
Binding of AuNP-mAb conjugates 7G5Au and 7G5AuPEG to RPB1 was first assayed using a classical immunocytochemistry procedure. HeLa cells cultivated on glass coverslips were fixed with 4% EM-grade paraformaldehyde for 20 min and their plasma membrane was permeabilised with 0.1% Triton X-100. The different 7G5 mAb conjugates were incubated with the permeabilised cells, excess probe was washed away and bound conjugates were detected with a secondary fluorescent goat anti-mouse antibody (Figure 3A). The 7G5 mAbs were found predominantly in the cell nucleus with reduced staining in the nucleolus, as expected for the nuclear RNA pol II enzyme. Enrichment of fluorescence was observed within foci in accordance with the hypothesis of transcription occurring mainly in factories or condensates containing several RNA pol II molecules [33]. A similar pattern was observed with the AuNP-mAb 7G5AuPEG component (Figure 3B). The intensity of the green fluorescence was nonetheless diminished as compared to the 7G5 mAbs signal. This feature might originate from the steric hindrance of PEGs [23] as well as from the fluorescence-quenching properties of AuNPs [34] leading to reduced association of the secondary Abs and dimming of the fluorescent signal, respectively. To map the distribution of gold nanoparticles, a similar immunolabelling experiment was performed using 7G5Au (Figure 3C) and 7G5AuPEG (Figure 3D), and a silver-enhancement procedure to reveal the AuNPs only (Supporting information, Figure S1 shows that the control HeLa cells remain unstained after incubation with the silver-enhancement solution). The 7G5Au probe showed a nuclear enrichment, but significant amounts of silver also stained the cytoplasm. Passivation of AuNPs with PEG 2 kDa diminished unspecific association to the cytoskeleton and provided a sharp nuclear gold immunolabelling in which dark spots are revealed. These dense spots might correspond to clustering of several RNA pol II molecules. The stealth property of PEGs surrounding AuZ was highly efficient since passivation of 7G5Au with the gold particle-stabilising CALNNG peptide [35] produced strong unspecific labelling of the cytoskeleton (Supporting information, Figure S2).

Immunofluorescence and gold immunolabelling analysis of the indicated monoclonal antibody anti-RPB1 (clone 7G5) derivatives on fixed and permeabilised HeLa cells. Images A and B: the mouse antibody was fluorescently detected with an AlexaFluo488-IgG anti-mouse conjugate. Images C, D and E: the gold particles were amplified with silver for detection by bright-field imaging. Conditions: fixation with 4% (v/v) PFA in 0.1 M phosphate buffer pH 7.6 for 20 min, permeabilisation with 0.1% (v/v) Triton X-100®, incubations with primary and secondary antibodies at 13 nM. Masquer
Immunofluorescence and gold immunolabelling analysis of the indicated monoclonal antibody anti-RPB1 (clone 7G5) derivatives on fixed and permeabilised HeLa cells. Images A and B: the mouse antibody was fluorescently detected with an AlexaFluo488-IgG anti-mouse conjugate. Images C, D and E: ... Lire la suite
Commercial monoclonal antibodies (listed in Table 1) targeting RPB1 of RNA polII (7C2) [36], nucleoporins (Mab414), the trimethyl lysine 20 of histone H4 (6F8-D9), α-tubulin (B-5-1-2), β-tubulin (TU27) and the green fluorescent protein (GFP)(2A3) were first checked for their immunofluorescence specificity (Supporting information, Figure S3). They were then conjugated to AuZ and passivised with PEG using the same protocol as for 7G5 (Supporting information, Figure S4). Gold immunolabelling experiments on adherent cell lines led to images with precise localisation of targets using bright-field microscopy. The anti-nucleoporins conjugated to pegylated AuNPs (MAb414AuPEG) labelled the Nuclear Pore Complexes embedded within the nuclear envelope (Figure 4A). The post-translational modification of histone H4 at lysine 20 was also detected in the nucleus (Figure 4B). Microtubules were also clearly revealed in the cytoplasm using the anti-α-tubulin B-5-1-2AuPEG (Figure 4C) and the anti-β-tubulin TU27AuPEG (Figure 4D). The monoclonal anti-RPB1 7C2AuPEG was not as effective as 7G5AuPEG in terms of labelling sharpness (Figure 5A). Finally, the anti-GFP 2A3AuPEG [12, 37] probe labelled extremely well the GFP-labelled histone H2B within the nucleus of stably transformed HeLa cells that express the GFP-H2B fusion (Figure 5C) but did not bind to wild-type HeLa devoid of GFP (Figure 5D). The importance of passivation by grafting PEG onto the gold surface to limit unspecific binding to cytoplasmic components was also confirmed for that antibody (Supporting information Figure S5).
