



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Fluorescence imaging has become a groundbreaking tool to study and image biological environments and processes, in domains ranging from medical applications to fundamental research in biology. In that context, a wide variety of fluorescent dyes and probes have been developed as tools for high contrast microscopy and cell imaging [1]. Ideal fluorescent probes should be bright, photostable and non-toxic. Additional key parameters are their absorption range and emission color. In terms of brightness, fluorescent organic dyes may be surpassed by luminescent nanoparticles [2, 3]. Indeed, in the last decades, inorganic nanoparticles have attracted growing attention in the field of biophotonics, in relation with various applications including new imaging modalities. Among them luminescent metal-, semiconductor- or oxide-based nanoparticles have been by far the most widely studied [4]. Luminescent metallic and semiconducting nanoparticles have been heralded as major nanomaterials due to their unique electronic and optical properties, giving rise to the blooming field of plasmonics in the case of metallic nanoparticles. In parallel, semiconductor quantum dots (QDs) have motivated many studies and have become extremely popular for bioimaging uses. Their luminescence properties can be tuned in the visible to the near-infrared (NIR) region by adjusting their size in relation with quantum confinement and they may show giant brightness [4] (both one-photon brightness [3]: up to 106 M−1⋅cm−1, and two-photon brightness [5, 6, 7, 8]: up to 104 GM). Yet, such luminescent inorganic nanoparticles suffer from several drawbacks: their inorganic core is inherently water-insoluble, requiring them to be coated by solubilizing agents (polymeric coating, surfactants, etc.) for use in bioimaging. In addition, several luminescent inorganic nanoparticles raise questions with respect to environmental issues (ecotoxicity, biodegradability) due to their content or composition (heavy metal or toxic components). In that perspective, fully organic nano-objects may offer interesting alternatives. The most widely used typically are polymeric such as dendrimers [9, 10, 11, 12], lipid-based [13] or “frozen” micelles systems where fluorescent probes are either encapsulated in [14, 15] or covalently linked [16] to the nanoparticle. In contrast, self-stabilized organic nanoparticles generated from the nano-aggregation of dedicated hydrophobic dyes in water represent a much less traveled route. Actually, this type of fully organic luminescent nanoparticles is often overlooked in the literature and some prominent reviews do not even mention them [3, 17]. Yet they offer promise for a wide range of applications, including biological applications [18, 19]. We propose to name these organic nanoparticles dFONs (for dye-based fluorescent organic nanoparticles) to distinguish them from the multitude of fluorescent organic nanoparticles, which otherwise create a somewhat fuzzy domain and some misunderstandings or confusion. We stress that we use the name dFONs to describe organic nanoparticles made only of pure dyes, requiring no stabilizers or additives, nor coatings to be made dispersible and stable in water or aqueous environments.
The ultimate molecular confinement of pure dyes may look like a counterintuitive approach as most of the time this generates aggregation-caused quenching of fluorescence (ACQ) due to symmetry reasons (in the case of π-stacking notably) or to competitive deactivation processes (such as photoinduced electron transfer (PET) favored by the close proximity of dyes within dFONs) that inhibit fluorescence. Yet by adjusting the electronic structure of the dye (to prevent PET) while adding suitable bulky substituents that hinder π-stacking, such effects can be circumvented. A supposedly similar strategy has been used extensively in the so-called “aggregation-induced emission” (AIE) [20] or aggregation-induced emission enhancement (AIEE) domain. In compounds designed for AIE, appending moieties preventing π-stacking and allowing for efficient non-radiative rotational deactivation processes in non-viscous environments are added to dyes, thus generating loss of fluorescence in solutions. When such dyes are placed in viscous solvents or in the solid state where rotations are restricted, their fluorescence is restored.
2. dFONs design: polar and polarizable dyes (PPDs) as dFONs building blocks
Key criteria for luminescent nanoparticles to be of interest for bioimaging purposes typically are large brightness for enhanced sensitivity (with the ultimate goal of detecting single emitters), photostability, colloidal stability as well as biocompatibility. In addition, for in vivo bioimaging in tissues or small animals, they should preferably absorb and emit light above 650 nm—i.e., within the biological transparency window(s) (650–950 nm and 1000–1200 nm) [21]. In that perspective, two-photon excitation has attracted much interest giving rise to two-photon laser scanning microscopy (TPLSM) [22], which has nowadays become a popular fluorescence bioimaging technique. Two-photon absorption (2PA) is a nonlinear optical phenomenon defined by the simultaneous absorption of two low-energy photons [23] by an excitable compound (Scheme 1). The wavelength used for two-photon excitation is thus favorably redshifted as compared to that used for one-photon excitation and allows for instance the replacement of excitation in the near UV–blue visible region by excitation in the deep red–NIR-I region. Dyes which have much higher two-photon absorption cross-sections (σ2) than endogenous chromophores (typically orders of magnitude larger, i.e., over 103 GM) are thus required not only to allow deeper imaging in biological tissues thanks to reduced scattering, but also to significantly reduce background fluorescence and provide more selective excitation compared to standard one-photon excitation [24]. In that perspective, we were interested in the design of biocompatible dFONs combining tunable emission, giant brightness (both for standard excitation, i.e., confocal fluorescence microscopy, and TPLSM), excellent colloidal and photo-stability in bioenvironments, aiming at various imaging modalities (including single particle tracking and two-photon imaging).
Two-photon excited fluorescence: principles and advantages in bioimaging.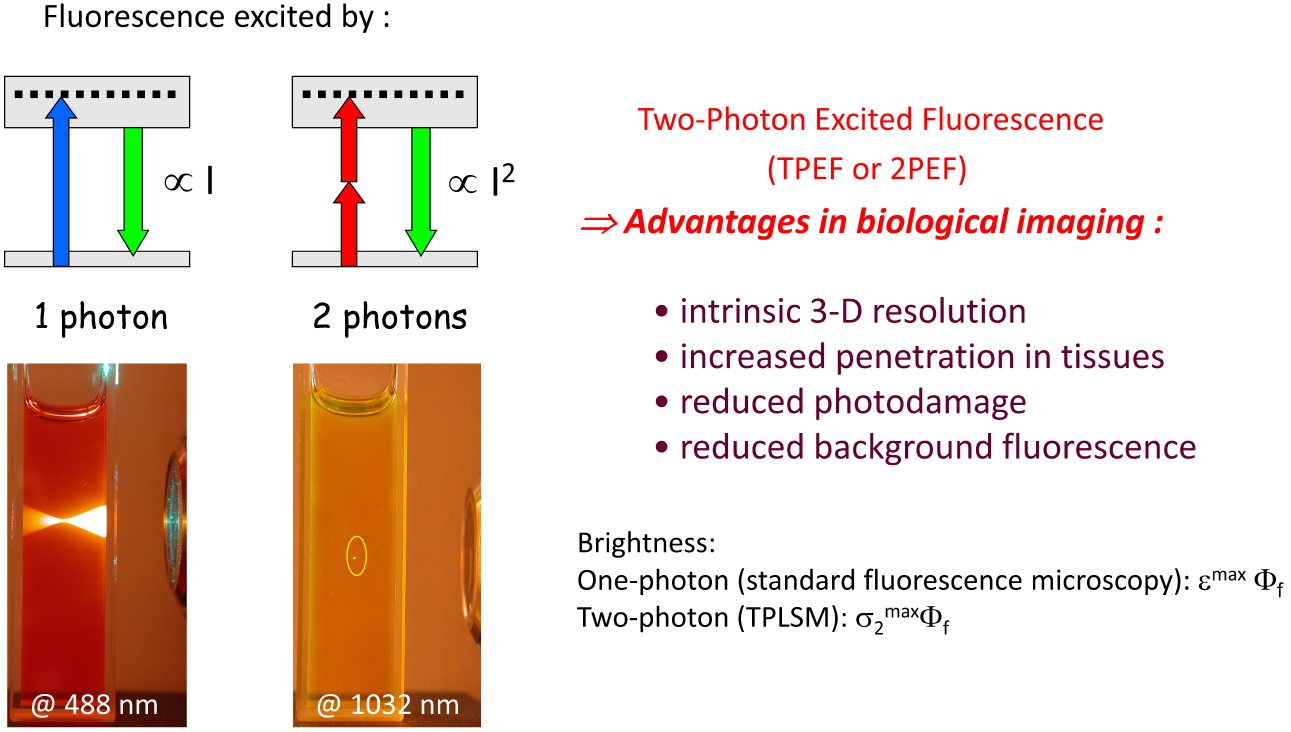
Two main strategies are used to prepare dFONs directly as a colloidal suspension in water (or aqueous media): (i) laser-assisted photofragmentation of a suspension of micro-crystals in water [25] or (ii) nanoprecipitation [26, 27] (Scheme 2). In the latter, a minute amount of a stock solution of the dye dissolved in a water-miscible organic solvent (typically THF, ethanol, acetone, DMSO …)—which acts as the good solvent for the hydrophobic dye—is added in a large volume of pure water (or buffer etc.) under either magnetic stirring or sonication. For bioimaging applications, no more than 1% (v/v) of organic solvent should be used and the residual amount can be eliminated by evaporation under vacuum or dialysis. As the good (organic) solvent for the hydrophobic dyes is dispersed in the bad solvent (water), dyes aggregate to form clusters then nanoparticles [26]. If nanoparticles are indeed formed, a colored (thanks to the dispersion of hydrophobic dyes in water) and non-turbid (thanks to the nanometric size of organic aggregates) aqueous suspension is obtained. We stress that this process is a kinetically controlled step contrary to crystallization and that hydrophobic interactions play a definite role as water is used to conduct nanoprecipitation. Hence, the organization of dyes within dFONs might be different from that in the crystal obtained by a thermodynamically controlled slow process. dFONs prepared by nanoprecipitation usually have a spherical shape. We emphasize that the colloidal stability of such dFONs suspensions is a major issue in view of their usefulness: their natural fate is to agglomerate, then deposit upon evolution towards the low-energy thermodynamic state. The nature of the dyes as well as their self-organization are extremely important in that respect as the kinetics of these processes may be rendered extremely slow depending on the type of dye and surface properties of dFONs.
