


 CC-BY 4.0
CC-BY 4.0
Introduction
Bioinorganic chemistry is a multidisciplinary field dealing with metal ions in biological systems. This field aims to understand and control inorganic molecules, mainly metal-based, within living organisms and cells, whether endogenous (e.g. metalloproteins) or exogenous (e.g. metallodrugs or probes). The goal is to explain how these inorganic molecules work in a biological context, and to design synthetic inorganic compounds with biological activity. These compounds may be systems with no endogenous equivalent, but which are likely to play a role in a biological environment (such as cis-Pt acting as anticancer drugs or contrast agents). In contrast, they may be inspired by biological molecules (in which case we call them bio-inspired), and reproduce the activity of a biomolecule (typically a metalloenzyme) of industrial or therapeutic value. In this last case, the metal-based compounds, meant to be metallodrugs, should be studied directly in the biological environment. Indeed, biological media display some specific physico-chemical characteristics, such as high viscosity, molecular overcrowding, compartmented environment, high content in Lewis bases and metal ions, that can impact kinetics and thermodynamics [1], and may interplay with the bioactivity of these metal complexes by modifying their nature (speciation), and their reactivity.
In addition, to be active a molecule must reach its target, that can be buried in a specific compartment of the cells: hence the knowledge of the sub-cellular distribution of a molecule of interest is also important to understand its bioactivity. To that extent, metal-based tags and probes are interesting tools to investigate the distribution of a molecule of interest or to develop organelles trackers for non-conventional bio imaging techniques appropriate for metal-based compounds. A very attractive feature of this kind of metal-based probes is the opportunity to perform multimodal imaging using a very compact structure that may not impact in a too large extent the physico-chemical properties of the labeled molecule. Towards this goal, metal carbonyl complexes [M(CO)x(L)(L′)] and, in particular Re complexes, are well-suited and will be discussed.
Below, we describe approaches in inorganic cellular chemistry developed in our group to design and apply bioinspired metal-based antioxidants and metal-based probes, with an interest for pathologies linked to oxidative stress (https://ens-bic.fr/). Sub-cellular metal imaging can be used to get access to the bio-distribution of the complexes, which is, as said above, a key parameter to their activity. Our approaches are highly integrated, from molecular design to study on cells, and require skills in organic (ligands) and inorganic (complexes) synthesis, physical chemistry, cell biology and imaging techniques.
1. Antioxidant protective metalloenzymes: a source of inspiration for catalytic antioxidants
Aerobic living organisms face a flow of species derived from dioxygen and that are much more kinetically reactive that dioxygen. These Reactive Oxygen Species (ROS), such as superoxide (), hydrogen peroxide (H2O2) or hydroxyl radical (HO∙) are involved in a range of physio-pathological situations [2]. In the eustress mode [3], these species play important physiological roles necessary to the well-functioning of the cell. The natural continuous flow of these ROS is controlled by an arsenal of protective antioxidant enzymes (SuperOxide Dismutase (SOD), CATalase (CAT) …). A distress mode [3] is activated when the natural defenses are overwhelmed and no longer able to maintain a flow weak enough to avoid irreversible oxidative damages to biomolecules. That occurs in pathological or therapeutic situations (for instance leading to side effect of some medicines).
SuperOxide Dismutases (SODs), metalloenzymes that catalyze the dismutation of superoxide , or CATalases (CATs), metalloenzymes that catalyze the dismutation of H2O2, are key players of the cellular antioxidant protective arsenal. Exogenous compounds can limit oxidative distress by acting on the flow of these ROS, either by converting them directly (direct action, as stoichiometric or catalytic antioxidants) or by activating endogenous defenses (indirect action, that will not be described further here but was commented in Ref. [4]).
To date, the conventional drug design consists of the quest for small molecules that stoichiometrically react with biomolecules, including proteins or DNA. One innovative alternative refers to catalytic drugs displaying therapeutic properties based on catalysis [5], which offers the opportunity to decrease the effective dose with promising therapeutic perspectives. Among catalytic drugs, Low Molecular Weight (LMW) complexes catalytically activated anticancer agents [6], complexes mimicking the rhodanese to detoxify cyanide [7], and also complexes mimicking the activity of SOD [4, 8, 9] or CAT [10] are prominent examples [5, 11].
2. SuperOxide Dismutases (SODs) physicochemical characteristics: a guideline for chemists [8]
2.1. Main features of SODs, relevant to SOD mimics design
SODs are metalloenzymes with either Cu(II)/Cu(I) (Zn), Mn(III)/Mn(II), Fe(III)/Fe(II) or Ni(III)/(II) ions at the active site for Cu-only SOD, Cu–Zn SOD, Mn and Fe SODs and Ni SODs. They are very efficient enzymes, reacting with superoxide with a kinetics close to the diffusion limit [12], and the parameters responsible for this efficient activity have been carefully analyzed [13]. All SODs show [8]:
- A redox potential in a very narrow range [13] (Figure 1). Interestingly, this redox potential is tuned to the optimal value for superoxide dismutation, id est the midpoint between the two redox potentials for the redox couples involving superoxide /H2O2 and O2/, as indicated in Figure 1.
- An electrostatic guidance of the anionic superoxide, with an overall negatively charged protein (surface) but a positively charged funnel of access for superoxide [13] (except for Cu-only SOD in which the active site is close to the protein surface and directly accessible to solvent [14]).
- Compartmentalization into specific organelles. Note that in this context of unresolved oxidative stress or distress, mitochondria appear as the fragile organelles to be protected. This is the main cellular location where superoxide is produced through uncoupling of the respiratory complexes involved in the reduction of dioxygen to water. The imbalance in the production of superoxide and the capacity of the protective pathways, associated with other ROS and Reactive Nitrogen species (RNS), induces oxidative damage leading to mitochondria and cell dysfunction.
(a) Half-reactions of the redox couples involving superoxide (1) and (2), and standard redox potentials E°′ pH 7 (V versus NHE). These are apparent potentials at pH 7 and with a concentration of dioxygen of 1.23 mM, which is the one found in water under standard conditions, 25 °C and 100 kPa [15] and Emidpoint = 0.36 V versus NHE [16, 17]. Midpoint potential optimal for catalysis of the dismutation (Emidpoint) [17], redox potentials reported for several SOD. (b) Spontaneous superoxide dismutation (3), and (c) simplified ping-pong mechanism for the catalysis of superoxide dismutation. The kinetics are from [12] and [18].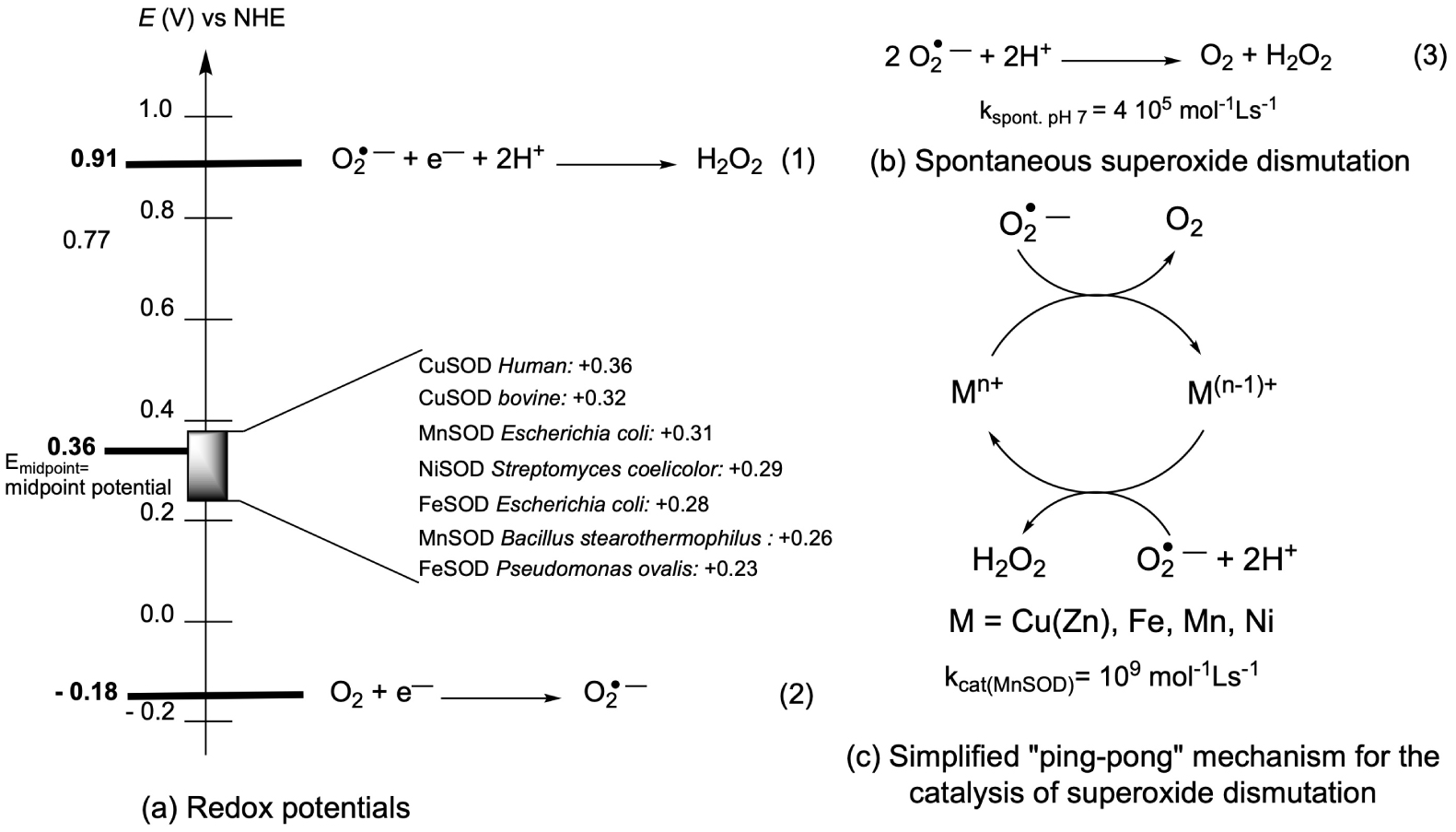
2.2. SOD mimics design
What metal ion to choose for the design of metal-complexes as SOD mimic? Mn ion is described as the “stress reliever” [19, 20], as less prone to Fenton chemistry than Fe and Cu, associated with the catalysis of the Haber–Weiss reaction (Figure 2), with potential oxidative stress exaltation by the production of HO∙ [4, 8, 9, 21]. This is why, although some neurotoxicity has been described [22], Mn ion is now the leading metal ion in the field of SOD mimics design for therapeutic use. In the literature, peptides-based Cu SOD-mimics are nevertheless developed (Sections 4.2 and 4.3), probably because Cu(II)-peptides complexes are easier to characterize than Mn-peptides, as they are more stable.
Fenton and Haber–Weiss reactions: oxidation of low-oxidation state metal ion (Fe(II) and Cu(I)) by hydrogen peroxide and cycling back to low-oxidation state with superoxide. Haber–Weiss equation, catalysis by M(n+1)+∕Mn+ with (M = Fe or Cu). For (1)–(3), see Figure 1.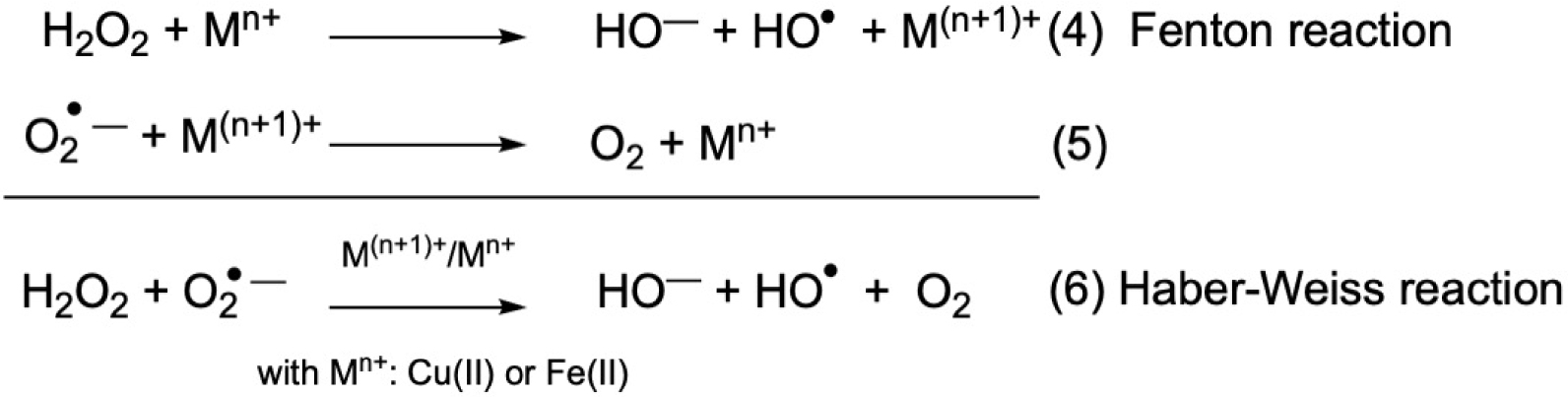
Mn(II), as a d5 ion with no ligand-field stabilization energy, has a low association constant for most ligands, one of the lowest within the Irvin and Williams series [23, 24]. However, in biological systems, the endogenous ligands competing for Mn(II) binding also have low association constants, rendering the criteria on the association constant within a complex paradoxically less drastic for Mn(II) than for other metal cations [8, 25]. This is also why Mn(II) is one of the most bio-available metal ion [26]. In this context, one has to keep in mind that, even if biological media abound with metal ions, the pool of loosely-bound metal ions, able to exchange, although important for biological function such as signaling, is way less abundant.