List of antibodies used for conjugation
| Name | Target | Source |
|---|---|---|
| 7G5 | RPB1 of RNA polII [TSPSYSP]3 at C ter | IGBMC |
| 7C2 | RPB1 of RNA polII [YSPTSPS]3 at C ter | IGBMC |
| Mab414 | Nucleoporins (Nuclear pores) | Biolegend |
| 6F8-D9 | Histone H4 trimethyl Lysine20 (H4K20me3) | Biolegend |
| B-5-1-2 | α-tubulin (C-t) | ThermoFisher |
| TU27 | β-tubulin (C-t) | Biolegend |
| 2A3 | Green fluorescent protein (GFP) | IGBMC |

Gold immunolabelling analysis of the indicated monoclonal antibodies (mAbs) conjugated to 1.4 nm AuNP passivised with 2 kDa PEG. H2B-GFP HeLa cell line stably expresses the histone H2B fused to GFP. The gold particles were enhanced by silver for bright-field imaging.

Gold immunolabelling analysis of the indicated monoclonal antibodies (mAbs) conjugated to 1.4 nm AuNP passivised with 2 kDa PEG. H2B-GFP HeLa cell line stably expresses the histone H2B fused to GFP. The gold particles were enhanced by silver for bright-field imaging.
Altogether, those first experiments demonstrated that 1.4 nm AuZ can be conjugated to several mAbs at the hinge region to produce effective gold labelling probes for classical immunocytochemistry procedures. Shielding the 1.4 nm gold nanoparticle with 2 kDa PEG was beneficial and considerably limited unspecific binding to cell components.
2.2. Fate of AuNP-mAb conjugates following cytosolic delivery into living HeLa cells
The ability of the AuNP-mAb conjugates to diffuse inside living cells, bind to their targets and label them was assayed (Figure 6). To introduce the probes into the cytoplasm of living cells, transient holes in the plasma membrane of freshly trypsinised cells were created using an electroporation device and optimised pulses [26, 38]. After the electric pulses and re-closure of the plasma membrane, HeLa cells containing cytoplasmic mAbs and AuNP-mAb conjugates were generally cultivated for 20 h to let them adhere onto glass coverslips. Cells were then processed for detection of antibodies and of AuNPs (Figure 6A). After delivery into the cytoplasm of HeLa cells expressing H2B-GFP, the unconjugated anti-GFP 2A3 accumulated into the cell nuclei and distributed similarly to the GFP, suggesting an effective binding of the antibody to its target (Figure 6B). Entry of 150 kDa Abs into the cell nucleus was unexpected since nuclear pore complexes usually prevent nuclear entry of proteins with a molecular weight above 60 kDa unless they are equipped with Nuclear Localisation Signals (NLS). The anti-GFP antibody is likely transported into the nucleus after binding to the NLS-containing GFP-H2B protein when it is synthetised in the cytoplasm by a piggyback mechanism [39]. When delivered into the cytoplasm of wild-type HeLa cells devoid of GFP-H2B, the anti-GFP 2A3AuPEG probe diffused in the cytoplasm but was excluded from the nucleus (Figure 6C). In contrast, the anti-GFP 2A3AuPEG delivered into the cytoplasm of HeLa cells expressing H2B-GFP accumulated in the nuclei in a fashion resembling 2A3 (Figure 6D), indicating that AuNP conjugation does not impair nuclear import of this specific mAb anti-GFP. The observed nuclear accumulation patterns relied on the presence of conjugates in the cytoplasm of living cells since electric pulses for electroporation were required (left-hand panels in Figure 6C and D). In cellulo labelling with anti-nucleoporins Mab414 and