Typical nanoprecipitation protocol for the preparation of dFONs.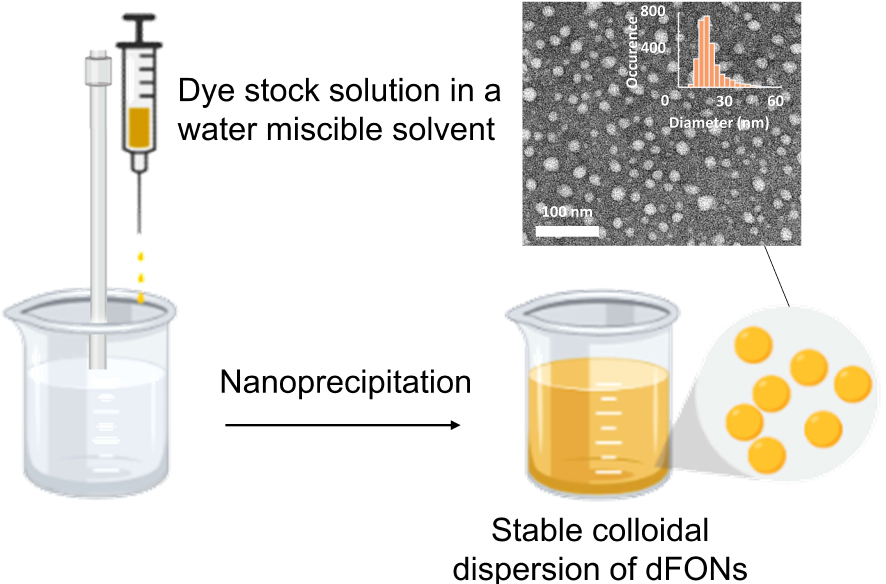
To achieve dFONs that meet the requirements mentioned above for bioimaging, we developed a successful bottom-up strategy based on the molecular engineering of specific chromophores, namely Polar and Polarizable Dyes (PPDs) as dFONs building blocks. Such dyes bear electron-releasing (D) and electron-withdrawing (A) moieties or substituents that interact via π-conjugated linkers, giving rise to intramolecular charge transfer(s) (ICT), i.e., electronic redistribution from the D to the A moieties upon excitation. The ICT phenomenon is responsible for the strong absorption of PPDs as well as for their unique nonlinear optical properties, especially two-photon absorption [28, 29, 30, 31]. PPDs may have different symmetry including dipolar, quadrupolar, or octupolar dyes (Figures 1–2) [32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42].
Examples of dipolar, octupolar and bis-dipolar dyes developed in the team for the bottom-up molecular engineering of bright dFONs for bioimaging. Adapted from Refs. [32, 33, 34, 35, 38, 40, 42].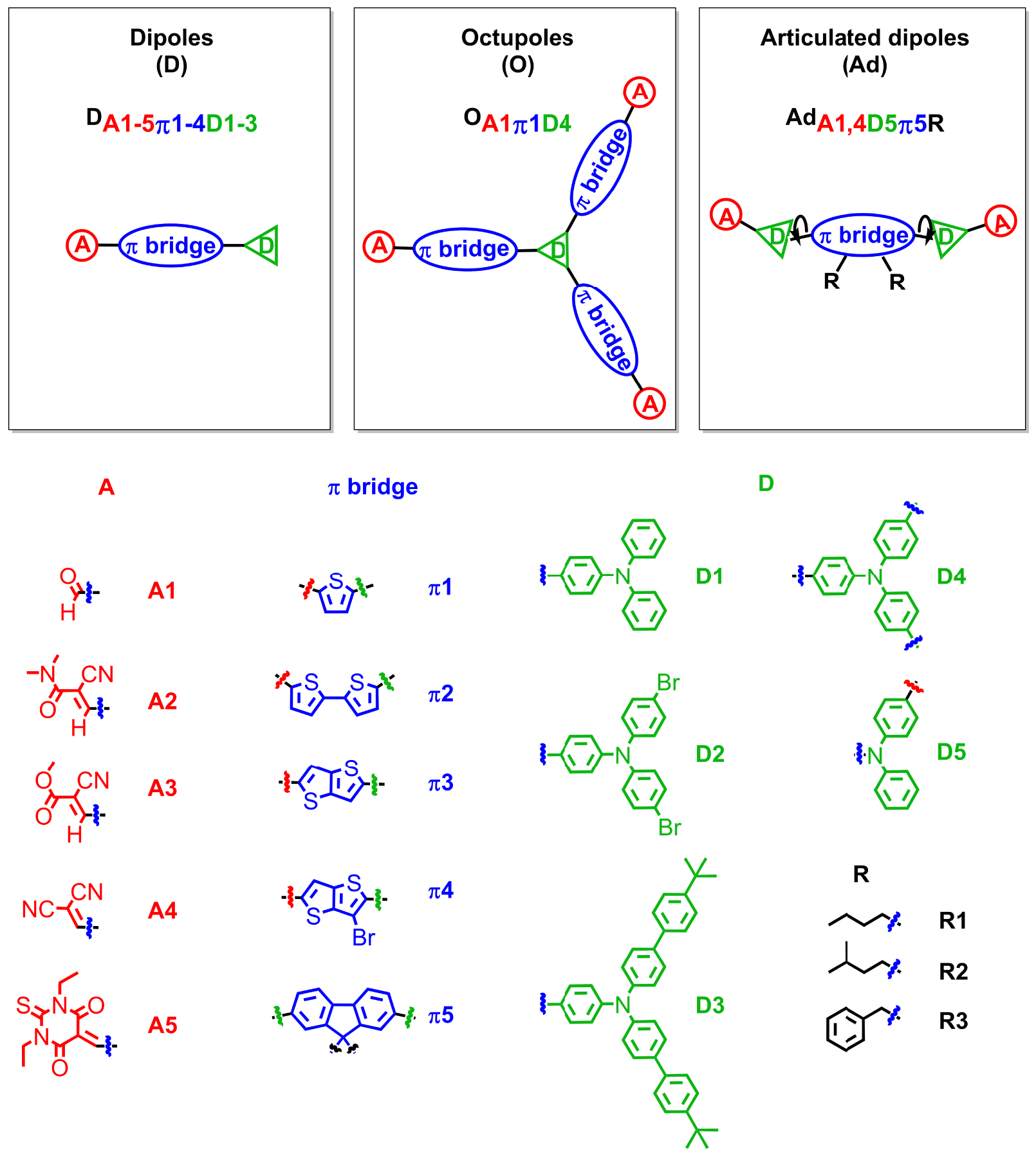
Examples of quadrupolar dyes developed in the team for the bottom-up molecular engineering of bright dFONs for bioimaging. Adapted from Refs. [36, 37, 39, 40, 41].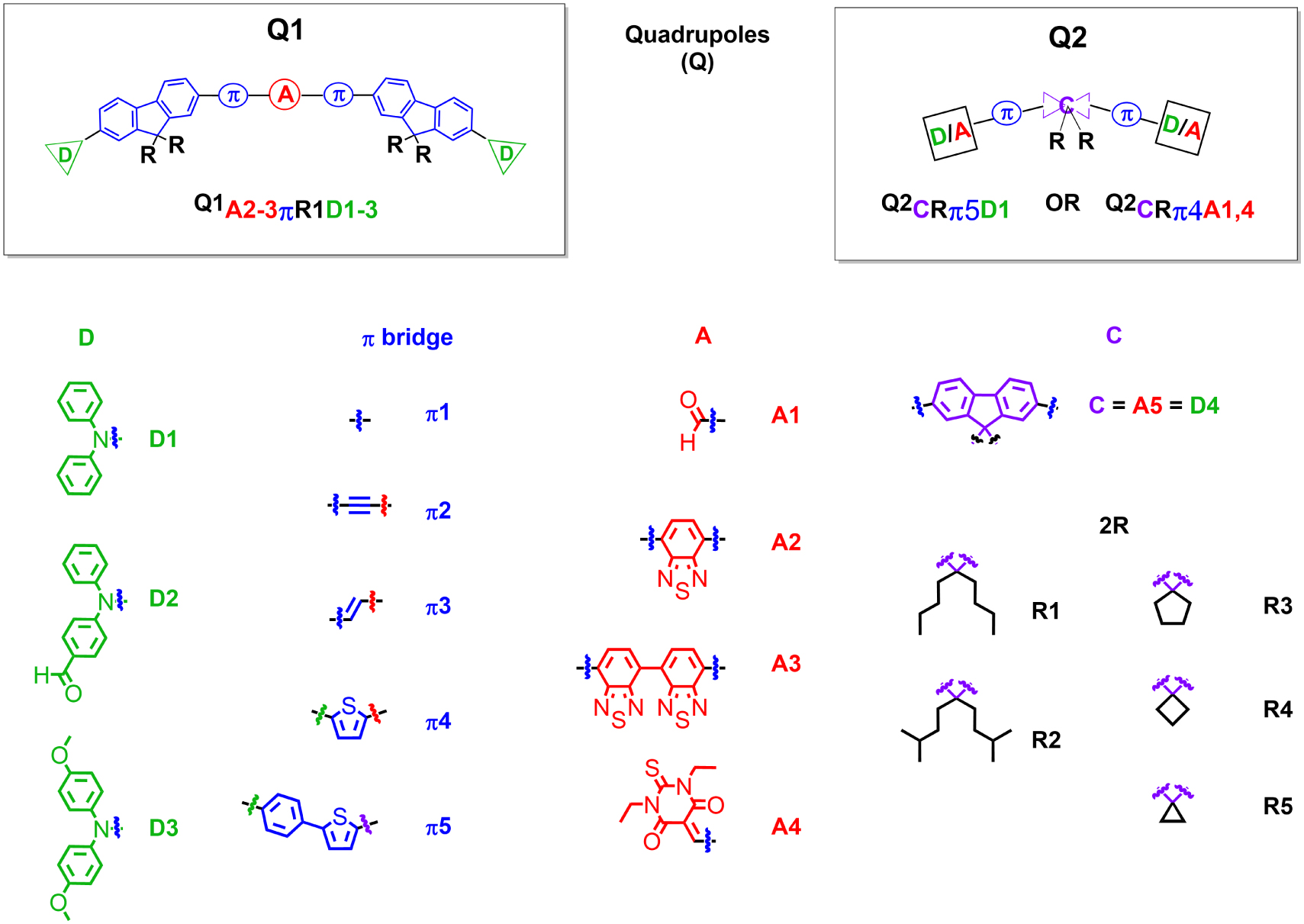
The presence of dipolar subunits in those dyes has strong implications that explain the specificity and peculiarities of dFONs made from PPDs:
- The dipolar interactions between dyes are strong and longer-ranged (∝1∕r3) than Keesom interactions (∝1∕r6) due to the dyes’ immobilization and ultimate confinement within dFONs. These electrostatic interactions play a crucial role in the structural cohesion of dFONs and explain their structural stability though there is no covalent bonding between dyes, contrary to polymeric organic particles.