Nevertheless, exchange with other metal cations, or reaction with an ancillary ligand leading to a ternary complex can occur (for example see [27, 28]). Then, characterizing the nature of the Mn-complexes in cells, or manganese speciation, is key.
Most of bioactive molecules are developed for their toxicity, possibly selective towards cancer cells, bacteria or fungi (for example see [29, 30]). Therefore, most bioanalyses, even for organometallics bioactive compounds, are meant to evaluate their toxicity. In contrast, SOD mimics must be non-toxic redox catalysts meant to restore the basal activity in cells under oxidative stress. Their characterization requires thus the development of specific non-routine strategies to evaluate their bioactivity.
The main challenge for chemists in the field of antioxidants mimicking SOD [4, 8, 9, 10, 31] is thus to design non-toxic stable and inert complexes, with a tuned redox potential and positive charge for superoxide attraction, which react quickly with superoxide, showing a good cellular availability and possibly an accumulation at the mitochondria, the most fragile organelle in the context of oxidative stress. Even more valuable, a SOD mimic should be a catalyst for superoxide dismutation and, the faster, the better. Indeed, there is a positive correlation between the catalytic kinetic constant for the superoxide dismutation, or kcat measured outside of any cellular context (referred below as intrinsic activity [8, 32]), and the biological and potential therapeutic effects [8, 22]. Characterization in cells, with information about speciation and location, will also be important to the understanding of the bio-activity (Section 5.5).
3. Bio-inspired design of SOD mimics
3.1. Tuning the ligand to improve SOD activity
To obtain Mn-complexes with an appropriate redox potential for SOD activity, we have chosen to use an approach bio-inspired from Mn-SOD. In the active site of this protein, the Mn ion is in a Trigonal BiPyramidal (TBP) geometry in an N3O/water coordination environment, with one monodentate carboxylate from an aspartate and two imidazole moieties from histidine residues in the median triangle/equatorial plane (in green in Figure 3), and a farther histidine with a water molecule (H2O or HO−) in the two apical positions.
Active site of SOD, structure from Ref. [33].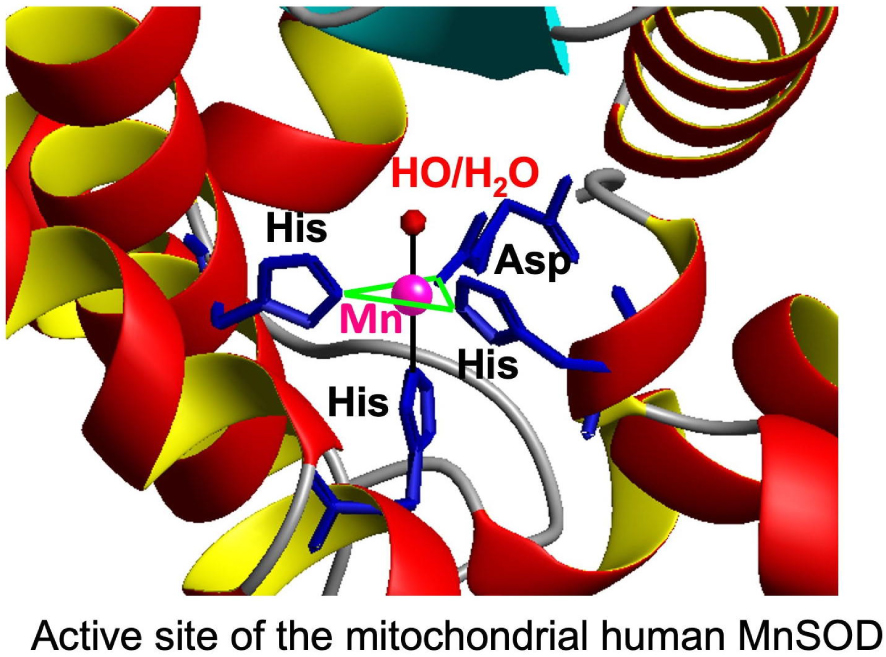
The particular axially distorted TBP geometry at the active site of SOD probably geometrically favors Mn(III), a d4 ion S = 2 subjected to a Jahn-Teller effect, over Mn(II), a d5 S = 5∕2 ion with no electronic geometric preferences. This contributes to the tuning of the redox potential (see above and Figure 1) [34, 35], which must be lowered from 1.51 V versus NHE (for hexa-aqua Mn(III)/Mn(II)) down to ca. 0.36 V versus NHE.
Starting from a tripodal amine, functionalized with two imidazole and one carboxylato moieties, we have modified the ligand to tune the redox potential of the Mn(III)/Mn(II) (Figure 4, variations on the nature of the coordinated Lewis bases). Figure 5 shows the kcat for superoxide dismutation plotted against the redox potential of the Mn(III)/Mn(II) redox couples of different SOD mimics. Porphyrins offer a large range of possibilities for redox and charge variations [36, 37] and we here compare them with the bio-inspired series centered on a central amine or 1,2-diaminoethane [25, 28, 32, 35, 38, 39, 40]. These two series of structurally unrelated complexes show a positive slope for E < 0.4 V/NHE and a negative slope for E > 0.4 V. The two “lines” cross at about 0.4 V, which is close to the optimal value for SODs (Figure 1), in line with this value being the optimal one for SOD-like activity.
Structure of the complexes discussed in the Section 3.1. S: possible solvent molecule. For the ligands, BIG stands for the bis-imidazole-glycinate, IPG for imidazole, pyridine, glycinate, TMIMA for tris-methyl-imidazole amine, BMPG for bis-methyl-pyridine glycinate. For the peptides: Fx = (L)-cyclohexyl alanine and r = (D)-Arg.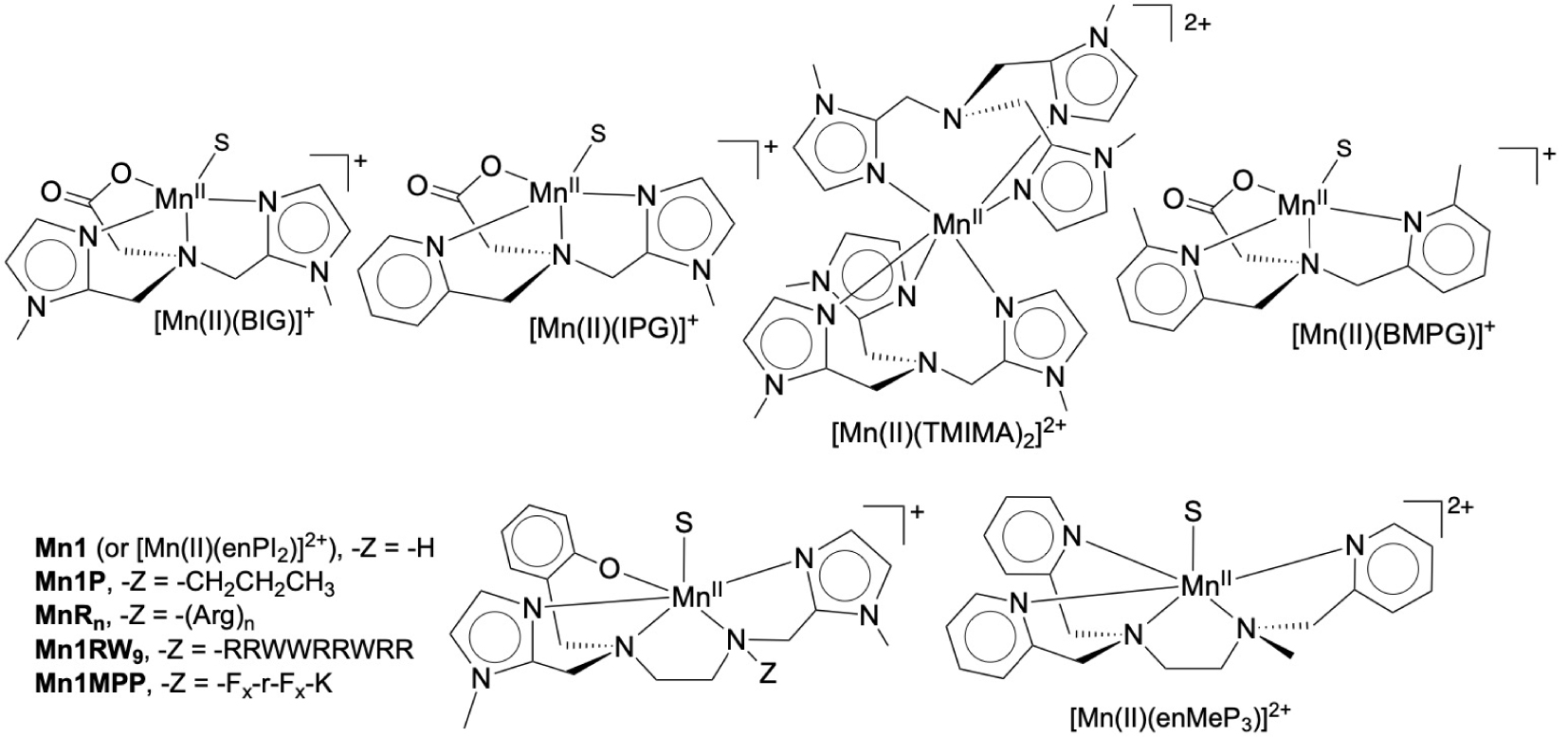
Plot of the log kcat(O2−) (kcat(O2−) mol−1⋅L⋅s−1) as a function of Mn(III)/Mn(II) redox potential (E1∕2 in V reported against NHE). Triangles, up-ward: Mn(II) complexes directly inspired from SOD active site (see Section 3.1); blue triangle, down-ward: Mn(II)(ClO4)2 (Mn(II)-hexa-aqua); open-squares, purple (N): negatively charged porphyrins; square with a cross, pink: Mn(III)-porphyrins, 5+ charge. Inspired from [8, 22, 35].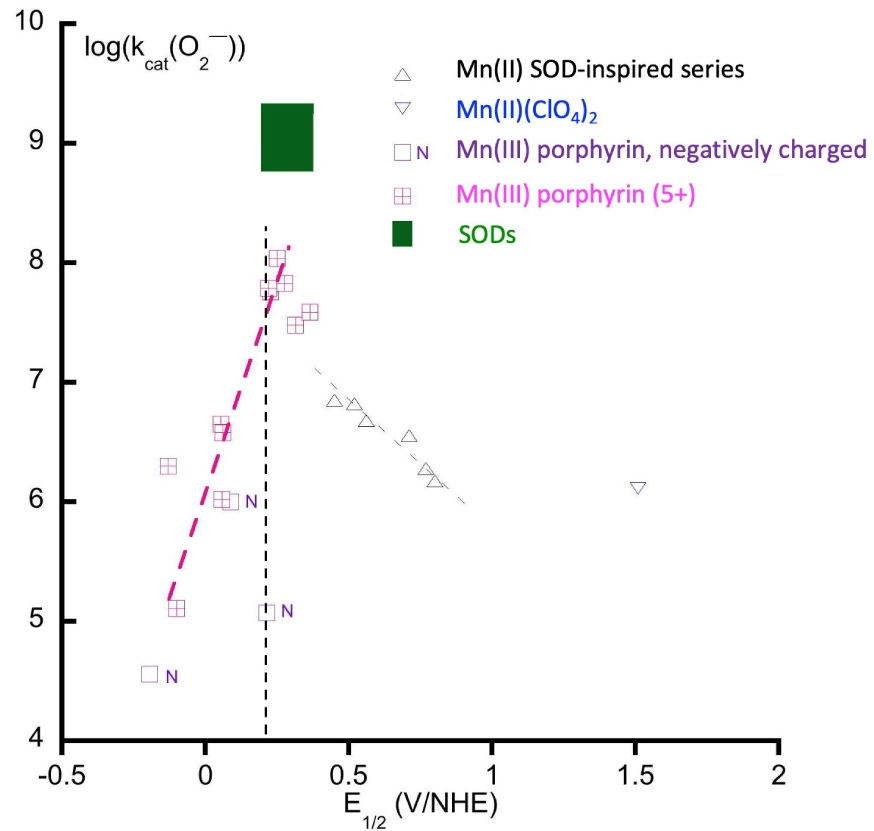
We also explored the effect of an increased bulkiness around the metal ion in the complex (picolyl moieties in the Bis-Methyl-Pyridine-Glycinate (BMPG) ligand, hexacoordination in [Mn(TMIMA)2]2+ Figure 4) to disfavor dimerization into Mn(III)–Mn(IV)-di-μ-oxo that is a dead-end for superoxide dismutation [35].
The optimal ligand in the series is enPI2, which displays pentadenticity with the highest association constant for the Mn(II) complex [25, 41] and a Mn(III)/Mn(II) redox potential very close to that of SODs (0.36 V/NHE, Section 2 and Figure 1) [42].