Mab414AuPEG yielded images (Figure 6E) which were highly reminiscent of the classical immunocytochemistry images (Figure 4A). Cytoplasmic-delivered anti-α-tubulin TU27AuPEG and anti-β-tubulin B-5-1-2AuPEG did not reveal an extensive microtubule network but rather accumulated as aggregates. This type of labelling and accumulation might be related to the dynamic nature of the microtubule network and to the electroporation procedure that involves a transient suspension of the cells using a trypsin treatment. Reshuffling of the microtubule network might take place during re-adhesion of the cells and after the electric pulses which are also known disruptors of microtubules [40, 41]. The cytoplasm-delivered 7G5AuPEG and 7C2AuPEG also accumulated in the nucleus (Figure 6H) similarly to the unconjugated mAbs [38]. Similarly to the classical gold immunolabelling protocol, passivation of the AuNPs with PEG was also extremely useful for nuclear accumulation. Indeed, capping the AuNP of 7G5Au with CALNNG peptide did not lead to nuclear accumulation of 7G5AuC 24 h following electroporation (Figure S6). Finally, the nuclear accumulation appears not as with the anti-GFP conjugates. This property is likely related to the production of RNA pol II in the cytoplasm which is lower than that of H2B-GFP, limiting de facto the entry of Ab-bound RNA pol II [37, 38]. The 7G5AuPEG labelling was further investigated by transmission electron microscopy (TEM) (Figure 7). Twenty hours after electroporation, cells were fixed, the gold signal was amplified by silver enhancement for better visualisation and thin sections of resin-embedded cells were observed by TEM. An image of a cell section displaying the nucleus and cytoplasm was recorded. The enhanced AuNPs were almost exclusively found in the cell nucleus, confirming that the RNA polII-driven accumulation of the corresponding AuNP-mAb occurred in this cell.

The AuNP-mAb conjugates (0.2 μg⋅μL−1) were delivered in HeLa cells by transient permeabilisation of the cell membrane using 3 electric pulses at 517 V⋅cm−1. Distribution of the mAbs and conjugates inside living cells was evaluated after culture during 20 h unless stated. Fixation: 4% PFA, 20 min. Permeabilisation: 0.1% Triton X-100. AuNPs were silver-enhanced. (A) Scheme of the procedure. (B) Images of the targeted H2B-GFP in HeLa cells (left) and of the delivered anti-GFP 2A3 (right). The mAb was detected using AlexaFluor568 goat anti-mouse (in red). (C, D) Detection of AuNPs by silver enhancement in cells incubated with 2A3AuPEG without application of electric pulses (left images) and with application of electric pulses. (E–H) Delivered mAbs were detected in cells using the antibody or the gold domain. Masquer
The AuNP-mAb conjugates (0.2 μg⋅μL−1) were delivered in HeLa cells by transient permeabilisation of the cell membrane using 3 electric pulses at 517 V⋅cm−1. Distribution of the mAbs and conjugates inside living cells was evaluated after culture during 20 h unless ... Lire la suite

Electron micrograph of a HeLa cell section containing 7G5AuPEGs in the nucleus. Cells were incubated with 7G5AuPEG (0.2 μg⋅μL−1) and the plasma membrane was transiently permeabilised with 3 electric pulses at 517 V⋅cm−1. Cells were cultivated for 20 h, fixed with 2% glutaraldehyde and processed for classical TEM observation. Gold particles were enhanced with silver.