- The dipolar interactions also play a major role in the self-organization of PPD dyes within dFONs during the nano-aggregation process [43, 44]. In that respect, molecular engineering of PPDs is crucial to prevent antiparallel stacking of dipolar dyes or π-staking of quadrupolar dyes [45] that are deleterious to fluorescence. To prevent such processes, we have used dedicated appending moieties such as diphenylamino end-groups (which have a non-planar propeller shape and are also electron-releasing moieties), as in dipolar and quadrupolar dyes shown in Figures 1 and 2. Alternatively, conjugated linkers (or cores) including fluorenyl moieties bearing alkyl substituents that extend above and below the plane of the π-conjugated system have been used. These substituents act as bulky spacers of tunable size, as in bis-dipolar and quadrupolar dyes shown in Figures 1 and 2. The geometry of the dyes, as well as the position and size of the bulky appending moieties, are very important in tuning their self-organization upon aggregation and offer a powerful handle for bottom-up engineering of the dFONs’ fluorescence properties (Figure 3).
- The dipolar subunits create strong local electric fields in their close vicinity. As a result, due to the intrinsic high polarizability of PPDs, these local electric fields may change the polarization of the proximal dyes resulting in a modification of their electronic distribution (and polarization) and subsequently of their optical properties (i.e., modification of the electronic gap [46, 47, 48, 49, 50, 51]) and even more markedly nonlinear optical properties [30, 52, 53]. Hence, depending on the self-orientation of the dipolar subunits within dFONs and on the distance between PPDs dyes (which can be tuned by adjusting the geometry of the dyes as well as the bulkiness and position of substituents—Figure 3), sizeable variations in the optical properties can be expected for dyes with similar optical (and NLO) properties in solution as a result of confinement effects.
- Finally, the nature of the PPDs may significantly influence their orientation at the surface of dFONs resulting in short-range order [54]. We stress that almost all push–pull dyes that we investigated bear electron-withdrawing end-groups that are susceptible to favor electrostatic interaction or H-bonds with water molecules surrounding the surface of the dFONs. Such orientations may result in large surface potentials (as in lipid membranes), emphasizing the specificity of dFONs made from PPDs in terms of the nature and properties of their surface.
Tuning the organization of dipolar (arrow) and quadrupolar (double arrow) dye subunits in dFONs and its impact on fluorescence intensity.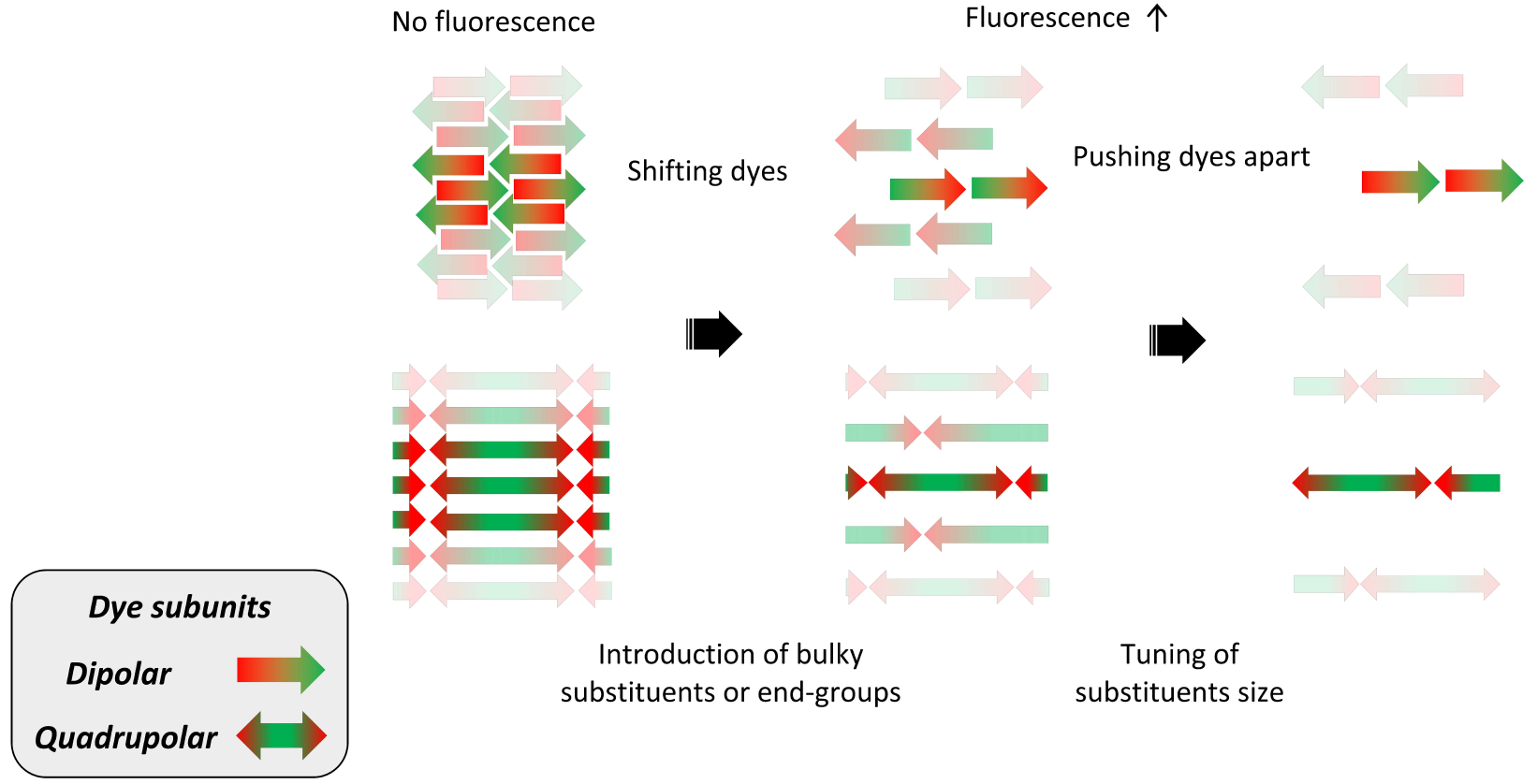
3. Bottom-up engineering of optical properties
In view of optical bioimaging, “redder is better” due to improved penetration depth and easier detection. In that perspective, we have been keen on red-shifting the absorption and emission of dFONs (bathochromic shift) and increasing their one- and two-photon response (hyperchromic effect) as well as their one- and two-photon brightnesses.
3.1. Adjusting the strength of D and A moieties
A classical strategy involves the increase in the D/A strength or the extension of the π-linker. Such a strategy is illustrated in Figure 4A for dipolar dyes. We note that a bathochromic shift is observed both in solution and in dFONs when increasing the strength of the donor or acceptor. A hyperchromic shift is also observed, such that for dFONs of similar size (i.e., same amount of dye subunits), the ones with a stronger donor or acceptor have a larger absorptivity. A bathochromic shift of the emission is also observed such that dFONs made from dipolar dyes DD3π2A1 and DD3π2A5 are respectively red–orange and NIR-I emitters that show huge brightnesses in water, larger than those of QDs without the need for surfactants or coating. We also note the effect of the size of substituents on fluorescence intensity: dFONs made from DD3π2A1 have an almost twice larger quantum yield than dFONs made from DD1π2A1, emphasizing the role of substituent size on the organization of PPDs within dFONs (Figure 4A).