3.2. Modulation of the charge
The electrostatic attraction of the superoxide anion down a positively charged funnel to the positively charged active site is also key to the fast kinetics of SODs. Can such an effect be seen for SOD mimics? This is obvious along the vertical dash line in Figure 5, with the couple showing a same potential but a negative charge (N), for which the kinetics is much slower than the positive 5+ porphyrin. But the charge is not all, and the topology of its distribution is important. In the SODs, there is an overall negative charge for the protein with a positive channel and a positive area covering less than 1% of the total enzyme surface [43, 44, 45]: the negative charge of the surface drives the superoxide away to this positive area where the funnel exerts a directional attraction to the active site. Spasojevic et al. have studied a series of porphyrins bearing pyridinium moieties on the meso-position, with atropoisomers either with all positive charge as a crown on one side of the porphyrin plane or alternating charges. They have shown that the optimal distribution is that with the positive charges on the same side of the porphyrin plane, hence with a positive crown, as a smaller 3D version of the positive charged funnel in SODs, over the center metal ion [46]. In the N-centered series, we have functionalized Mn1 with an oligoarginine (positive charged amino acid) to modulate the overall charge (Figure 4). Interestingly, an incremental favorable effect on the kinetics was recorded for Arg1 (R) and Arg3 (R3), but no additional effect was observed for Arg6 (R6) and Arg9 (R9). This makes sense as the oligoarginine are not structured and, if too far away and not organized in space as a funnel, they should not display any effect [47].
3.3. Effect of the hydration level
The complexes in the N-centered series showed different hydration levels when they were isolated in the solid state, with complexes bearing zero, one, or two water molecules coordinated to the Mn(II) ion, as evidenced by crystallography [35, 48, 49]. The consumption of superoxide as a function of this hydration level in anhydrous dimethylsulfoxide (DMSO) showed that the number of superoxide consumed was correlated with the amount of water on the Mn ion (Figure 6) [35]. This indicates the capacity of the complexes to cycle between the Mn(II) and Mn(III) redox states in the course of the reaction with superoxide (for a detailed description, see also Ref. [8]). Interestingly, H2O introduced in anhydrous dimethylsulfoxide (DMSO) at the same concentration did not react efficiently with superoxide in the absence of complex.
Schematic rationale for the number of superoxide equivalents (with regard to the Mn-complex) consumed in anhydrous DMSO upon the number of bound water molecules. Adapted from Ref. [8]. For details, see Refs [48] and [8].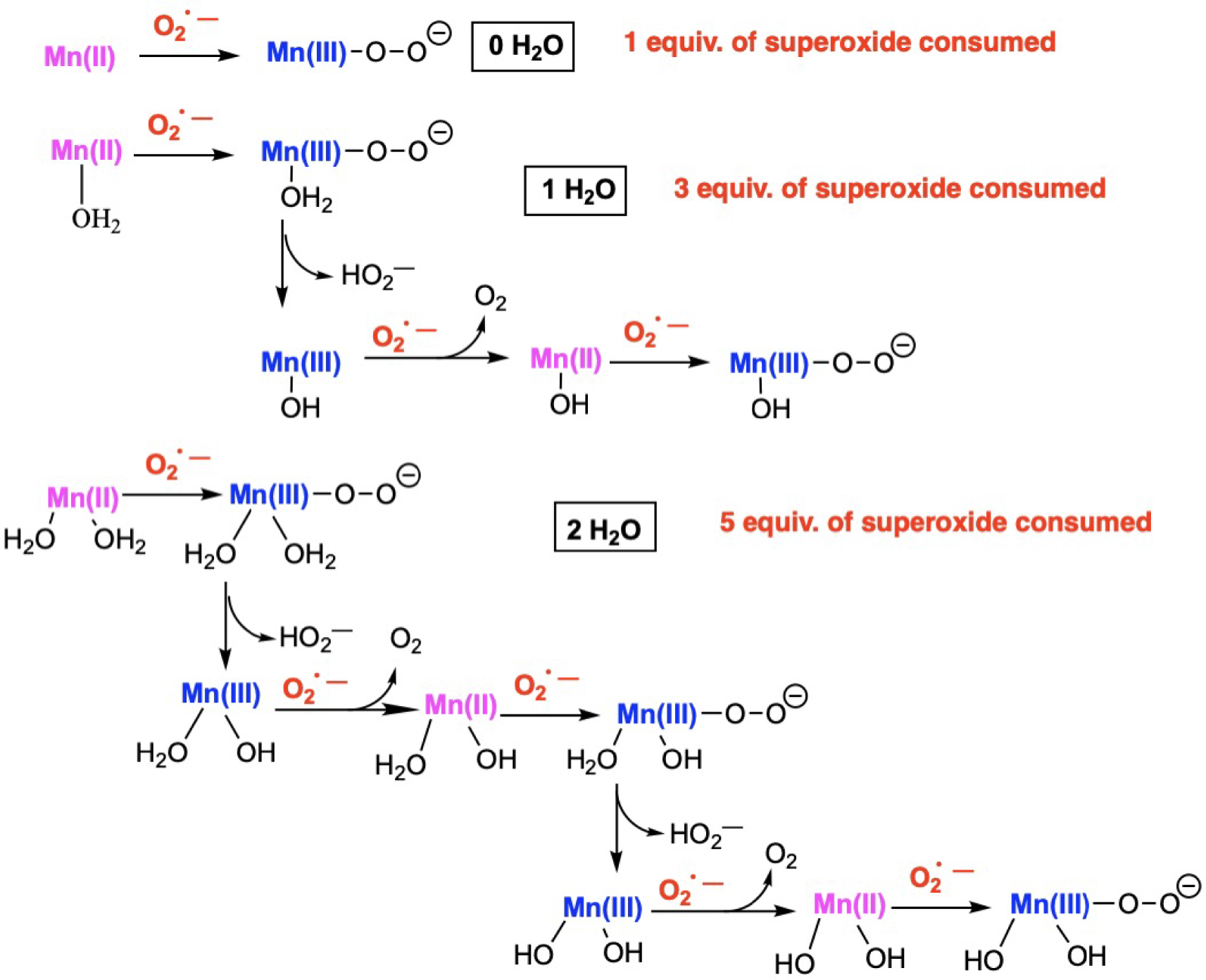
3.4. Quantification of the intrinsic SOD activity and in cells
3.4.1. Quantification of the intrinsic activity in water
The reactivity with superoxide can be studied in water using the indirect assay developed by McCord and Fridovich [8, 50, 51, 52]. The xanthine/xanthine oxidase system produces a slow and continuous flow of superoxide reminiscent of what is found in biological environments. All the complexes were found to be active [28, 35, 38, 47, 48, 53, 54], with kinetic constants kMcCF [8, 35] in the order of 106–107 mol−1⋅L⋅s−1. The kinetics was also studied using pulsed radiolysis to produce a high concentration of superoxide [39] and stopped-flow, with fast kinetic measurements with a highly concentrated solution of KO2 in DMSO or acetonitrile (MeCN) and rapid mixing in water [47, 53, 54, 55], leading to kinetic constants consistent with that from the McCord and Fridovich assay.
3.4.2. Studies in cells
To characterize the bio-activity of these SOD mimics, we have developed three cellular models of oxidative stress [56].
The SOD mimics from Figure 4 displayed an intrinsic SOD-activity (ca. kcat from a few 107 to a few 106 mol−1⋅s−1) that is ca. 1% of the enzyme MnSOD, which remains quite good since the kinetics of these enzymes is only limited by diffusion [8, 12, 13]. Among those SOD mimics, Mn1 was studied more thoroughly in cells. Mn1 was shown to limit ROS flow in macrophages [32] and to have an anti-inflammatory activity, both in intestinal epithelial cells, namely HT29-MD2 cells, and in mice [25]. In the rest of this article, we will mainly focus on the HT29-MD2 cells assay [25, 27, 28, 53, 54, 56]. This cell-model was constructed to increase the inflammatory response involving both ROS production, inflammatory cytokine secretion (interleukin 8 IL8) and expression of inflammatory markers (cyclooxygenase 2 COX2) upon bacterial LipoPolySaccharide (LPS) stimulation [57]. The strong inflammatory effect, reminiscent of what is observed in Inflammatory Bowel Diseases (IBD) [57], provides a large window to observe a return toward the basal state after a 6-hour LPS stimulation upon treatment with a SOD mimic. In this model, we have evidenced that Mn1 and its derivatives (Figure 4) (Mn1P, Mn1Rn) [25, 27, 54] are not toxic up to 200 μM, with an effective anti-inflammatory effect with a dose response in the range 10–100 μM (ca. 35% IL8 secreted in comparison to LPS at 100 μM; see Figures 7c and 20, downleft). The redox inactive Zn(II) analogues, when studied [25, 27, 28, 53, 54], were found ineffective, stressing the bioactivity is associated with the redox properties of Mn(II). Interestingly, upon LPS stimulation, cells overexpress mitochondrial MnSOD in an active form, as evaluated by Western Blot (WB), and such an effect has not been seen for the cytosolic CuSOD or CAT. This shows that LPS induces deleterious superoxide flux at the mitochondrial level. HydroEthidine assay (HE), a fluorescent marker oxidized by superoxide [58], showed that Mn1 reduces the basal superoxide content [25] but the link with the effect of LPS could not be made and no increase of HE oxidation upon LPS activation could be recorded, in contrast to what was reported before [58]. We believe that this is probably because of the cell antioxidant feedback upon oxidative stress [3]: LPS stimulation induces expression of an active MnSOD that reacts faster with superoxide than HE itself [25].
(a) Structures of the manganese(II) complexes; (b) kinetics study of the metal exchanges occurring between the manganese center of the SOD mimics and with Zn2+. The percentage of complexes that underwent metal exchanges was monitored by UV–vis; (c) Quantification of the inflammatory marker IL-8 in intestinal epithelial cells activated with LPS (0.1 μg/mL). IL-8 secretion was measured by ELISA in supernatant of LPS-activated HT29–MD2 cells incubated for 6 h with the SOD mimics at different concentrations. The IL-8 amount measured for LPS-activated cells is set at 100% for each independent experiment. Data represent mean ± SEM for at least three independent experiments: the number of independent experiments is indicated above each column. The p-values were calculated using the student test (bilateral test with equal variances not assumed). The mean ranks of each column were compared to that of the LPS control, each comparison stands alone. (∗∗∗) p < 0.001, (∗∗) p < 0.01 and (∗) p < 0.05 versus LPS control, ns non-significant.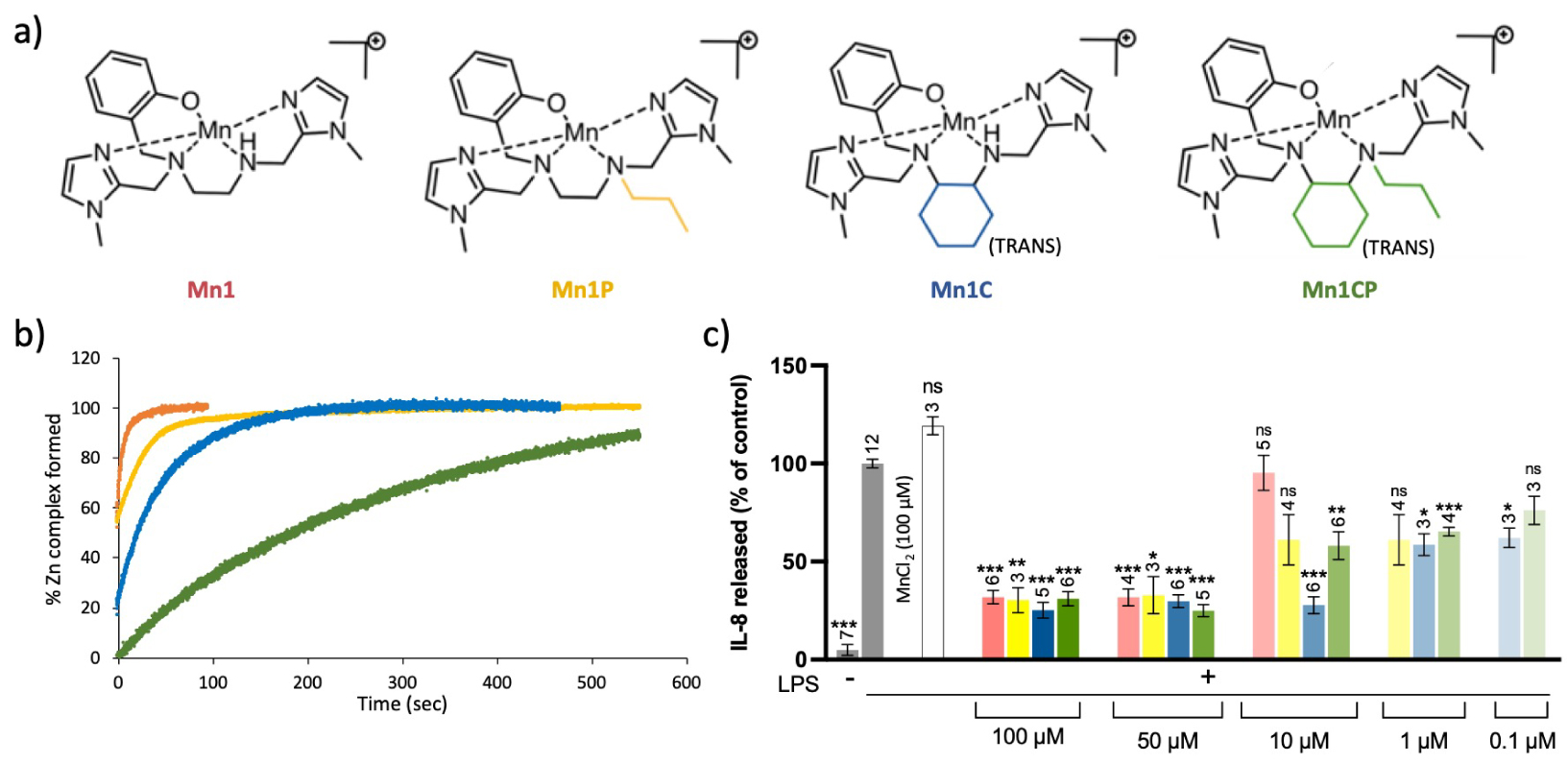
3.5. Towards higher inertia
Inertness and thermodynamic stability are two important features for metal-based drugs, since biological media are very rich in metal ions and Lewis bases that could compete with the native complexes. This is particularly true for complexes involving open-chain ligands such as Mn1 (Figure 4). Consequently, inspired by the literature [59, 60], in order to improve inertness of the SOD mimic we develop, we have replaced the 1,2-diamino group of the ligand by a (±)-trans-1,2-cyclohexyl diamine to rigidify the structure (Figure 7a) [28]. Stability, inertness, and more generally bioactivity of the corresponding complexes have been investigated. Note that the secondary amine was propylated (in Mn1P and Mn1CP) in view of a detection in cell lysates by mass spectrometry to avoid deprotonation that could occur and would lead to a more complicated mass spectrometry (MS) spectrum.