3. Conclusion
We hereby provide a synthesis protocol for preparing mAbs conjugated to 1.4 nm AuZ gold nanoparticles by taking advantage of the disulfide bonds which connect the hinge region of the antibody heavy chains. The reaction process can be easily monitored by non-reducing SDS-PAGE analysis which reveals the key reaction intermediates. Passivation of the gold surface with 2 kDa polyethyleneglycol provided probes with excellent gold immunolabelling properties such as solubility, absence of in vivo aggregation, good diffusion properties, and dramatically diminished the unspecific binding to cellular components. Moreover, these conjugates can be delivered into the cytoplasm of living cells by electroporation, where they diffuse and bind to their targets. The piggyback mechanism allowing nuclear entry of AuNP-mAb only upon binding to NLS-containing targets can be extremely useful as filter since unbound AuNP-mAbs remain in the cytoplasm. Our first proof of concept data demonstrated that AuNP-mAbs can be delivered into living cells, bind to their specific targets and accumulate at locations where their targets go. In principle, the anti-GFP antibody has the potential to become a rather generic gold immunolabelling probe. Standard immunolabelling procedure and silver enhancement will enable to monitor the labelling dynamic and efficiency, to adjust the amounts of the electroporated probes and the incubation time (and maybe other parameters) to ensure that the most specific labelling is observed later in cryo-EM. Specific monoclonal antibodies bind to a specific protein domain and then interfere with the functioning of their targets. After labelling with AuNPs and delivery inside living cells, these interfering probes might help decipher the function of protein/protein interactions and dynamics of exposure to Ab binding. Although the electroporation procedure is convenient and has demonstrated efficiency, the electric pulses are not neutral to cell physiology. Cells mostly recover but the cell cytoskeleton is perturbed. Alternatively, differentially disruptive liposomal formulations have been used as delivery solutions and can be employed as well.
4. Experimental section
4.1. Materials
The 2 kDa alpha-methoxy-omega-mercapto poly(ethylene glycol) (PEG-SH) was ordered from Iris Biotech. EM-grade paraformaldehyde (20% w/v solution) and glutaraldehyde (25% w/v solution) were purchased from Electron Microscopy Sciences. Other reagents were obtained from Sigma Aldrich, Carl Roth, VWR Chemicals, Euromedex or Honeywell. They were used without further purification unless stated otherwise. All solutions and buffers were made with water purified with a Millipore Q-POD apparatus. Precision Plus Protein Standard Dual Xtra (BioRad) was used as protein ladder for SDS-PAGE analysis. Monoclonal antibodies were purchased from commercial sources and were devoid of bovine serum albumin or gelatine. They were cleared from sodium azide or any other preservatives by size exclusion chromatography (Sephadex G-25) and eventually reconcentrated by ultracentrifugation.
4.2. Gold nanoparticle synthesis
The 1.4 nm AuZ was synthesised and characterised in a previous publication [18, 19, 20]. The concentration of gold nanoparticles was determined by UV–vis spectrophotometry using the absorbance at 520 nm and an extinction coefficient 𝜀520 of 2.7 × 105 mol−1⋅L⋅cm−1.
4.3. Reduction of monoclonal antibody
Before performing any conjugation, reduction of the monoclonal antibody at the hinge area was monitored by SDS-PAGE using a non-reducing loading buffer. The antibody (2 μg⋅μL−1 or 13.34 μM, 2 μL) was titrated with TCEP (final concentrations: 0.1, 0.25, 0.5, 1, and 2 mM). After 16 h incubation at 20 °C, the reaction mixtures were mixed with 30% (w/v) glycerol containing 0.1% (w/v) SDS and 0.05% (w/v) bromophenol blue (0.4 μL) and analysed by SDS-PAGE. The minimal TCEP concentration producing full disappearance of the band at about 150 kDa was selected for medium-scale synthesis.