Effect of donor and acceptor end-group strength and 𝜋-linker length on the optical properties of dFONs made from dipolar (“push–pull”) dye (A: top) and quadrupolar dyes (B: bottom). Adapted from Refs. [32, 34, 35] and [40, 41].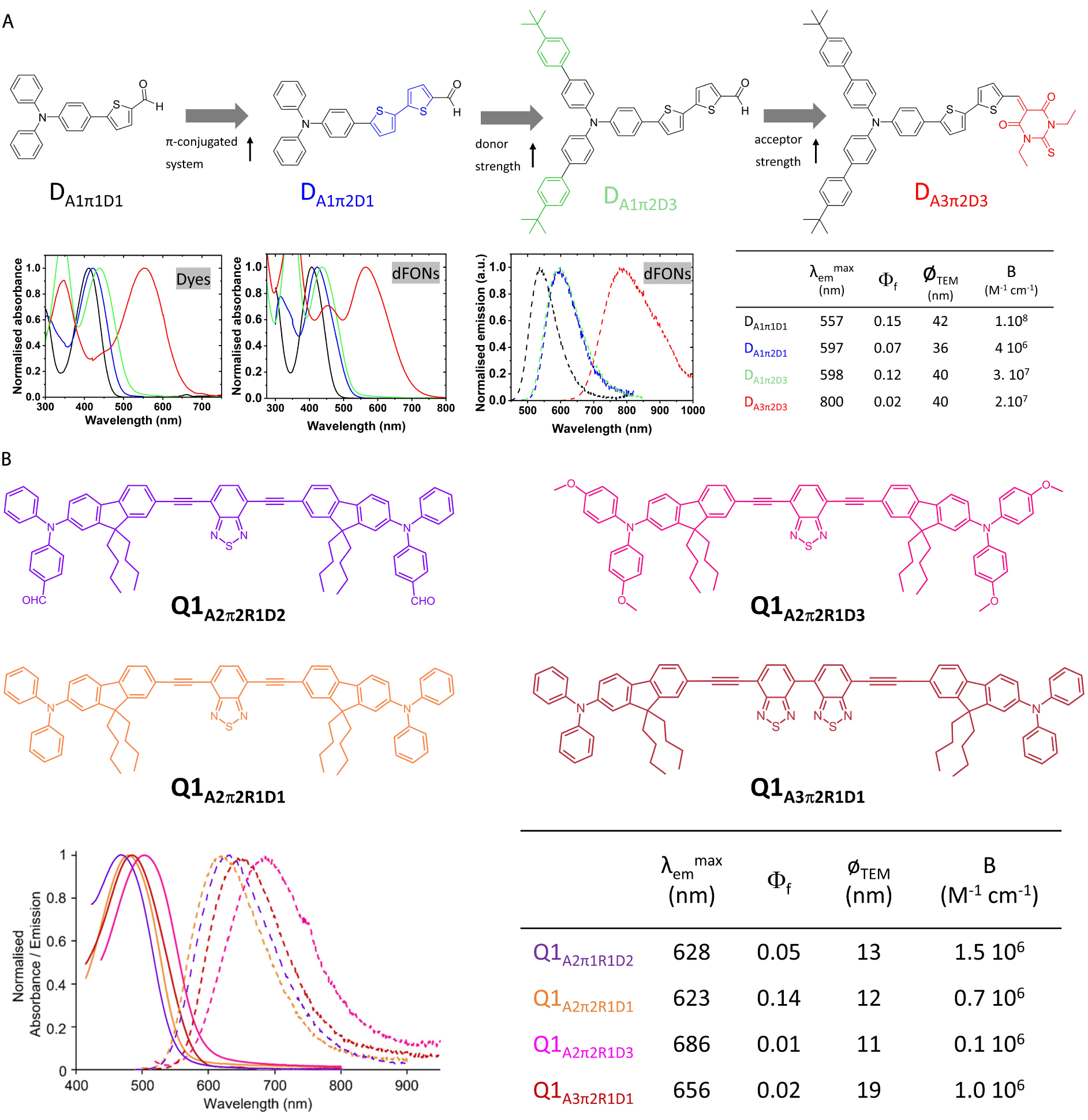
Subtle tuning can be achieved by tweaking the donor strength or the acceptor strength both for dipolar [33, 40] and quadrupolar dyes [41] (Figure 4B).
3.2. Adjusting the π-linker’s nature and length
Similarly, a hyperchromic and bathochromic shift can be achieved by adjusting the length and nature of the π-linkers as illustrated in Figure 4A for dipolar dyes and in Figure 5 for quadrupolar dyes [37, 40].
Effect of π-linker length and structure on the optical properties of dFONs made from quadrupolar dyes. Adapted from Ref. [37].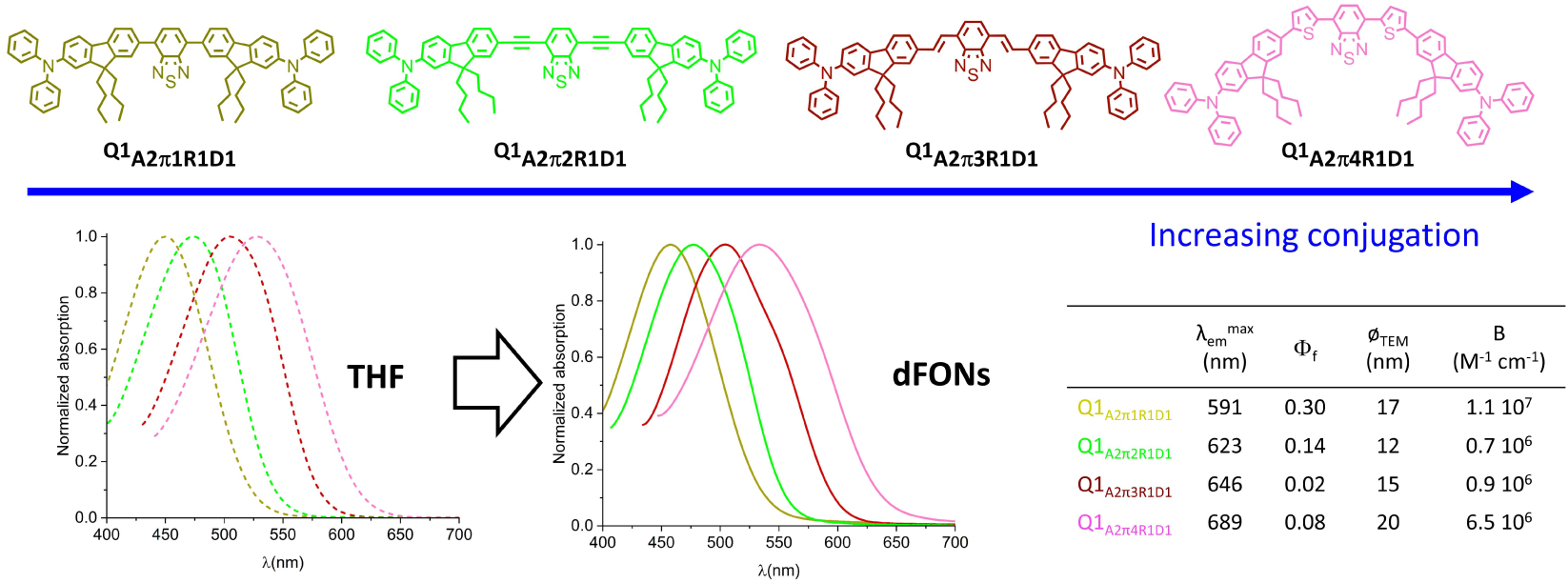
We note that similar trends are observed for dyes in solution and dFONs. On the other hand, the observed changes in fluorescence quantum yield emphasize the impact of dye size and geometry on dye packing within dFONs in the case of quadrupolar dyes. Indeed dFONs made from Q1A2π3R1D1 show a much lower fluorescence quantum yield than dFONs made from Q1A2π1R1D1 and Q1A2π2R1D1 and even than dFONs made from Q1A2π4R1D1 although the latter are significantly redshifted (Figure 5). As a result, dFONs made from quadrupolar dyes Q1A2π1R1D1 and Q1A2π4R1D1 are respectively orange–red and deep red emitters that exhibit a very large brightness.
3.3. Adjusting the geometry of the PPDs and the size of substituents to tune packing and fluorescence properties
Not only may the geometry of the PPDs significantly influence the fluorescence quantum yield (and thus affect the brightness) of dFONs, but it can also be used to change the emission color by controlling and exploiting the local environment created by local dipoles within dFONs. Indeed, whereas dipolar DA1π1D1 and related octupolar OA1π1D4 PPDs have identical fluorescence properties in solution (fluorescence quantum yields and lifetimes) and show the same emission solvatochromism (blue emission in toluene, green emission in chloroform and orange emission in DMSO), dFONs made from DA1π1D1 are green emitters whereas dFONs made from OA1π1D4 are orange emitters: the three-branched geometry of OA1π1D4 imposes a different packing from the rod-like DA1π1D1, generating a stronger local electric field such that OA1π1D4 dyes experience a larger polarity within dFONs (similar to DMSO) and its emission is redshifted [32].
Another way to influence the packing of PPDs and tune their luminescence properties is to adjust the size of the bulky substituents. A striking example is given in Figure 6 for a series of quadrupolar dyes built from a fluorenyl core. All dyes bear the same electron-withdrawing end-groups and π-linker, the only difference being the nature of the substituents; cyclopentyl, cyclobutyl and cyclopropyl for dyes with a spirofluorene core or two isopentyl chains for dyes with a bis-alkyl fluorene core. Whereas all quadrupolar dyes show identical optical properties in solution, dFONs fluorescence is strongly affected [55]. Decreasing the size of the cycle in spirofluorenyl derivatives generates a redshift of the dFONs fluorescence, allowing one to tune the dFONs fluorescence from blue (cyclopentyl), to cyan (cyclobutyl) and to red–orange (cyclopropyl). This illustrates the effect of subtle tuning of the side-chain size on the packing of the dyes and thus on fluorescence. We note that the quadrupole with two isopentyl chains leads to dFONs whose fluorescence is both redshifted (green emission) compared to its spiro (cyclopentyl) analog and enhanced by a factor of 2, demonstrating the effect of substituent size and bulkiness on dye packing and its consequences on optical properties.