Thanks to their rigidified skeleton, Mn1CP, and to a lesser extent Mn1C, were found to be more resistant in the series to metal cation exchanges that may lead to the formation of redox-inactive analogues in a biological medium (Figure 7b). The modifications of the SOD mimics led to an improvement of their anti-inflammatory and antioxidant activities in epithelial intestinal cells HT29-MD2 under LPS-mediated inflammation. Indeed, Mn1C displays the best bioactivity, with significant effects when incubated at 10 μM (Figure 7c). These improved efficacies at low incubation concentrations could be correlated to the higher bioavailability of the new SOD mimics in the cellular environment associated with a higher inertness. Note that, at this stage, we cannot exclude the possibility of a different subcellular location that could also be responsible for the better activity of Mn1C. This is now under exploration.
Interestingly, thanks to higher inertness, the intact complex Mn1CP could be identified and quantified in cell lysates using Mass Spectrometry (MS), which was not possible with its Mn1P analogue, more prone to metal exchange (Section 5.5). After controlling that no exchange occurred during analysis, a known quantity of internal standard (here the cobalt complex having a phenol where the six carbons are 13C in order to distinguish them by mass) was added. Using a previously established calibration curve, we were able to determine the amount of Mn1CP in lysates from LPS-activated HT29-MD2 cells incubated for 6 h at 100 μM. The peaks corresponding to Mn1CP (m∕z = 504.2406), the internal standard Co1CP (514.2557 m∕z), and the dissociated ligand are clearly identifiable in the mass spectra (Figure 8). We also found that Mn1CP partially exchanged with Zn within the cells to give Zn1CP, but to a much weaker extent than Mn1P (Section 3.6).
Mass spectrometry analysis of a cell lysate of HT29-MD2 activated cells incubated with Mn1CP for 6 h and ultra-centrifugated at 10,000 rpm for 20 min. The lysate was diluted in a solution of 20% NH4CO3/80% MeCN and the internal standard Co1CP (13C) (0.4 μM) has been spiked before analysis.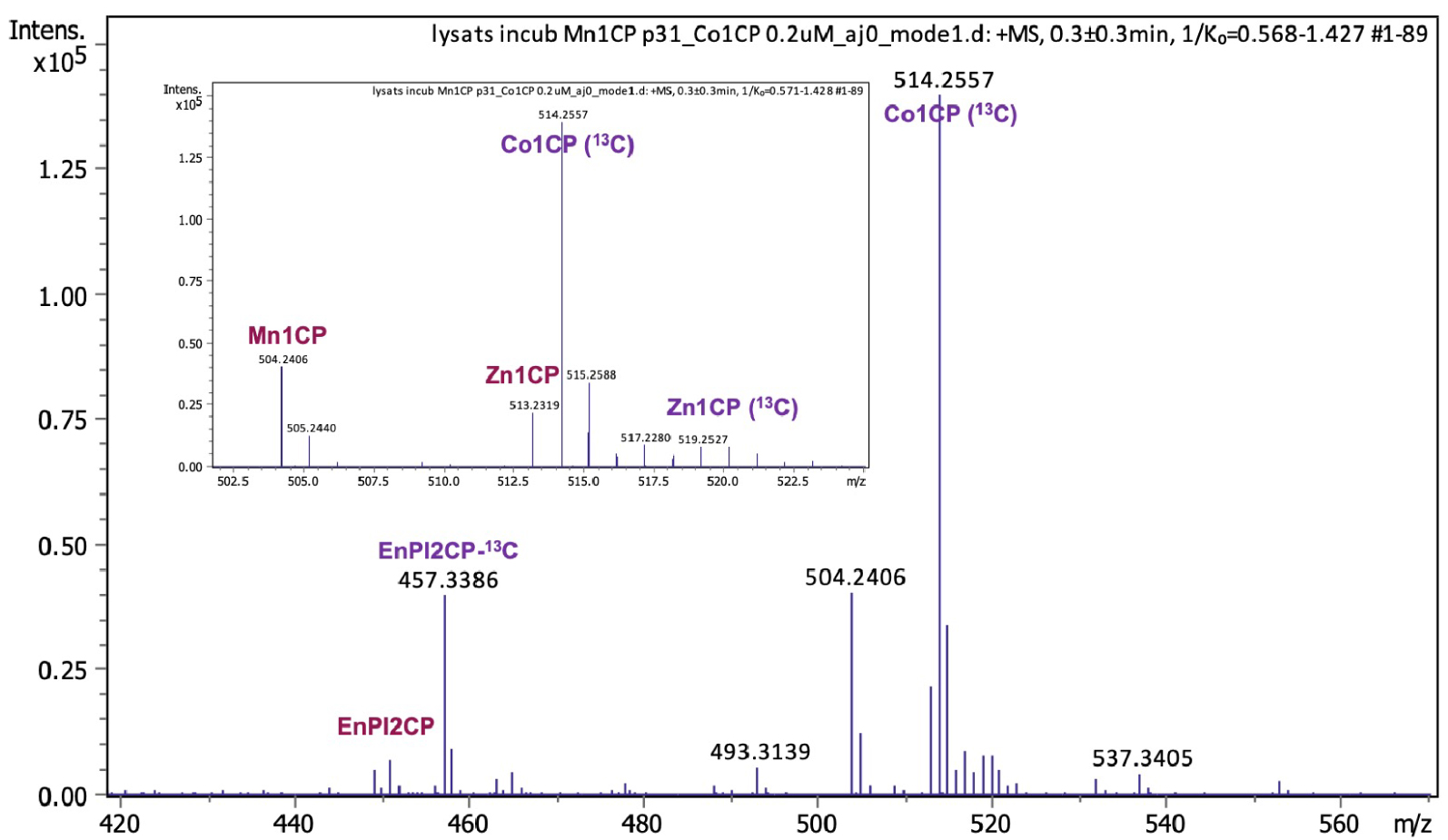
3.6. Ion mobility spectrometry (IMS) coupled to detection in mass spectrometry (MS): an efficient technique to detect and quantify LMW labile complexes in cell lysates
For the more labile compounds, Mn1 (m∕z = 408.1465) and Mn1P (m∕z = 450.1935), MS had to be coupled with a separation technique. Liquid chromatography (LC)-MS was not effective as metal ions exchanges occurred in the system. We then turned to Ion Mobility Spectrometry (IMS). This technique consists of the separation of the ions according to their size, or Collision Cross-Section (CCS), using an electric field to accelerate ions and a gas flow to push them further or slow them down (depending on the set-up).
Interestingly, we have shown that IMS is capable of separating low molecular weight (LMW) metal complexes built on the same ligand with different divalent metal cations (Mn, Fe, Co, Ni, Cu, Zn) although they exhibit only small differences in ionic radii. The collision cross-section (CCS) correlated with ionic radii [61], as shown in Figure 9. In other words, when a LMW ligand folds around a divalent transition metal cation, the weak difference associated to that of the ionic radii of the metal cation changes the CCS enough for the complexes to be discriminated in IMS (differences down to 1% of the mass can be discriminated). We applied that with a detection in MS, to demonstrate the intracellular presence of two SOD mimics Mn1 and Mn1P in the lysates of cells incubated with the complexes. We were able to estimate the intracellular concentration of Mn1P using a similar standard analogue with a heavy ligand (13C) and a Co(II) cation.
Collision cross-section of complex Mn1P in Å2 plotted against the ionic radius of metal cations in Å. The rion of any M(II) corresponds to a high spin pentacoordination, except for Fe(II) (high-spin hexa-coordination). The rion for Cu(II) is also shown (in yellow) for a high-spin tetra-coordination. The rion values were taken from http://abulafia.mt.ic.ac.uk/shannon/ptable.php [27].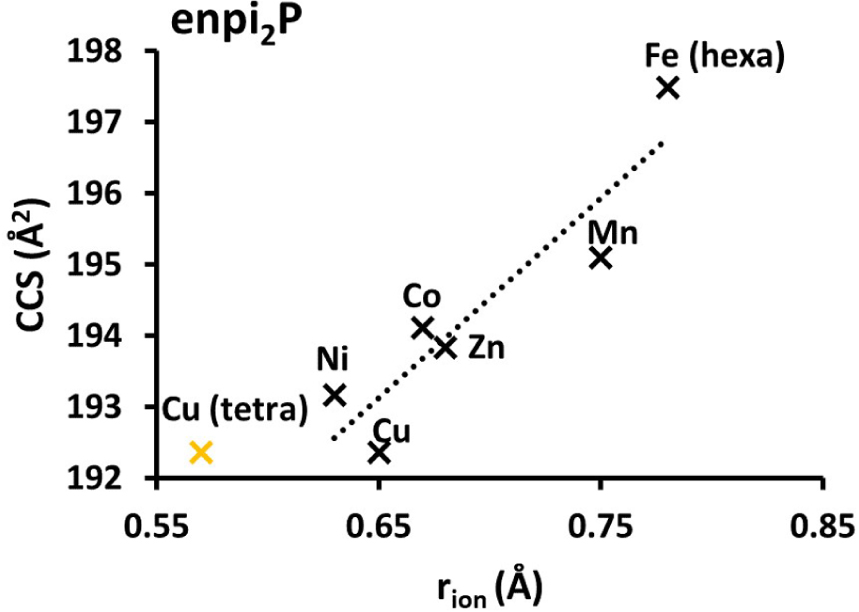
4. Peptides-based catalytic antioxidants for SOD and CAT activity
4.1. Conjugation of Mn1 with peptides
A path explored to improve the biological activity of the SOD mimics was their functionalization with peptides [54] and in particular cell penetrating peptides (CPP) [62] -R9 (or -Arg9), and -RW9 (or -RRWWWRRWRR), and a mitochondria targeting peptide MPP designed by Kelley et al. (-Fx-r-Fx-K, with (L)-cyclohexylalanine (Fx), (D)-arginine (r), and (L)-lysine (K) residues) [42] (Figure 4). As indicated previously, mitochondria have been considered as the organelle to be protected against oxidative stress, which is the rationale for choosing these Arg-based CPP and MPP.
These complexes Mn1-R9, Mn1-RW9 and Mn1-MPP (Figure 4) have intrinsic SOD activities, and dissociation constants similar to Mn1. However, the cell-penetration of Mn1, already good probably because this is a lipophilic charged compound [25], was deceptively decreased by the functionalization with the CPPs or MPP. The complexes Mn1-R9, Mn1-RW9 and Mn1-MPP also showed a cellular distribution different from the homogeneously distributed Mn1 [25], although this was difficult to follow due to the low intracellular concentration. Their bioactivity in epithelial intestinal cells (HT29-MD2) was similar to that of Mn1 when assayed at a concentration of incubation of 100 μM. Interestingly, Mn1-R9, Mn1-RW9 and Mn1-MPP showed a better anti-inflammatory activity than Mn1 when incubated at 10 μM. Importantly, the redox-inactive analogue Zn1-R9 was inactive. The situation was less clear for Mn1-RW9 and Mn1-MPP for which a contribution of the peptidyl moiety to the anti-inflammatory effect independent of redox metal-based regulation was observed as Zn1-RW9 and Zn1-MPP showed a weak activity [54].
Overall, the functionalization with peptides improved the anti-inflammatory activity: indeed, it was possible to reduce the incubation concentration by an order of magnitude, while retaining a significant anti-inflammatory effect and enabling a better activity/concentration ratio, which is interesting for future bio-applications.
4.2. Metallo-oligopeptides
Peptides are very attractive as ligand for metal complexes because: (1) peptides are easily synthesized on solid support and their synthesis is very versatile, (2) proteolytic instability and rapid clearance of peptides which were considered as main drawbacks, are nowadays seen as key advantages since peptides do not accumulate in tissues, are not metabolized in the liver avoiding drug-drug interactions [63, 64]; (3) peptidyl ligands can be easily conjugated with peptides or molecules in order to address them to specific organs, cells or organelles and (4) peptidyl ligands can be genetically encoded and thus produced by bacteria. However, the design of a sequence that binds selectively a metal cation and leads to a stable complex with the appropriate redox potential—redox potential enabling the targeted antioxidant activity—is very tedious. That is why a combinatorial approach has been combined with activity-based assays to develop efficient catalytic drugs that will be directly usable for oxidative stress resolution (Figure 10). Two antioxidant activities have been targeted leading to the development of SOD mimics and catalase mimics involving copper as the redox center. Catalase is an enzyme able to catalyze the dismutation of H2O2 into O2 and H2O. For both mimic types, a combinatorial peptide library has been prepared by a split and mix approach on solid support (bead) using a classical Fmoc strategy. This leads to the production a variety of peptides within the beads collection, with one given sequence per bead, each bead bearing several copies of the same peptide. After peptide elongation, the amino acids side chains have been deprotected, and the beads were incubated in a solution containing the metal salts (typically, Cu(II), Fe(II/III), Mn(II), Ni(II), etc.) to generate the complexes. At this stage, washing the beads with 2,2′,2′′,2′′′-(ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA) enables further increasing the selective pressure and select only stable complexes. The beads bearing peptidyl complexes have been then immobilized in an agarose gel and screened by two different colorimetric assays, one specific to SOD activity [65] and the other to catalase activity [66, 67]. This original approach yielded peptide-Cu(II) mimics of SOD [65] or catalase [66, 67] that are efficient in test tubes but also in biological contexts. A Mn(II) hit was also obtained but so far the Mn-peptide complex could not be properly characterized and remains unpublished. This is probably due to the weaker affinity constants of peptides for Mn(II) than for Cu(II) that stem from the intrinsic electronic properties of the metal as previously mentioned and the flexibility of the peptidyl ligands.