4.4. Synthesis of 7G5AuPEG
The monoclonal antibody in PBS containing 1 mM EDTA (2 μg⋅μL−1 or 13.34 μM, 100 μL) was reduced by addition of 0.1 M TCEP, pH 7.0 (1 μL) to a final concentration of 1 μM. After a 12 h incubation at 20 °C, an aliquot was withdrawn for analysis and the mixture was then quickly added and mixed with Auz (25 μL of 80 μM solution). After overnight incubation at 20 °C, the crude mixture was purified by size exclusion chromatography (Biorad P100, 2 mL of resin, elution with PBS). Thiolated polyethyleneglycol 2000 (2.7 μL of a 20 mM aqueous concentration) was then added. The mixture was incubated at room temperature for 16 h and the crude mixture was purified by ultracentrifugation (7 cycles of 0.5 mL sterile PBS) using a 0.5 mL Microcon® centrifugal filter device with a 100 kDa cutoff (Merck, Molsheim). At the end of the process, the gold nanoparticle-antibody conjugate was recovered in 60 μL PBS and analysed by SDS-PAGE to estimate the concentration and conjugation efficiency. The other gold nanoparticles-antibody conjugates were similarly prepared.
4.5. Electroporation protocol
Transient permeabilisation of the plasma membrane of cells was conducted as previously described using the Neon® transfection system and three 10 ms pulses at 1550 V [20, 26]. Typically, HeLa cells (2 × 105 cells in 10 μL of the Neon Buffer R) were mixed with each antibody (2.5 μL of 2 μg⋅μL−1 solution in PBS). After the electric pulses, cells were diluted in prewarmed antibiotic-free cell culture medium (1 mL). The cells were recovered by gentle centrifugation, suspended into the warmed antibiotic-free complete cell culture medium and plated in 24-wells plates for cell adherence and growth onto glass coverslips and left in a cell culture incubator, usually for 18 h.
4.6. Cell culture
Cells were cultured in a 37 °C humidified incubator supplied with 5% CO2. HeLa, CaSki, and stably transformed H2B-GFP HeLa cells were maintained in Dulbecco’s modified Eagle medium containing 2 mM L-glutamine, 10 mM HEPES buffer, pH 7.0, 10% heat-inactivated foetal bovine serum (FBS) and 50 μg/mL gentamycin. For immunocytochemistry, immunofluorescence and pre-embedding immuno-EM experiments, cells (25,000 cells/well) were seeded into 24-well plates in 0.5 mL cell culture medium in which 13 mm diameter glass coverslips were deposited. The cells were left to adhere overnight by culture in a 37 °C humidified incubator supplied with 5% CO2.
4.7. Immunocytochemistry for bright-field and fluorescence imaging
Coverslip-adhered cells were fixed with 4% PFA in 100 mM Sorenson’s buffer, pH 7.6 for 20 min at 20 °C. The coverslips were then washed with PBS (3 × 0.5 mL, 5 min) containing 50 mM glycine (0.5 mL, 20 min) and cell plasma membranes were permeabilised with 0.1% Triton X-100 in PBS (0.5 mL, 5 min). Coverslips were soaked in PBS containing 10% (w/v) BSA for 1 h, washed with 0.2% acetylated BSA (BSA-c) in PBS (2 × 0.5 mL, 5 min) and then incubated with 6 to 10 nM of the conjugates in 0.2% BSA-c containing 10% FCS, 0.5 mL for 1 h. Next, cells were washed with 0.2% BSA-c (2 × 0.5 mL, 5 min) and with 80 mM citrate buffer pH 6.7 containing 2% sucrose (3 × 0.5 mL, 5 min). Gold was revealed by silver enhancement according to a modified Danscher protocol [42] using silver acetate and propyl gallate [43]. For fluorescence imaging, the primary antibodies were labelled using secondary fluorescent anti-mouse conjugates (AlexaFluor568 goat anti-mouse, Invitrogen A11031 and AlexaFluor488 goat anti-mouse, Invitrogen A11029). Fluorescence observations were carried out using a Leica DM5500B microscope equipped with an HCX PL Apo 63 × 1.40 oil PH3CS objective and a Leica DFC350FX camera.