Influence of the substituents on the fluorenyl core of quadrupolar dyes on the fluorescence emission of corresponding dFONs in water.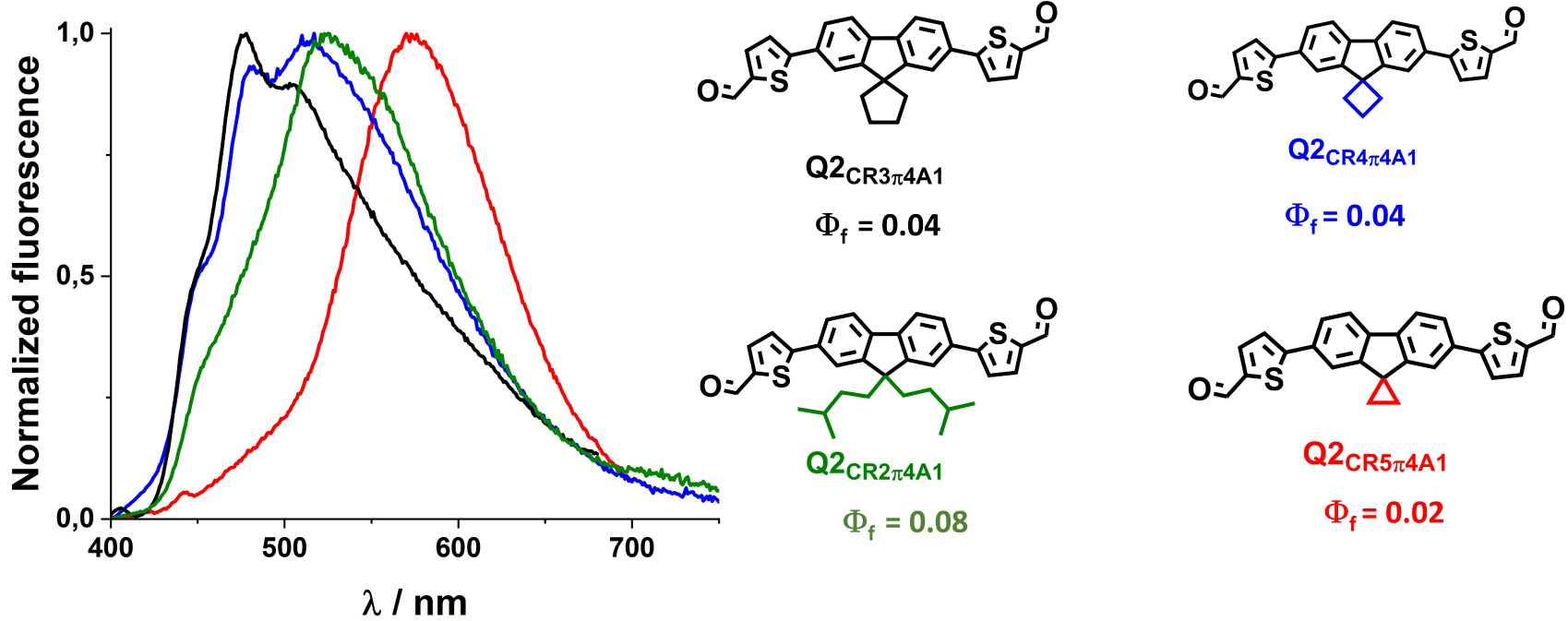
3.4. Enhancing two-photon absorption. Effects of confinement
The two-photon absorption of dipolar and quadrupolar dyes can be modulated by molecular engineering, adjusting the donor and acceptor groups as well as the length and nature of the π-conjugated linker [28, 29, 31, 52]. Increasing the strength of the donor or acceptor groups in dipolar thienothiophene [33] or bisthienyl [40] dyes allows one to significantly increase their response in solution as illustrated in Figure 7A. Yet, the molecular confinement of dipolar dyes within dFONs has a major effect on their 2PA response; either a major decrease (by a factor of 3 or 2 for DA4π3D1 and DA4π3D3) or an increase (DA1π3D1 and DA4π3D2) of the 2PA cross-section of the dipolar dyes is observed upon confinement within dFONs (Figure 7A). This phenomenon can be ascribed to electrostatic interactions between dipolar and polarizable dyes depending on their relative orientation and proximity [11, 30]. The striking difference between the 2PA response of dyes DA4π3D1 and DA4π3D2 into dFONs illustrates the positive role of the Br substituent in tuning the packing and allowing a cooperative enhancement of the 2PA [11], as is also the case for DA1π3D1 (Figure 7A). Although the fluorescence quantum yields of NIR-I-emitting dFONs made from dipolar dyes DA4π3D2 and DA4π3D3 remain low in water, their large 2PA response and number of dye subunits in dFONs lead to a very large two-photon brightness at 1070 nm (Figure 7A). As such, they provide interesting nanoprobes for NIR-II to NIR-I bioimaging in living tissues.
Molecular engineering of the two-photon absorption of dFONs made from dipolar [33] (A: top) or quadrupolar [37] (B: bottom) dyes.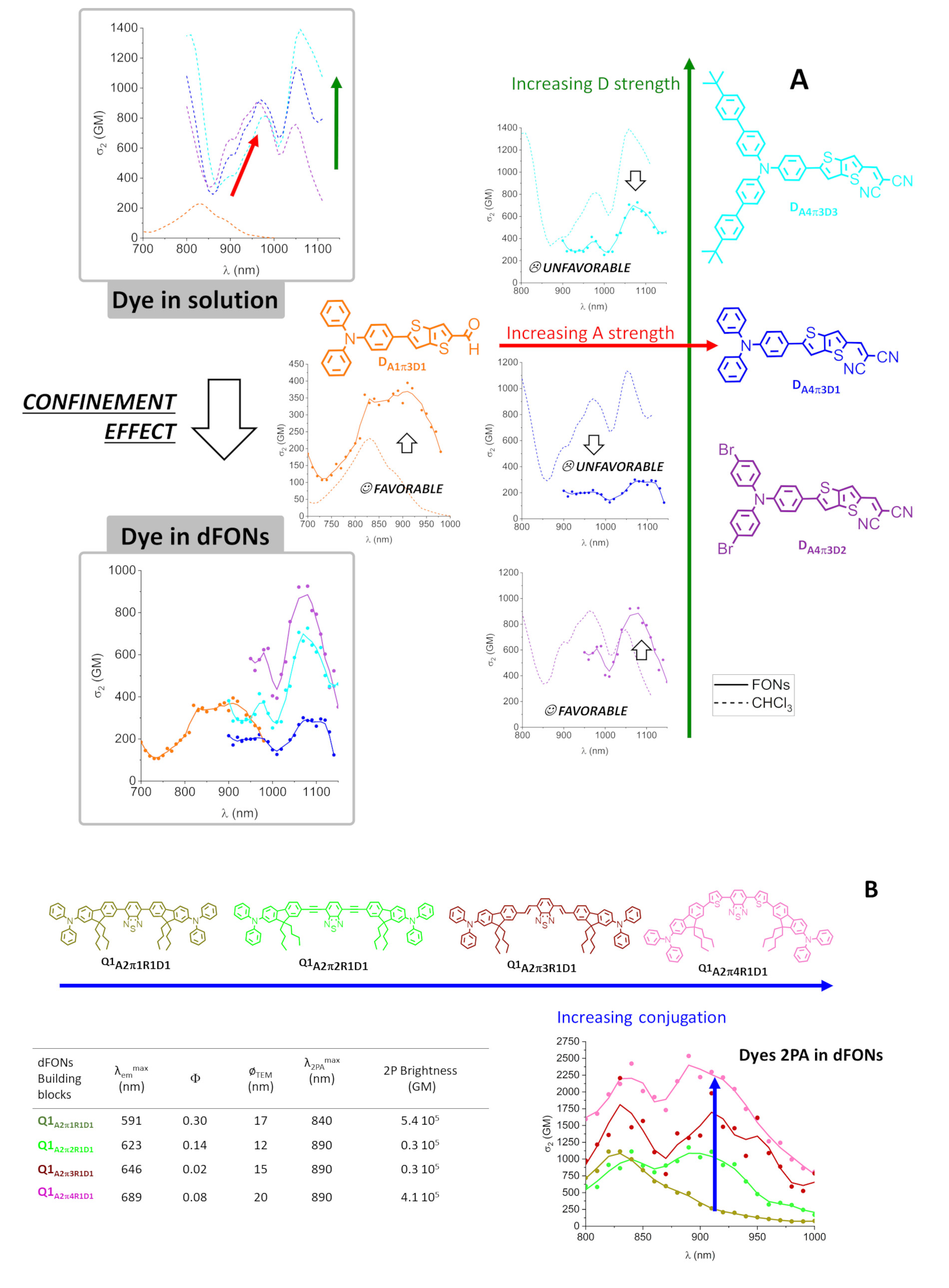
The increase in length leads to a redshift of the 2PA as was the case for the 1PA, but it also induces a major increase in the two-photon absorption response of the quadrupolar dyes, both in solution and as dFONs building blocks. (Figure 7B). The nature of the elongating moieties (ethynyl, vinyl or thienyl) influences not only the fluorescence quantum yield and the emission color (see Section 3.2) but also the 2PA response: an enhancement is observed upon increased conjugation [37]. Interestingly, in all cases, confinement does not reduce the 2PA response of the dyes but rather leads to broadening of the 2PA spectrum [37]. Thanks to the combined variation of the 2PA responses and of the fluorescence quantum yields values, the orange–red and deep red emitting dFONs made from Q1A2π1R1D1 and Q1A2π4R1D1 also show the largest 2P brightness. As such, they represent bright nanoprobes for NIR-I to red bioimaging [37].
3.5. Enhancing brightness by increasing size
The brightness of dFONs made from PPDs can be enhanced simply by increasing the size of the dFONs, as is the case for QDs. This can be achieved by adjusting the nanoprecipitation conditions, notably the dye concentration of the stock solution in the organic solvent [27]. Contrarily to QDs, this does not lead to a change of the luminescence properties (emission spectrum, fluorescence quantum yield) as shown in the case of dFONs made from dipolar dye DA1π2D1 [35] and quadrupolar dye Q1A2π1D1 [36]. Hence, dFONs with a similar emission color but different sizes (and thus brightnesses) can be obtained from PPDs, as can dFONs with a similar size but different emission colors [32, 35, 37].