Illustration of the workflow for the development of efficient SOD and CAT mimics involving a peptidyl ligand and active on cellular models.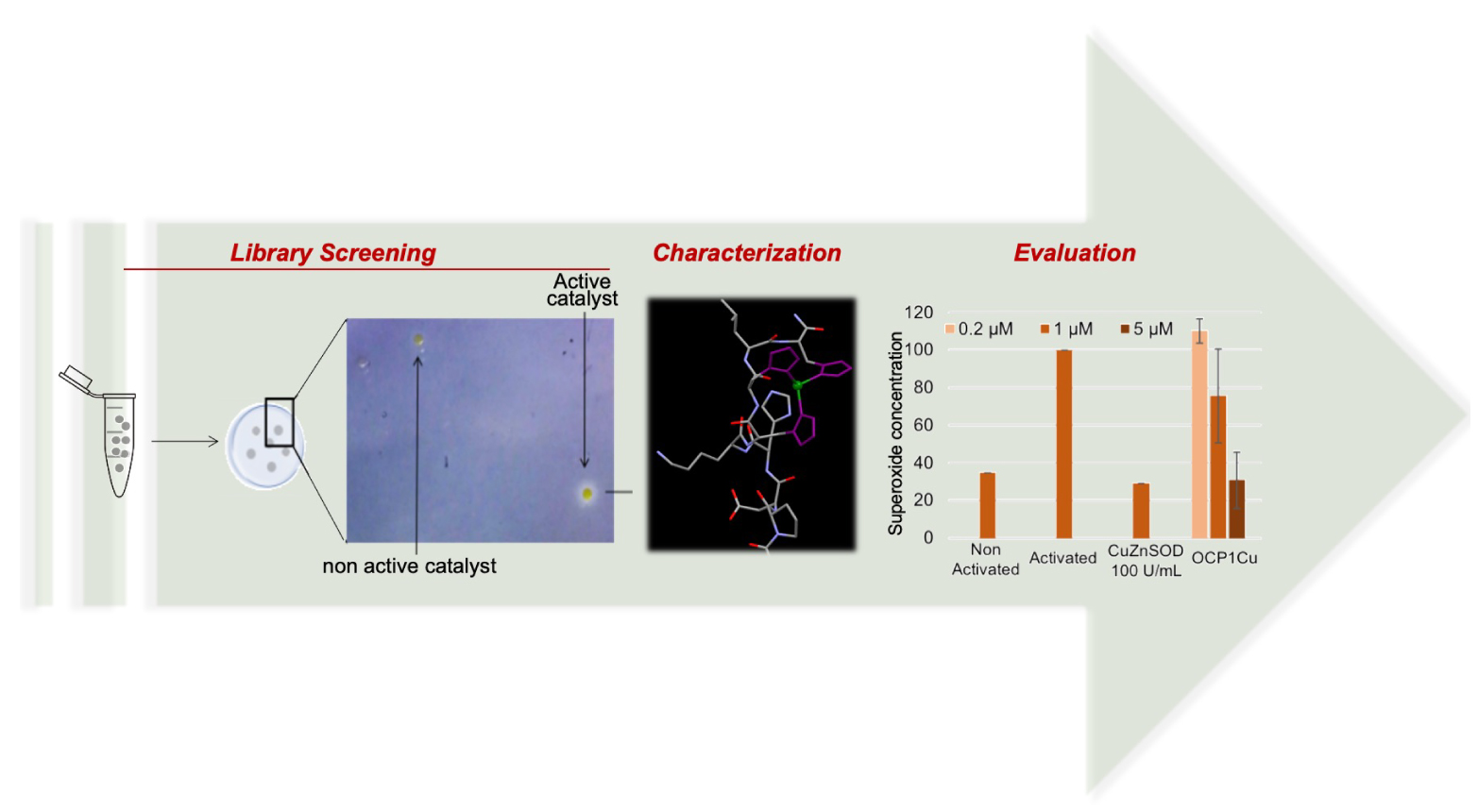
Interestingly, even by starting with the same combinatorial library, the two different CAT or SOD assays lead to the selection of different peptide sequences. In addition, in the case of the SOD mimic, the active species for catalysis is the complex involving one ligand for one copper whereas for the CAT mimic, the active species is the one ligand—two copper complex. This is consistent with the fact that superoxide dismutation involves the transfer of a single electron (Figure 1, SOD cycle) whereas H2O2 dismutation requires two electrons (Figure 11).
Half-reactions of the redox couples involving hydrogen peroxide H2O2.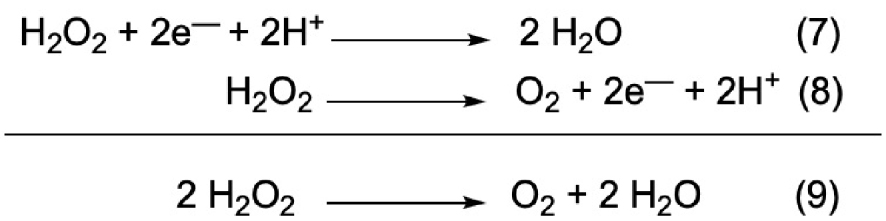
We can thus hypothesize that for the CAT mimic, each copper center brings one electron and cycles between the +II and +I redox states.
Importantly, the CAT mimic was found to have a poor SOD activity and the SOD mimic no CAT activity. This means the screening approach was specific of the activity selected.
4.3. De novo SOD mimics: rationale design of artificial proteins mimicking SODs
Expanding the possibilities beyond low-molecular-weight complexes and oligopeptides, rationally designed self-assembling peptide scaffolds with well-defined secondary and tertiary structures are valuable tools for mimicking enzyme structure and activity, leading to artificial de novo enzymes [68].
Because of the low association constant of Mn(II) for any ligand (Section 2), we decided to go for a mimic of Cu-based SOD. Designing heteroditopic de novo scaffold with specific Cu and Zn sites was not straightforward, as in the native CuZnSOD, and we decided to go for a mimic of Cu-only SOD [14] and not of the Cu–Zn SOD. For that, we used the α3D family of proteins, originally designed by DeGrado et al. and that consists of seven amino acid repeats in which the first and fourth residue of each heptad is hydrophobic [69]. This sequence favors the folding into an antiparallel three helices bundle [69]. A metal-cations binding site can be introduced inside the bundle, at the expense of thermodynamic stability [70] and an elongated version with an additional heptad, called Grα3D, was proposed by Pecoraro et al. to compensate for this loss in thermodynamic stability [70, 71, 72, 73].
A series of four structures featuring chelation sites for Cu(II) was generated, with histidine and aspartate residues [73], as in the active site of the Cu-only SOD [14]. More precisely, the chelation sites were H3 (His3), H4 (His4), H2DH (His3Asp with 2 His and 1 Asp in the same plane and one His above) and H3D (His3Asp with 3 His in the same plane and one Asp above). The corresponding complexes were characterized by various techniques (UV-Xis, EPR and XAS), showing they are good structural and spectroscopic mimics of Cu-only SOD. All the complexes show SOD activity with kcat in the range 1–3 × 106 M−1⋅s−1 and, although still three orders of magnitude lower than Cu-only SOD, this makes them the first de novo Cu-SOD mimics [73]. Note that a Mn-based de novo SOD mimic had been previously published, but with a very weak activity (ca. four order of magnitude slower than SOD, ca. 105 M−1⋅s−1) [74].
5. Imaging metal-based compounds
5.1. Direct detection of metal-based compounds using micro-X-ray fluorescence or IR-imaging
Because biological media, and especially cells, are compartmentalized (cytosol, organelles, nucleus, etc.), the bioactivity of a compound depends strongly on its localization and ability to reach a specific target. This target can be a macromolecule (e.g. DNA to silence or modify, or specific protein to activate or inhibit). It can also be a small molecule (e.g. ROS). Note that superoxide because it is negatively charged is not able to cross membrane, whereas hydrogen peroxide can [75].
The development of an innovative cellular inorganic chemistry or inorganic chemical biology can be bolstered by the exploration of analytical techniques adapted to the mapping of metal complexes at the sub-cellular level. Fluorescence in the UV–vis is a widespread technique. A large variety of probes are available, and they can be excited in a range of energies (UV–vis); they can be used as conjugable tags or organelles trackers, conjugated to antibodies and then used to detect specific proteins and map their sub-cellular distribution. Many of the corresponding trackers/tags/antibodies are commercially available [76].
However, these fluorescent probes are mostly large hydrophobic moieties. If used to tag a macromolecule (protein, DNA, antibody …), the physico-chemical effect of the probe can be minimal and the location of the tagged “chemical object” not modified. But when it comes to tagging a low-molecular weight compound, such as those presented in Sections 3 and 4, a probe will most certainly modify the physico-chemical properties of the compound of interest (as exemplified in Section 5.5). This is why we envisioned to try to detect the metal complexes directly through the metallic core. This can be done using X-ray fluorescence imaging. Another interesting modality is IR absorption. In the mid-IR (4000–400 cm−1), molecules show specific signature (fingerprints) and chemical mappings can be generated.
We detail below some basic principles for X-ray Fluorescence Microscopy (XFM) and IR-microscopy (μ-IR), before explaining the design of multimodal probes for these two modalities and classical fluorescence, along with a few applications.
5.1.1. X-ray Fluorescence Microscopy (XFM)
XFM has emerged as a highly efficient technique to image heavy elements, including metal cations. Excellent reviews have detailed the underlying physics, technical characteristics, biological applications [76, 77, 78, 79, 80, 81] and possibilities offered by multimodal imaging (coupling of XFM with fluorescence [80] for instance).
In a nutshell, providing its energy is high enough, id est above the absorption edge of the core electrons, an X-ray beam can excite a core electron of a heavy element, possibly a metal cation, to a photoelectron. This leads to a more positive ion (+1) in an electronic excited state. Deexcitation pathways, to a cation in a less excited or in the ground state, can occur with an upper electron filling this core hole, with the emission of an X-ray fluorescence light, detected to perform micro-XFM [80]. Note that this emission is isotropic and, to limit incident beam light recording, the detectors are set at ca. 90° from the incident beam. The energy of the emitted light informs on the nature of the metal ion and on the two levels involved in the transition. Hence, each element emits a set of X-ray bands that are specific. For elements heavier than Si, these bands are clearly separated (Figure 12), and if there is an overlap, one can expect that at least one band in the emitted set is isolated.
X-ray Fluorescence (XRF) sum spectrum of A549 cells. Conditions: excitation energy: 13.5 keV, pixel size: 300 × 300 nm2, accumulation time: 4.8 s/pixel [82].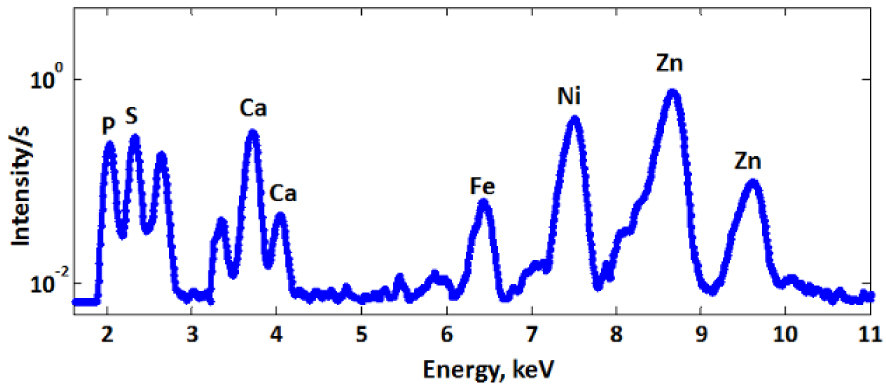
This makes X-ray fluorescence an excellent technique for detection of heavy elements in a sample. Synchrotron-based radiations in the hard X-ray (>2 keV, hence for elements heavier than Si) allow a brightness many orders of magnitude greater than other sources [77]. The incident beam can be focused to a size smaller than that of a cell (typical diameter of 20 μm for eukaryotic cells), while keeping enough power to detect diluted metal ions in the small voxel of observation. Raster scanning the sample through the beam makes subcellular bio-imaging of metal ions possible. Recently, resolutions better than 100 nm, down to 30 nm, have been achieved [80, 83].
It is possible to grow cells in a monolayer on a window transparent in the X-ray (typically, silicon nitride Si3N4 windows, sheets of Kapton® or Ultralene® sheets [78, 80, 81, 84, 85]) and map them to determine the metal ions content distribution at the subcellular level. The X-ray incident beam (excitation) and the emitted X-ray signal are penetrating enough to pass through the entire sample (thickness of one cell in case of a monolayer) to provide elemental distribution for the entire cellular depth [80]. Hence, at each pixel, the map describes an integration of the total content in the heavy element over the thickness of the cell. For example, the potassium (K) is homogeneously distributed in cells, and its map in Figure 13 shows its distribution within a cell in false colors indicating the content (red > black). The area with a higher content in potassium (red) corresponds to the location where the cell is the thickest. Interestingly, by rotating the slide in the beam, tomography of cells can be obtained [86], with a very long time of acquisition, but opening new 3D opportunities for the characterization of metal ions distribution in cells.