4.8. Preparation of cells containing gold nanoparticle-antibody conjugates for bright-field microscopy imaging
After the indicated time periods following application of the electric pulses, the adherent cells cultivated on glass coverslips were washed with PBS (2 × 1 mL). The cells were fixed with 4.0% paraformaldehyde (PFA) (1 mL) for 20 min or 2% glutaraldehyde in 100 mM Sorenson’s buffer, pH 7.6 for 1 h. After removal of the fixative solution and 3 PBS washes (1 mL), the plasma membrane was permeabilised using 0.1% (w/v) saponin or Triton X-100 (1 mL, 15 min) in 0.1 M Sorenson’s buffer pH 7.6. The phosphate-buffered solution was replaced by a 0.1 M citrate solution, pH 6.7 containing 2% sucrose (5 washes, 1 mL). Development of AuNPs [42] was then done in a dark room for 30 min using freshly prepared 6 mM silver acetate solution in 0.16 M sodium citrate, pH 6.7 containing 20% gum arabic and 2 mM propyl gallate [43]. The silver-promoted enhancement of AuNPs was stopped with 0.16 M sodium citrate solution, pH 6.7 (1 mL, several washes). The glass coverslips were finally mounted onto 3 × 1 inch microscope slides (knittelglass.com) using Fluoromount-G (Southern Biotech, Ref. 0100-01, batch I2819-WC79B). Observations were carried out using a Leica DM5500B microscope equipped with an HCX PL Apo 63 × 1.40 oil PH3CS objective and a Leica DFC350FX camera.
4.9. Cellular specimen preparation for EM
20 h after the electroporation treatment, the adherent cells were washed with warmed (37 °C) Sorenson’s buffer (3 × 2 mL). The cells were then fixed with 2.0% glutaraldehyde (1 mL) for 24 h. They were then washed with TEM storage solution (5×, 1 mL). Enhancement of the gold particles with silver was performed using the SE-EM kit (Aurion, the Netherlands) according to the manufacturer’s protocol. The cells were then contrasted with 0.5% osmium for 20 min followed by 2% uranyl acetate both diluted in water. The specimens were dehydrated in a solution containing increasing concentrations of ethanol and flat-embedded in Epon (Ladd Research Industries, Williston, Vermont, USA). The resin-embedded specimens were sectioned into 100 nm thick slices which were deposited on an electron microscope grid.
4.10. TEM and image analysis
Cellular specimens were imaged on a Hitachi H7500 transmission electron microscope (Hitachi High Technologies Corporation) equipped with an AMT Hamatsu digital camera (Hamatsu Photonics). For EM imaging of 7G5AuPEG we used nickel EM grids covered with a thick holey carbon film (Quantifoil R2/2). The grids were topped with a thin continuous carbon layer and rendered hydrophilic by a 90s treatment in a Fischione 1070 plasma cleaner operating at 30% power with a gas mixture of 80% argon:20% oxygen. The 7G5AuPEG solution was diluted to a final concentration of 50 μg/mL in Buffer A (Tris 10 mM, pH 7.4, NaCl 50mM) and crosslinked for 30 s at a final glutaraldehyde concentration of 0.3%. Three μL of this solution were deposited on the glow-discharged EM grid and the particles were allowed to adsorb for 1 min. The grid was washed once on a drop of buffer A and negatively stained with a 2% uranyl acetate solution. The sample was then imaged in a Tecnai F20 (FEI) operating at 200 kV and equipped with a 2K CCD camera (Gatan). Micrograph montages were recorded at an image magnification of 50 kX with a 2.01 Å pixel size at −2 μm defocus using the SerialEM software [44]. For single particle analysis, micrographs were imported within the Scipion framework for image processing software [45] and Fourier band-passed filtered using the xmipp3 protocol [31]. Reference-free picking was performed using the eman2 protocol to generate a dataset of 80431 particles [46]. The particle set was aligned, masked, 2-D classified, cleaned to obtain a total of 36671 molecular images and classified again into 100 classes using xmipp3 protocols.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.