4. Bottom-up engineering of surface properties
4.1. Molecular engineering of dFONs stability
Strikingly, molecular engineering of the PPDs also allows tuning of the surface properties of dFONs. dFONs made from PPDs show good colloidal stability in relation with large surface potentials. This kinetic stability can be further enhanced by adjusting the structure of the PPDs. As illustrated in Figure 8, increasing the size of the π-conjugated linker [35] or adding bulky substituents on the donor end-group [33] or the nature of the acceptor end-group [34] of dipolar dyes allow increasing the colloidal and structural stability of dFONs. The evolution of the fluorescence quantum yield values over time is of major importance for the usability of dFONs for bioimaging but also the signature of the structural stability of dFONs made from dipolar dyes: the decrease in fluorescence over time is related to the exchange of dyes from the surface to the bulk water back to the surface, leading to surface defects that are responsible for fluorescence quenching. Hence, the larger the interaction energy of the dyes with the surface (i.e., the energy required to extract a surface dye from the surface to the bulk), the slower the process [54]. Therefore stronger electrostatic interchromophoric interactions within dFONs and addition of hydrophobic substituents allow improving their colloidal and structural stability. As a result, dFONs made from DA5π2D3 show very high colloidal and structural stability [34].
Molecular engineering of the colloidal and structural stability of dFONs made from dipolar dyes. Adapted from Refs. [33] and [35].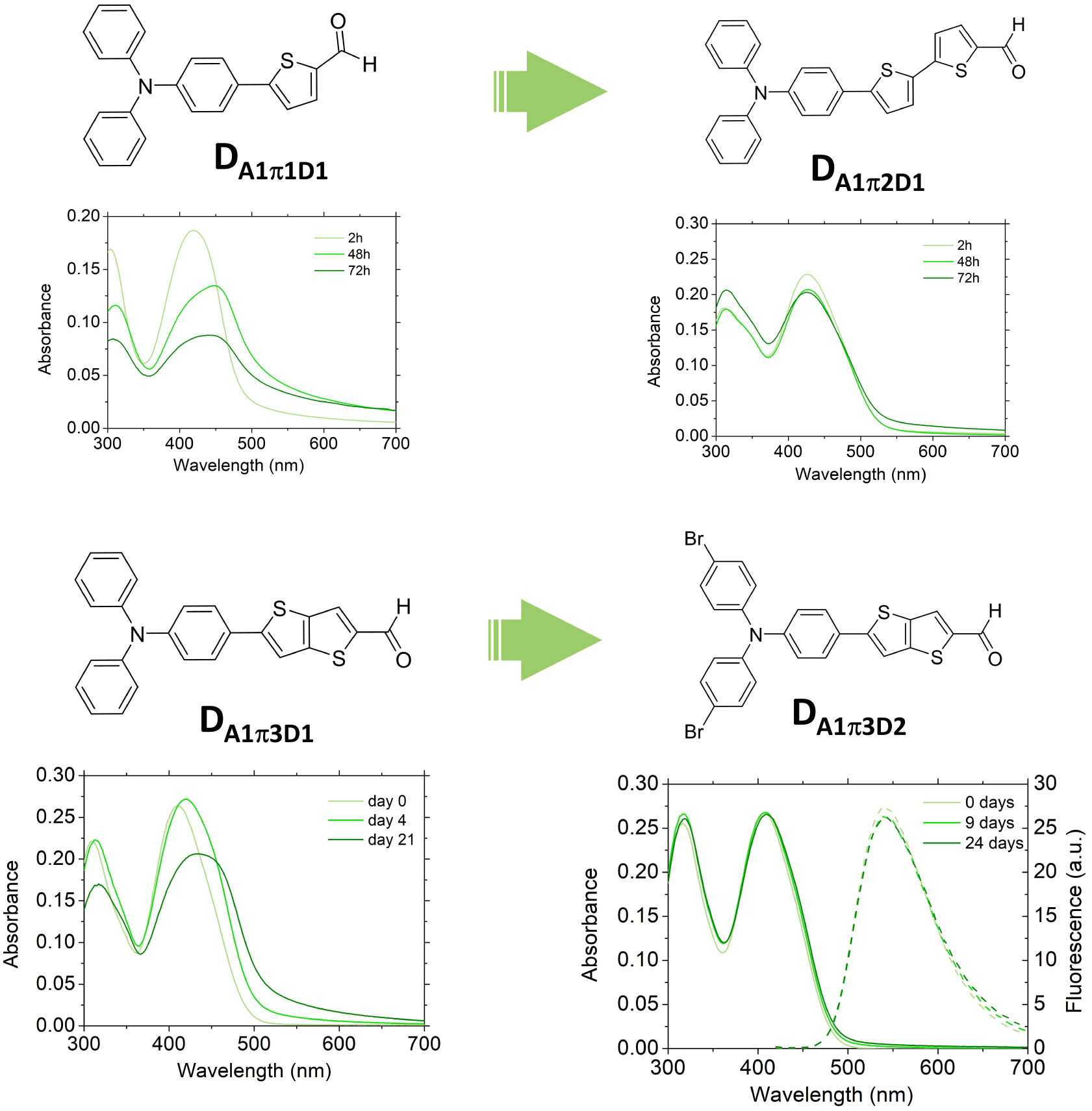
4.2. Molecular engineering of dFONs cell internalization and stealthiness
Strikingly, the interaction of dFONs with cell membranes can also be “bottom-up-engineered” by adjusting the structure of the PPDs. dFONs made from the dipolar dyes DA5π2D3 were found to be non-toxic and to internalize within cells while retaining their structural integrity [34]. In contrast, articulated dipoles (AdA1D5π5R, Figure 1) bearing two long hydrophobic chains were found to show no non-specific interactions with cell membranes, exhibiting an intrinsically stealth behavior [42]. Hence, such dFONs made only of pure PPDs do not require specific coatings (PEG or zwitterionic) to be made stealth. As such, thanks to their high brightness and intrinsic stealth character, they hold promise for imaging of specific receptors in cells once surface-functionalized with specific targeting moieties.
Similarly, dFONs made from quadrupolar dyes built from a spiroflurenyl core (Q2CR3π5D1 and Q2CR3π4A4) were found to internalize within cells [39] while retaining their structural integrity. In contrast, quadrupoles Q1A2π1R1D1 [36], Q1A2π2R1D1 [37], Q1A2π2R1D2 [41], Q1A2π21RD3 [41] and Q1A2π4R1D1 [37], with a rigid elongated conjugated system bearing four hydrophobic alkyl chains were found to be non-toxic and to exhibit a stealth behavior. Interestingly Q1A2π3R1D1 does not show a stealth behavior indicating that both the presence of several lipophilic chains and the nature of the π-conjugated system are decisive in controlling the nano-bio interface [37, 41].
Furthermore, thanks to their highly negative surface potential, these dFONs can be surface-coated by a polycationic polymer, resulting in a reversal of the surface potential. This change in surface potential triggers the internalization of the dFONs within cells, opening a promising route towards innovative drug delivery protocols [37].
5. Bioimaging applications (in vitro and in vivo)
The unique combination of high colloidal stability, small size, giant brightness and tunability of dFONs made from PPDs makes them ideal candidates for bioimaging in dilute conditions (i.e., nM range).
5.1. Two-photon angiography in small living animals [32]
As discussed earlier, the molecular design of PPDs is crucial for dFONs properties with consequences on bioimaging applications. A typical case is the difference between dFONs made from dipolar dye DA1π1D1 and from its three-branched (octupolar) version OA1π1D4 when used as a contrast agent for angiography [32]. Both dFONs show a very large two-photon brightness (106 and 2 × 105 GM respectively for dFONs of about 35 nm in diameter) [32], one being a green emitter and the other an orange emitter. These dFONs were injected in the blood circulation of living Xenopus laevis tadpoles. The tail of the tadpole was then imaged by two-photon-excited fluorescence (TPEF) 20 min and 90 min after injection. Both dFONs allow the high contrast and high resolution visualization of blood vessels in the tiny animal (Figure 9) by two-photon excitation in the NIR region (820 nm) and detection of the green or red–orange fluorescence emitted by dFONs made from DA1π1D1 or OA1π1D4 dyes. Thanks to their nanoscale, the dFONs do not permeate through the walls of the blood vessels, thus allowing their imaging with high resolution. Furthermore, no toxicity is observed for the dFONs made from DA1π1D1. Yet, this is no longer true for the dFONs made from OA1π1D4, which quickly agglomerate into large micro-agglomerates resulting in a deadly clogging of the blood vessels. Hence, not only does the geometry of the dye influence the optical properties of dFONs (see Section 3.3), but it also dramatically impacts the nature of their nano–bio interface and consequently their usability as a contrast agent for angiography by TPEF.