X-ray fluorescence mapping on cryofixed cells (human epithelial intestinal cells, HT29-MD2). Potassium (K) and manganese (Mn) maps by X-ray fluorescence on cryofixed and freeze-dried cells HT29–MD2 incubated with Mn1 (100 μM). Images recorded on the 2-ID-D beamline of APS synchrotron (excitation at 6.8 keV; integration time, 2 s pixel−1; pixel size, 200 nm) (from Ref. [25]).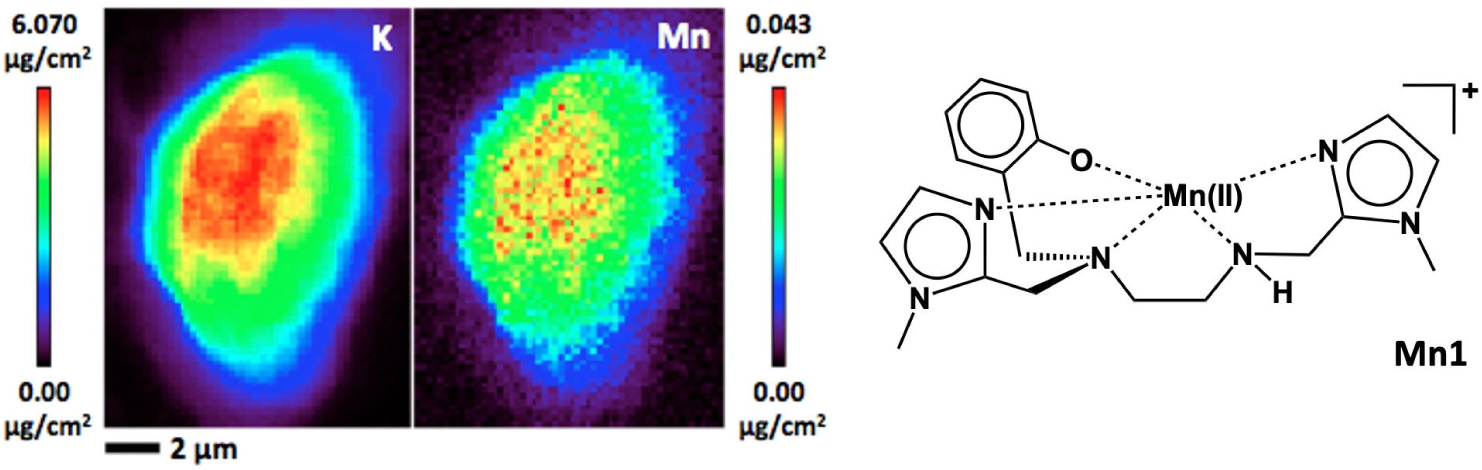
Sample preparation is a key aspect, for any kind of spectroscopy and imaging. For classical optical fluorescence, several methods of chemical fixation of cells have been developed, such as paraformaldehyde, that are suitable for XFM a priori. However, these techniques show issues associated with possible re-distribution of a molecule of interest. Let us take the example of fixation using paraformaldehyde (H2C=O, PFA) that reacts with amines present on proteins, peptides, or any other type of molecule. In a way, fixation with PFA casts some sort of a molecular net on the cell, but leaves large holes in which unfixed molecules can move and diffuse away. This can lead to redistribution, either a more even distribution within the cell or an accumulation in some organelles. For X-ray fluorescence, it is necessary to work with dry cells, and the drying process after chemical fixations has been shown to lead to leakage of metal ions, with a significant lowering in the apparent metal content [84]. This has been shown to mostly affect highly diffusible metal ions (Ca, K, Cl), but also the loosely bound transition metal ions pool that is not sequestered at the active site of metalloproteins, whereas metal cations such as Zn or Cu, highly controlled by proteins coordination, are not much redistributed by the fixation-drying procedure [81, 87].
Cryofixation, with shock-freezing through plunging in liquid hydrocarbons (ethane, typically), possibly associated with lyophilization (freeze-drying), has been shown to better preserve both the sub-cellular structure and the distribution of metal ions [84, 87]. After cryofixation, the frozen sample can be mapped at low temperatures. An alternative is to lyophilize the sample at low temperature, leading to a sample that can be handled at ambient temperature. But the lyophilization process must be performed steadily to ensure non-transition of ice from amorphous to crystals that would disrupt cell membranes (as crystalline water shows a lower density than liquid water or amorphous ice, such a transition would lead to a dilatation responsible for the damaging of the sub-cellular structure).
Nowadays, the consensual idea is that the less the sample is manipulated before being mapped the better, so as to avoid the effect of re-distribution but also contamination with metal ions, that in our modern world, can be found everywhere.
We have applied XFM to the imaging of Mn-based SOD mimics. We have used cryofixed/lyophilized cells incubated with the complexes at 100 μM, the typical concentration at which we evaluate their anti-inflammatory activity. As shown in Figure 13 for Mn1, the Mn element is distributed over the whole cell, as potassium, whereas a perinuclear distribution is seen for Mn1Re (further discussed in Section 5.3).
5.1.2. Infrared microscopy (μ-IR)
μ-IR is extensively used for its ability to perform detection using molecular chemical fingerprints in the mid-IR (4000–400 cm−1, energy range where all of the molecular vibrations are excited). IR-maps, or chemical imaging, can be recorded to provide local chemical endogenous information by determining distribution of specific chemical vibrational signatures, for instance, IR-absorption of proteins at ca. 1600–1700 cm−1 (amide I), or lipids at ca. 2900–3000 cm−1 ( and ) (Figure 14). IR bio-imaging has been used, for instance, to follow cellular events, such as proteins acetylation [88].
Typical IR-spectrum of a cell showing the main features mentioned in the text. Inspired by C. Sandt, SMIS beamline, synchrotron SOLEIL.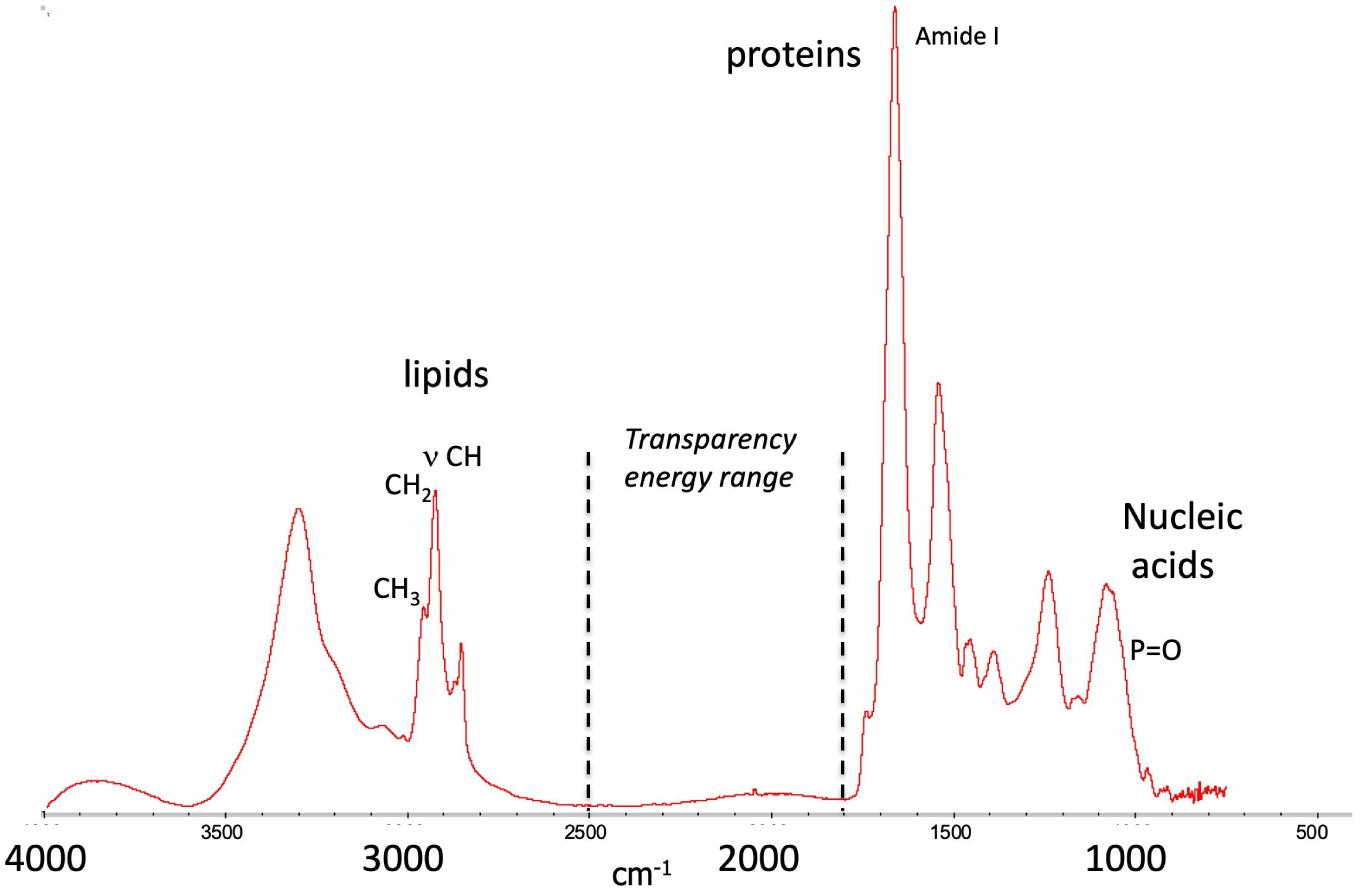
In the course of our work, we have used specific bands (amide, phosphate, lipids), or association of bands (e.g. high amide + phosphate for the nucleus for instance) [89, 90], or ratio of bands to identify specific organelles (membrane-rich organelle with very intense CH2 stretching versus nucleus with more intense CH3 stretching) [91]. One asset is that this chemical mapping of biological endogenous compounds is label-free, avoiding issues associated with the requirements of tagging [92], and can be implemented easily for bio-medical applications [93], with an easy process using bench IR-microscopes [94]. Synchrotron sources for IR-light are two to three-order of magnitude brighter than thermal source, making SR-FTIR mapping sensitive, with a lateral spatial resolution in the range of the μm (whereas bench-IR-microscope with a thermal source the spatial resolution is no better than 5–10 μm) [95]. The resolution with an optical detection is indeed physically limited by the light diffraction and the Abbe criteria (about λ/2, which is 2.5 μm at 2000 cm−1).
Sub-micrometric resolutions suitable for intracellular mapping in the IR can be achieved using a near-field technique, which involves exploring mechanically a sample with a probe of sub-cellular dimensions. This was made possible by photothermal induced resonance, coupling an Atomic Force Microscope (AFM) with an IR pulsed-laser. A pulsed irradiation in the IR at a wavelength tuned to the sample IR absorption induces an excitation in the vibrational levels. The only dissipation is heat-release in the near environment. This induces a local dilation [96, 97] and the AFM-tip receives a “punch” at every IR laser-pulse. The amplitude of its oscillation has been shown to be proportional to the deformation, and in fine to the amount of IR-absorbing compound below the tip. The resolution of the technique is that of the AFM (ca. 10–50 nm).
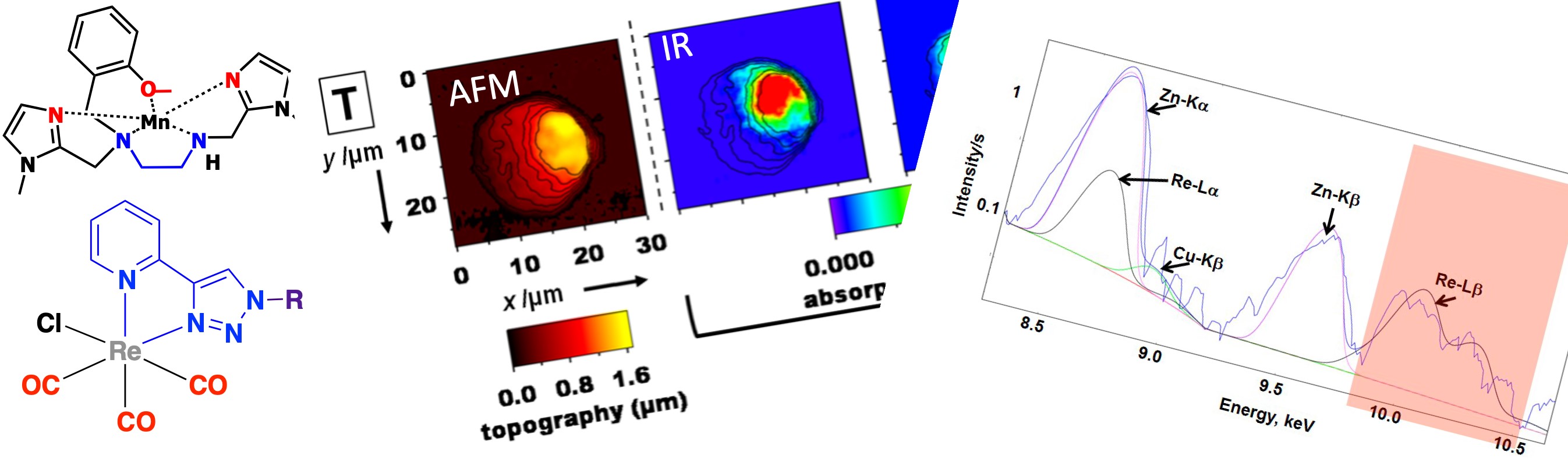
Interestingly, we were able to map the distribution of the [Re(LCp)(CO)3] compound indicated in Figure 15 in MDA-MB231 cells. Its distribution was colocalized with high spots of amide and phosphate, pointed at a nuclear accumulation which is an organelle dense in proteins and DNA [89]. The colocalization with the nucleus was checked in correlative imaging (SR-FTIR imaging and classical fluorescence of DAPI, a nucleus tracker) [98].