Two-photon angiography in small animals. Left, 3D projection Two-Photon Excitation Fluorescence (TPEF) and SHG (λ2P @820 nm) image 90 min after injection of dFONs made from DA1π1D1. Right, 3D projection TPEF (λ2P @820 nm) image 90 min after injection of dFONs made from OA1π1D4. For all images, the red color corresponds to the endogenous SHG signal (second harmonic generation at 410 nm) that arises from muscle tissue, while the green corresponds to the TPEF signal generated in vivo by the dye (460–590 nm). Scale bar: 100 μm. Adapted from Ref. [32].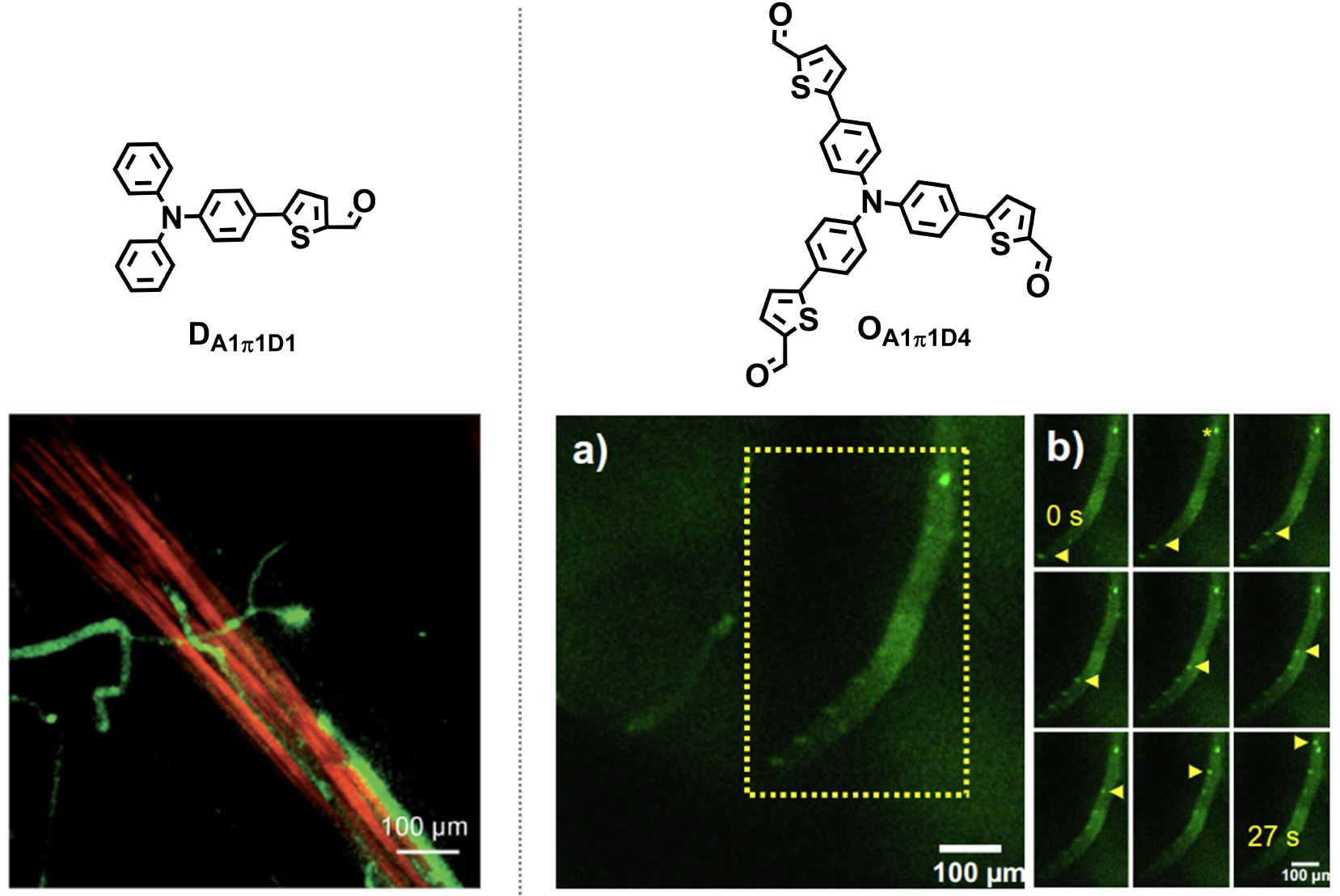
5.2. Single-particle tracking in solutions, cells and in the brain
Single-particle tracking (SPT) is a class of techniques that allow one to dynamically follow the fate of single molecules in living tissues. In this respect, QDs are the most widely used nanoparticles thanks to their unprecedented brightness and photostability. Due to the giant brightness of dFONs made from PPDs (similar or higher than those of QDs), they represent promising candidates for SPT and their usability for SPT in aqueous solutions was investigated. A critical parameter in that respect is their photostability. Organics are recognized to be much less photostable than inorganics. Yet depending on the structure of the PPDs, we found that the brightness of dFONs made from dipolar (DA5π2D3) or quadrupolar (Q2CR3π5D1 and Q2CR3π4A4) dyes allows performing single particle tracking with adequate [34], good [39] to excellent photostability (up to 3 min) [39]. The reconstruction of the trajectories of single particles gives access to the hydrodynamic diameter of the nanoparticles. The hydrodynamic size of dFONs made from articulated dipole-based (AdA1D5π5R1, AdA1D5π5R2 and AdA1D5π5R3) was found to be similar to the dry diameter measured by transmission electronic microscopy [42]. Interestingly, SPT experiments conducted in saline aqueous buffer containing proteins (i.e., PBS with fetal bovine serum) reveal that the hydrodynamic size of articulated dipole-based (AdA1D5π5R1, AdA1D5π5R2 and AdA1D5π5R3) dFONs increases by ∼30–40% (Figure 10) as compared to that determined in pure water [42]. Such an increase reveals the formation of a protein corona at the surface of the nanoparticles, thus preventing aggregation of dFONs despite the ionic strength of the solution. The presence of such a corona is definitely an advantage for bioimaging in live cells when dFONs are used as tracers.
Single-particle tracking of dFONs made from DA5π2D3, AdA1D5π5R1 and AdA1D5π5R3 in water. Single-particle tracking of dFONs made from DA5π2D3 (left), AdA1π5D5R1 (middle) in water and AdA1π5D5R3 (right) in phosphate buffer saline (with 10% of fetal bovine serum). Examples of traces of single particles with corresponding histograms of hydrodynamic diameter are given. Adapted from Refs. [34] and [42].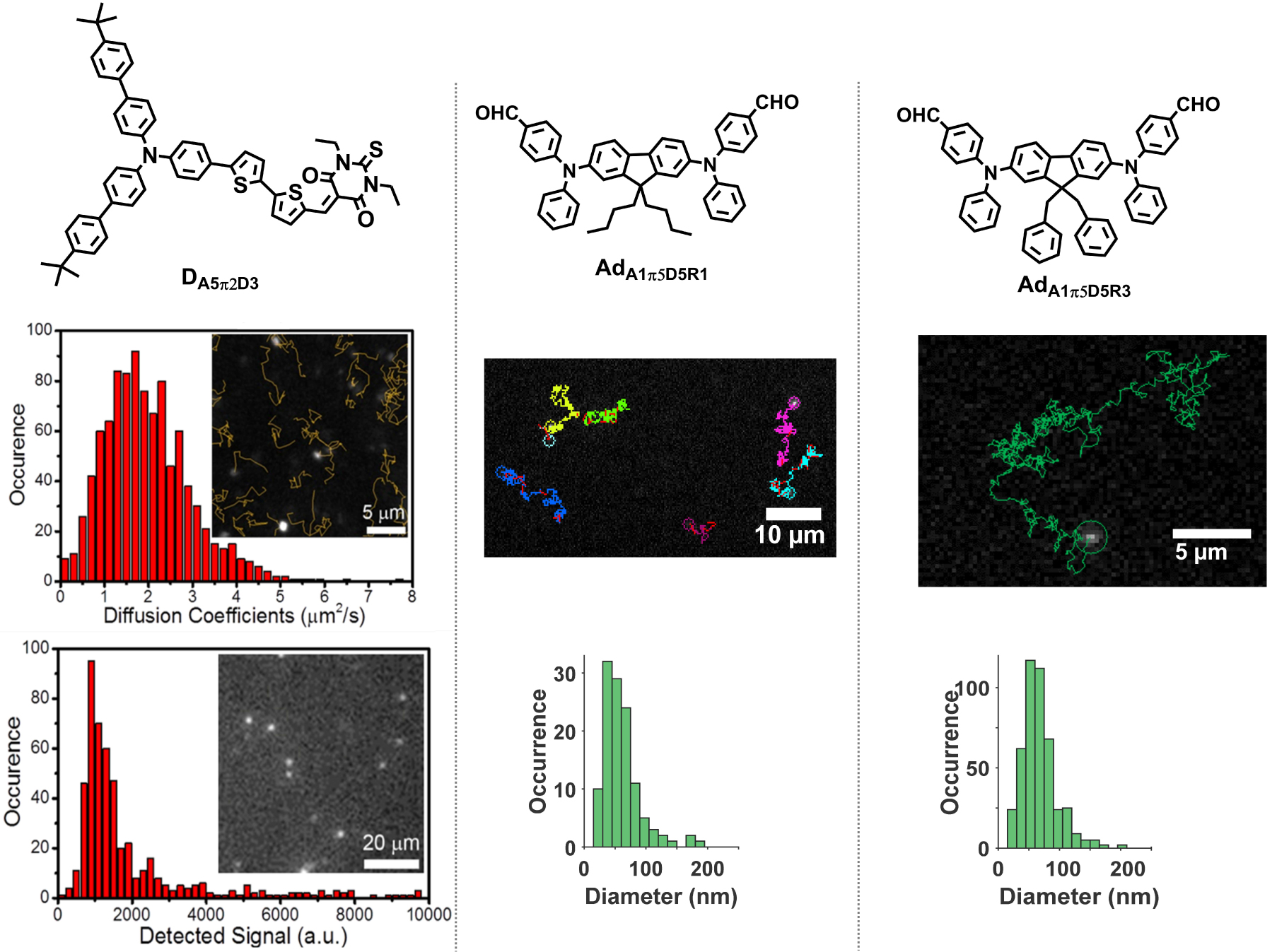
SPT experiments can also be performed with quadrupolar dye-based (Q2CR3π5D1 and Q2CR3π4A4) dFONs within cells. Interestingly, due to their different sizes (43 nm and 14 nm for Q2CR3π5D1 and Q2CR3π4A4, respectively) and different absorption spectra ( 400 nm and 503 nm for Q2CR3π5D1 and Q2CR3π4A4, respectively), the two dFONs show similar and large brightnesses at 488 nm (wavelength of the blue laser). Yet their emission can be discriminated easily as dFONs made from Q2CR3π5D1 are green emitters while dFONs made from Q2CR3π4A4 are NIR-I emitters. Thanks to these unique properties, these dFONs that internalize into cells (see Section 4.2) can be tracked at the single-particle level and used together for simultaneous bicolor SPT within cells (Figure 11) [39].
Simultaneous bicolor single-particle tracking within COS cells. Simultaneous bicolor single-particle tracking of dFONs made from Q2CR3π4A4 and Q2CR3π5D1 in live COS7 cells; A. wide field, COS7 cell contour is drawn in black; B. Merged single particle tracks on green and red channels; C. Live tracking of Q2CR3π4A4 dFONs (boxes) in COS7 cells on red channel; D. Live tracking of Q2CR3π5D1 dFONs (boxes) in COS7 cells on green channel; Scale bar = 5 μm, excitation at 488 nm (P = 306 W⋅cm−2). Adapted from Ref. [39].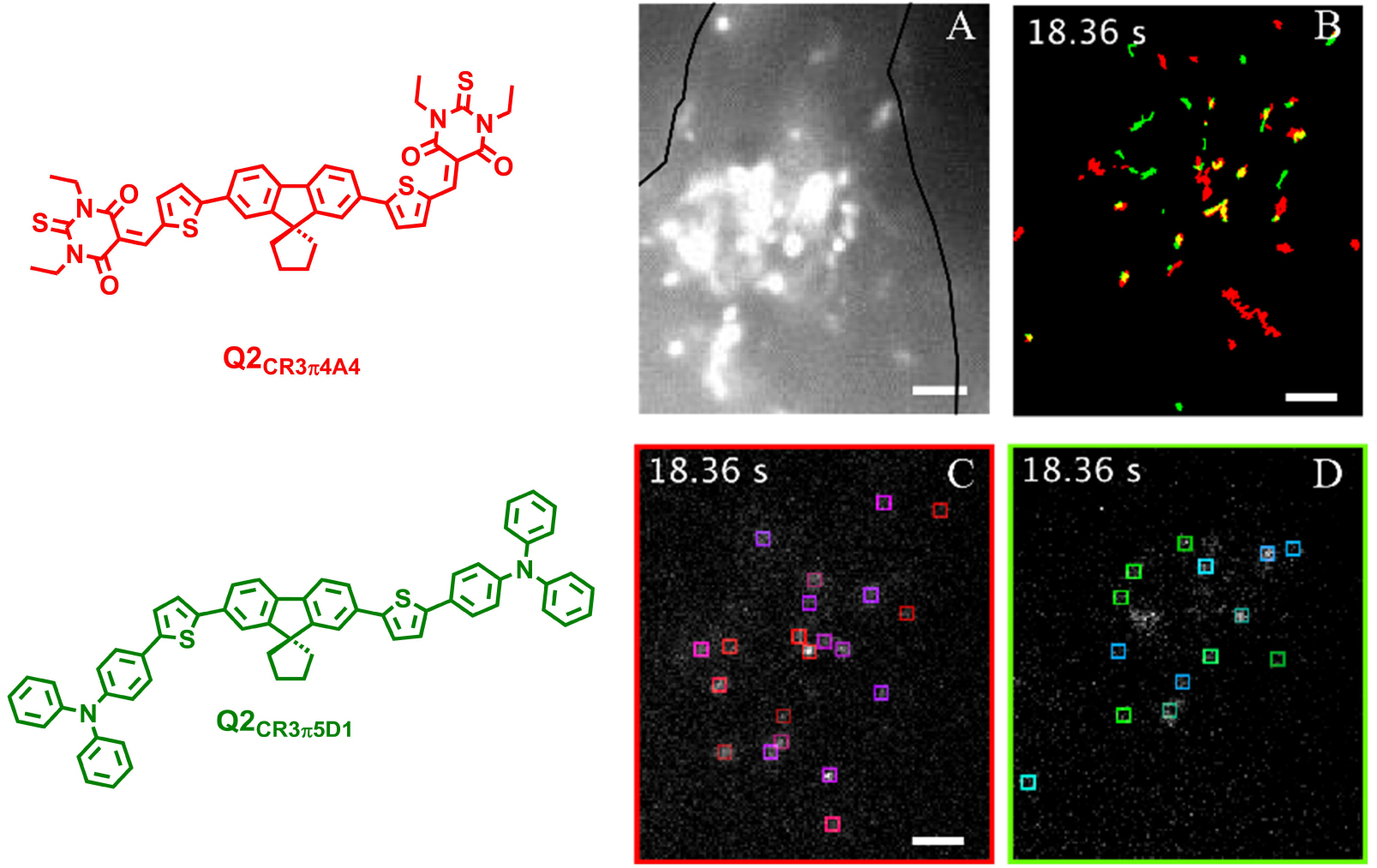
To date, the wealth of information provided by SPT was mostly obtained in simplified systems such as cell monolayers. However, the microenvironment of a cell highly regulates its behavior and its molecular dynamics. There is therefore a need to perform SPT deep in intact tissues. Using small (10 nm in diameter), bright red-emitting and stealth dFONs made from quadrupolar dyes Q1A2π1R1D1, it was possible to explore the extracellular brain space deeper into tissues than with the equivalent QDs (i.e., QD655). The small dFONs diffuse deep into organotypic brain slices and can be tracked down to 150 μm depth while QD655 are no longer detected [36] (Figure 12).
Single-particle tracking deep into brain tissues. Single-particle tracking of small (10 nm) dFONs made from Q1A2π1D1 in organotypic brain slices. (A) Organotypic brain slices preparation after extraction of the mouse brain and dissection of the hippocampus. (B) Trace of single-particle tracking for 11 s, 150 μm below the surface of an organotypic brain slice. (C) Fluorescence images of single QDs-655 and Q1A2π1D1 dFONs recorded at the indicated depths (an intensity profile of single particle is provided on the right). Fields of view are 6.5 μm wide. Adapted from Ref. [36].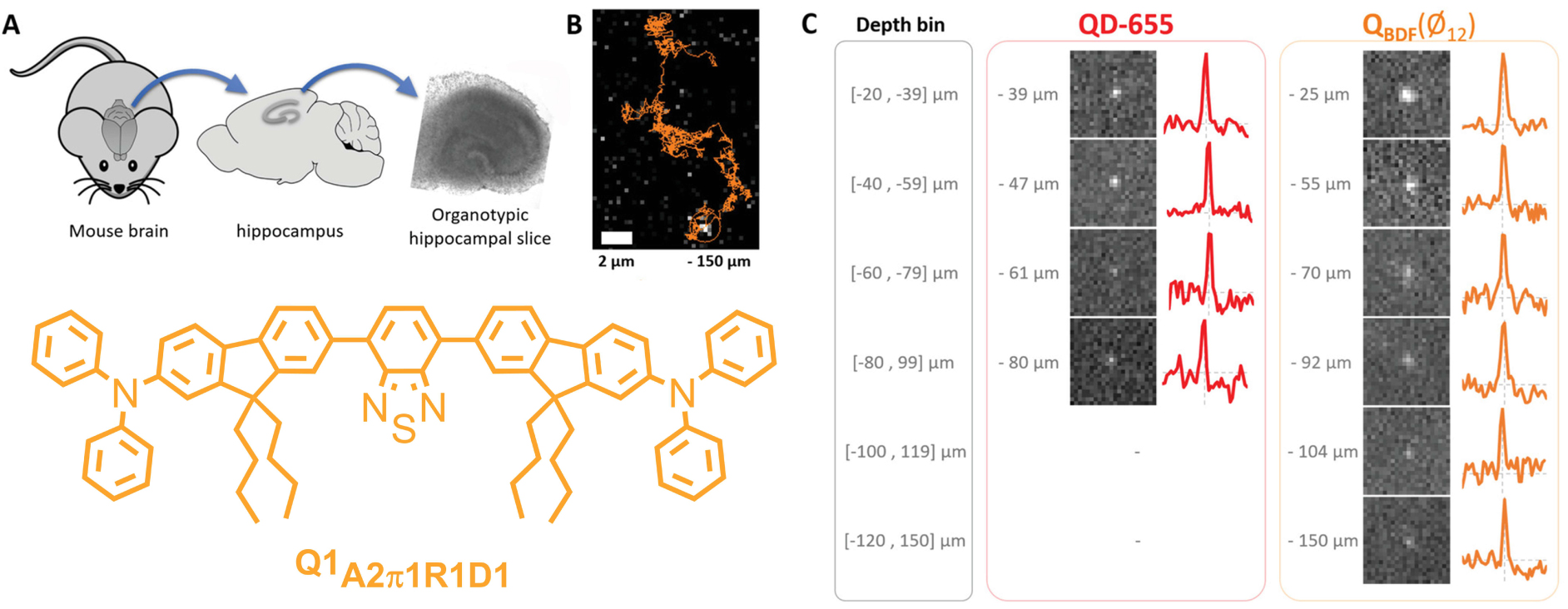
6. Conclusion and perspectives
The molecular engineering of dedicated polar (“push–pull”) and polarizable dyes as dFONs building blocks has proven to be a successful approach to achieve ultrabright small luminescent organic nanoparticles that compete with QDs for bioimaging purposes. Not only can their optical properties be modulated, but their (photo)stability and surface properties can be tuned by adjusting the structure of the PPDs. This demonstrates that organics can also play an important role in the field of Nanobiotechnologies, opening new routes for nanotools for drug delivery or super-resolution imaging. dFONs made from PPDs is a highly versatile approach benefiting from the wide wealth of organic dyes. Compared to inorganics, such dFONs may provide a safer and more sustainable alternative: their toxicity can be tuned by adjusting the structure of the dyes, while their synthesis can benefit from green protocols.
An important aspect is their surface functionalization for applications such as early diagnostic, ultrasensitive sensing and super-resolution imaging. In that respect, two main approaches are developed: non-covalent (electrostatic-based) functionalization [37] and covalent functionalization [56]. We are currently investigating these routes.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The Conseil general d’Aquitaine is acknowledged for financial support (Chaire d’Excellence Grant to MBD). Financial supports for ODP and EK is provided through the MESRI via the University of Bordeaux. Continuous support from Univ. Bordeaux, CNRS and Bordeaux-INP is acknowledged. This work was also supported by the ERC COMET (101077364).