AFM-IR Top: images of MDA-MB-231 cells. MDA-MB321 treated with a 10 μm solution of [Re(LCp)(CO)3] for 1 h at 37 °C. (1) AFM topography; (2–6) AFM-IR mappings at different wavelengths, with the AFM contours superimposed as black lines; the wavelengths correspond to the following bands: phosphate (2), amide (3), tricarbonyl (4 and 5), and out of band (6). Bottom: structure of [Re(LCp)(CO)3]; IR-spectra of (a) pure [Re(LCp)(CO)3], (b) control cells, (c) cells treated with [Re(LCp)(CO)3] (50 μM, ca. 50,000 cells), and (d) AFMIR spectrum recorded at the nucleus for a single cell treated with [Re(LCp)(CO)3] (1 h, incubation concentration: 10 μm). See Ref [89] for details.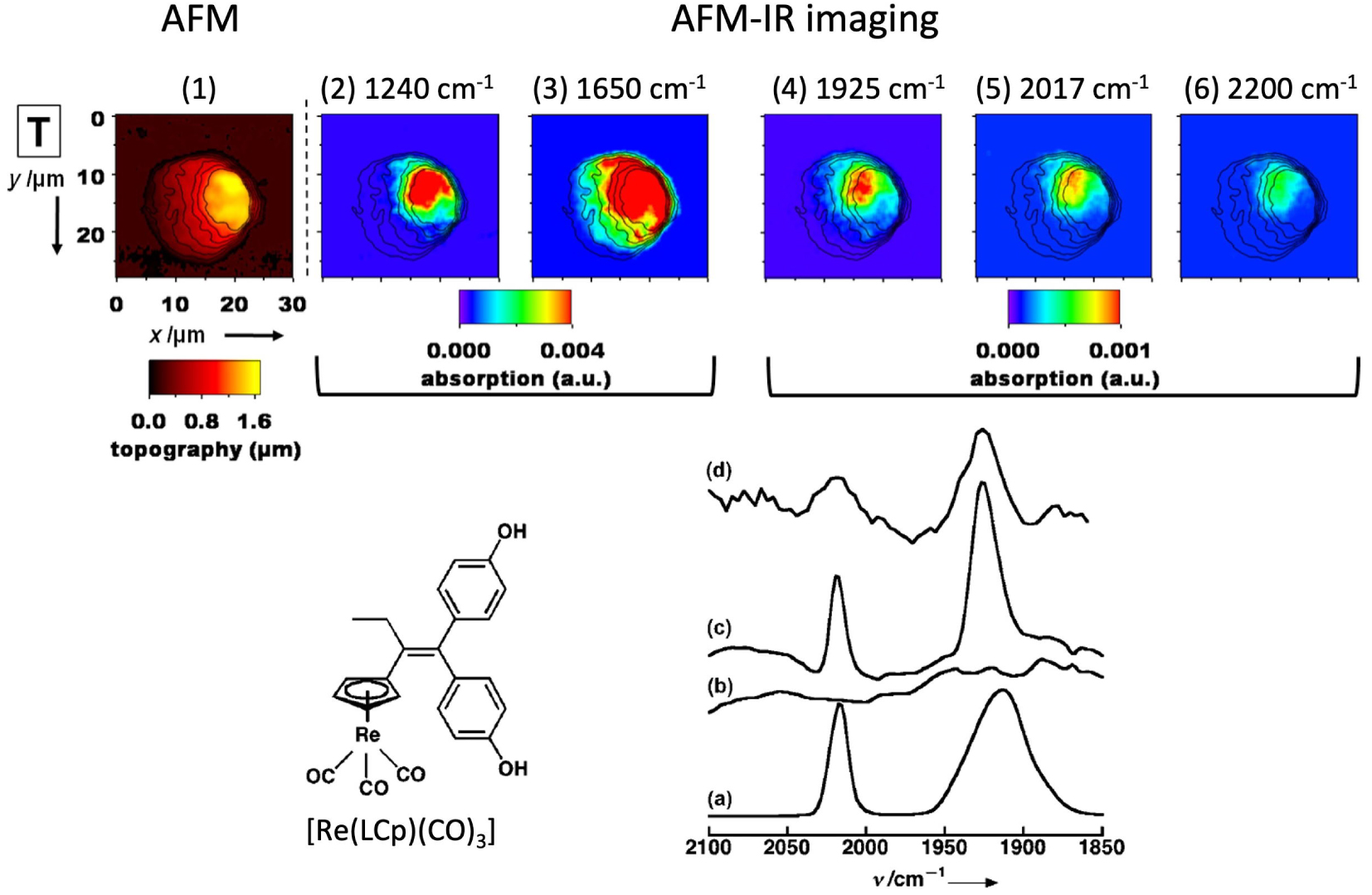
For sub-cellular imaging in the IR, the sample has to be on a slide transparent in the IR (thin section of a bio-sample or monolayer of cells grown on the slide):
- CaF2 slides with chemical fixation (too fragile for plunging and cryofixation). Note that for multimodal imaging in optical fluorescence, optical IR-imaging and AFM-IR [99], thin CaF2 slides (100–200 μm) can be used. In the case of AFMIR the thin slides can be laid on the sample holder with paraffin oil to ensure a good optical contact and avoid air gap [99]. For instance, we have performed IR-imaging on tissue sections [100, 101] or cells grown on the slide [53, 90, 99, 102, 103, 98], with multimodal imaging (FTIR, AFMIR, and classical X-ray fluorescence) in several cases.
- Si3N4 windows than can also be used for multimodal imaging in classical and X-ray fluorescence [104] with chemical or cryofixation.
- ultralene thin film that can be used for multimodal imaging in IR and X-ray fluorescence [101].
Note that mid-IR is absorbed by bio-samples and thin samples (10 μm max) should be used in the case of optical detection (the IR-light passes through the sample). Interestingly, for AFM-IR a set-up (with irradiation from above the sample) [105] allows thick samples. But then they are probed only at the surface (absorption of the IR-light or depth of the penetration of the evanescent wave and the thickness probed depends on the recording parameters [105]).
5.2. Design of IR or X-ray fluorescence probes
A wider availability of probes, as tags or organelles trackers, would contribute substantially to the development of these two unconventional bio-imaging techniques to answer key biological questions [106]. Probes that can be detected by several complimentary techniques offer the possibility of cross correlative comparisons. The conventional way to develop a multimodal probe is to covalently conjugate several molecular moieties, one for each modality, leading to large molecular entities, with risk of discrepancy if a moiety is cleaved. In contrast, we wanted to develop a “swiss-knife” probe, more compact, as a single molecular core for multimodal imaging (SCoMPI). To that extent, rhenium(I) tricarbonyl complexes are interesting. Indeed:
- Metal carbonyls (L)M(CO)n feature intense elongation vibration bands at ca. 2000–2200 cm−1 in the transparency window of biological sample in the mid IR (1800–2500 cm−1, see Figure 14). They have been used as markers for immunoassay, for example as pH or cation probes [107]. One can also note that for the IR modality, only vibrational modes are excited and it does not involve transition into an electronic excited state, whose reactivity, different from the ground state, may lead to non-fluorescence species. In other words, unlike what happens for fluorescence probes, no photobleaching of the probes is at stake in IR-imaging [108]. The sole deexcitation pathway after IR excitation is heat release, which opens up possibilities to use them for all imaging techniques relying on the photothermal effect such as IR nanospectroscopy (AFM-IR).
- Rhenium tricarbonyl complexes bearing heteroaromatic ligands with low lying π∗ orbitals [Re(N^N)(CO)3]n+ display interesting electronic properties at the origin of their development as luminescent probes. They show additional interesting features for use in a biological context such as low toxicity and good chemical inertness. The emission of such complexes mostly arises from triplet metal-to-ligand charge transfer (3MLCT) excited states and they display large Stokes shift. However, their quantum yield is low (usually around 1%) and their excitation usually occur in the UV-A range.
- Re gives access to detection in X-ray fluorescence, with bands that can be identified in a biological environment at its L-edge (one band, the L-α is superimposed with K-α of Zn, but the L-β is well-separated, as seen in Figure 16).
- Rhenium tricarbonyl complexes with the ligand pyta are easily synthesized and functionalized. The pyta (4-(2-pyridyl)-1,2,3-triazole) ligand seen in Figure 17 is formed through Cu-catalyzed Alkyne-Azide Cycloaddition (CuAAC) chemistry and there is a large variety of possible functionalizations of a R-N3.
Cu, Zn and Re XRF lines in the mean spectrum of a single Chinese Hamster Ovarian (CHO) cell incubated with an homeodomain trafficking protein tagged with a Re-probe. Recorded data in blue, fits for Cu (green), Zn (violet) and Re (black). For details, see Ref. [82].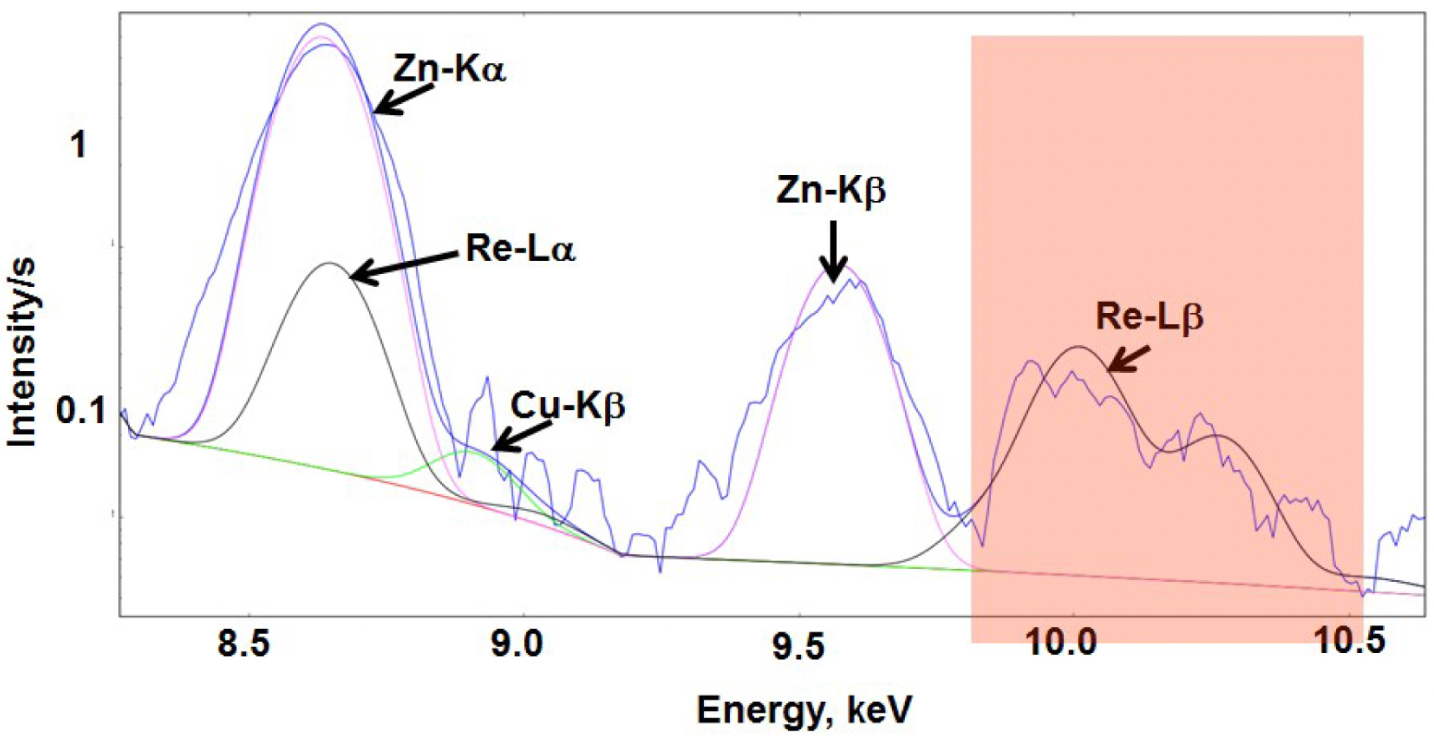
5.3. Variation of the ligands to control the physico-chemical properties of the probes
The photophysical properties of these [Re(N^N)(CO)3X] compounds have been well characterized, particularly with the N^N ligand being a bipyridine or a phenanthroline ligand. Their electronic properties can be tuned by modifications of the di-imine or the axial X ligand. We have explored the impact of diverse structural modifications around rhenium tricarbonyl complexes based on a pyridine–triazole N^N ligand (see Figure 17).
Schematic structures of the [Re(pyta)(CO)3Cl] and [Re(tapy)(CO)3Cl] complexes and their typical luminescence in aqueous medium.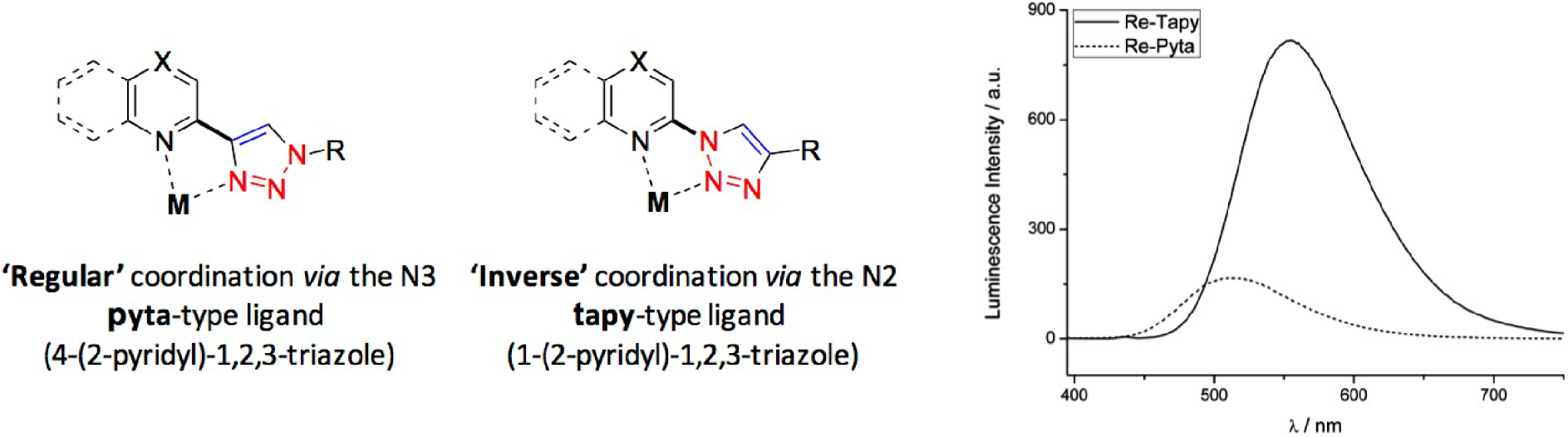
To explore questions of cell penetration and toxicity of [Re(pyta)(CO)3Cl] complexes, we have prepared and studied complexes with pyta ligands functionalized with aliphatic chains of increasing length (C4, C8, and C12) [109].
We showed that increasing the lipophilicity of the complex by the increase in the carbon chain length correlated with an enhanced cellular uptake, as quantified by a correlative luminescence and infrared (IR) study, and thus an increased cytotoxicity. This quantitative cellular study stressed out that cytotoxicity values (IC50) reflect intrinsic toxicity but also the ability of the compound to penetrate inside cells. Correlating the toxicity of a compound to its internal concentration and not to an incubation concentration as commonly performed requires the use of such analytical techniques. We also showed that the appendage of a long aliphatic chain (C12) on the pyta ligand strongly impacted the photophysical behavior of the complexes in aqueous medium compared to organic medium (such as acetonitrile) as opposed to shorter chains. The folding of the carbon chain around the metal core in aqueous buffer was proposed to shield the metal center from the emission quenching due to water.
In an attempt to optimize quantum yield and excitation wavelengths—aiming for a bathochromic shift of the Metal-to-Ligand Charge Transfer (MLCT) bands in the visible range that is better adapted for bioimaging—in these complexes, we studied structural variations of the pyta ligand (with X axial ligand as an halogen Cl or Br) [110]. Extending the aromatic surface (from a pyridine to a quinoline) in the quinta series, led to the expected bathochromic shift but it was associated with a decrease in the quantum yield. A satisfying compromise is challenging to reach. We identified the tapy (1-(2-pyridyl)-1,2,3-triazole) ligand as a very interesting scaffold for the design of rhenium tricarbonyl complexes. The common pyta ligand displays a normal or regular coordination through the N3. Inverting the bridge between the pyridine and triazole rings, we generated the tapy ligand showing an inverse coordination through the less donor nitrogen N2. DFT calculations confirmed the poorer electron-donor ability of the tapy ligand in comparison with pyta, with a stabilized LUMO level, leading to a smaller gap and a bathochromic shift of the MLCT absorption band. The enhanced luminescence was proposed later to arise from slower rates of non-radiative decay. The corresponding complexes displayed a strongly enhanced luminescence—particularly with long aliphatic chains—compared to the pyta counterparts, with quantum yields up to 3.5% in aqueous medium. Despite their reduced stability in coordinating solvents, the inverse coordination has interesting potential in organometallic chemistry.
We further modified Re(CO)3 complexes bearing pyta-C12 or tapy-C12 ligands (with long alkyl chains) replacing the axial X ligand [111]. The introduction of benzenethiolate π-donating ligands results in high lying filled orbital on sulfur and benzenethiolate leading to a strong absorption band of mixed MLCT/LLCT (LLCT: Ligand-to-Ligand Charge Transfer) character around 330 nm. The emission of such complexes depends both on the diimine and on the axial benzenethiolate, offering an opportunity to tune the emission wavelength across a wide range extending to the near IR region (Figure 18).
Structural variations on the pyta and tapy ligands, and variation of the fluorescence response.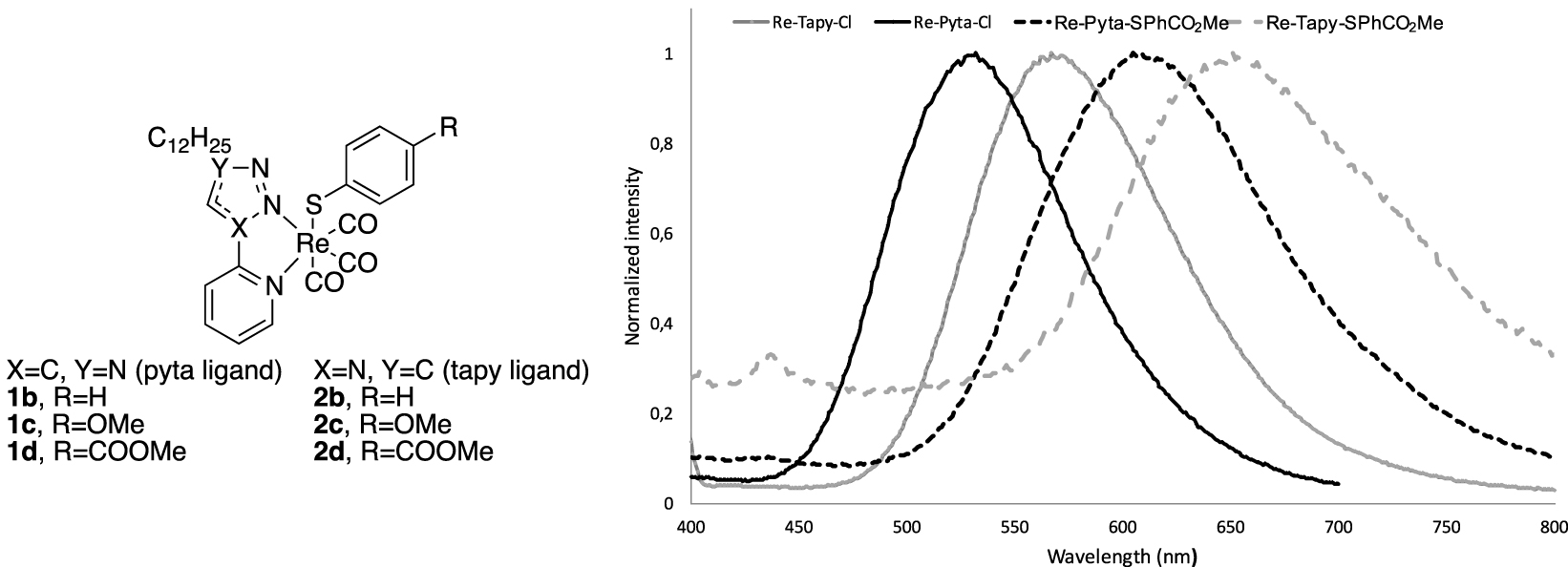
5.4. LMW organelle trackers working with PFA standard fixation
For co-localization studies by X-ray Fluorescence (XRF), some endogenous elements can be indicative of a subcellular compartment—e.g. phosphorus, sulfur, copper and zinc reveal nucleus location [112]. Manganese has been shown to accumulate in the Golgi apparatus in neural cells [113]. But this relative metal-specificity cannot be made general for all the organelles. Hence the design of probes for organelle tracking responsive in X-ray fluorescence is of great interest [106]. Sealed carbon nanotubes filled with heavy metals and decorated with organelle-specific peptides have been designed for nucleus targeting. But the attempt to track endoplasmic reticulum failed [114], showing this strategy of peptide-targeting for specific organelle tracking is not easy to extend. Metal-tagged antibodies have been developed, for instance with lanthanides [115, 116] and some used for mitochondria tracking—involving an antibody targeting a protein specific of this organelle [117, 118]. But the procedures involve permeabilization with leakage of the loosely-bound metal pool, that can be undesirable. Low-molecular weight complexes targeting organelles can also be used for their tracking [119, 120, 121] with compatibility with cryofixation. Recently, we have conjugated the typical [Re(pyta)(CO)3X] core, with trisphenylphosphonium cations of various lipophilicities (see Figure 19). Trisphenylphosphonium cations are known to accumulate at the mitochondria, due to their delocalized positive charge over a lipophilic trisphenyl core [62]. We chose here a procedure with PFA fixation, more accessible to users than cryofixation, that was hopefully successful. Indeed, using their fluorescent properties, the [Re(L)(CO)3X] probes were first co-localized with a fluorescent mitotracker. Then XFM with the same PFA fixation showed that the Re of the probe was easily detectable for the two more lipophilic probes C2 and C3, and an accumulation of Re (probe) around Zn (nucleus) was clearly seen [121]. This example also shows here the interest of the multimodality of the probes such as the metal-carbonyl probe: the multimodality of a probe means that it can be validated using a tried-and-tested technique, with a view to implementation using a less conventional technique.
Structures of the [Re(pyta)(CO)3]+ conjugated with a trisphenylphosphonium moiety and elemental distribution of Zn and Re (XFM) in A549 cells incubated with C2 (4 h, 20 μM) before PFA fixation and air drying (excitation at 14 keV; integration time, 300 ms per pixel; pixel size, 500 nm). XFM recorded at synchrotron SOLEIL, on nanoscopium beamline [121].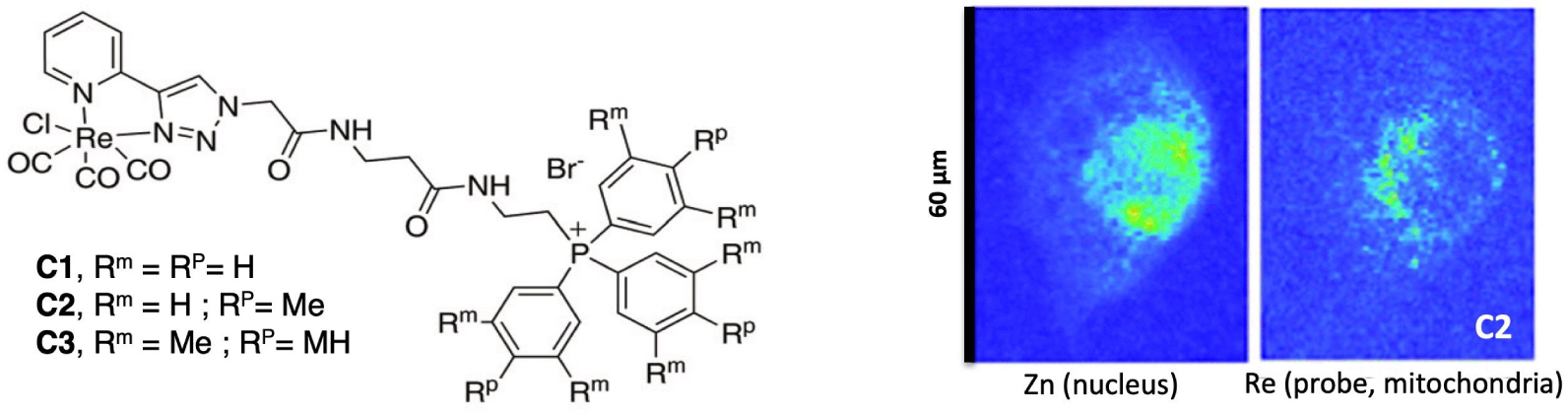
In another example, we have tagged a TrisPhenylamine-based (TP) mitotracker with a Br (TP-Br) or with a [Re(pyta)(CO)3]+ (TP-Re) [122]. TP-Br can be present in cells in a non-emissive off-state and, as such, fluorescence imaging could not reflect accurately the distribution of the probe. The TP-Br was found to be co-localized with a fluorescent mitotracker, whereas the TP-Re was co-localized with a lysotracker. In contrast with what happened with the trisphenylphosphonium functionalization, the [Re(pyta)(CO)3X] changed the localization of the TP probe. Interestingly, this makes TP-Br and TP-Re potential new organelle trackers for XFM.
Remembering that some of the [Re(L)(CO)3X] compounds we have studied have been shown to be accumulated at the Golgi [102] or at the nucleus (see Figure 15) [89], these Re(CO)-based compounds constitute a series of multimodal organelle trackers that can be followed by classical fluorescence, in IR-imaging, and X-ray fluorescence.
5.5. X-ray fluorescence imaging: an original direction to explore metal-complex integrity in cells through distribution
As mentioned earlier, one challenge faced when studying metal complexes in a biological environment is to ensure that the complex L-M remains intact, despite the abundance of Lewis bases, and the presence of other metal ions, even if they are mostly found coordinated by proteins. One way to interrogate this is to use a ligand tagged with a heavy atom (L∗) and check if the two moieties (heavy atom as a covalent tag of the ligand L∗ and metal ion) are present in cells at the same location [79]. This can be done by recording maps of cells incubated with a L∗-M complex in μ-X-ray fluorescence. This has been performed in the literature with a ligand tagged with Br [123] or I [124, 125].
As an example, the conjugate of Mn1 with a multimodal Re-tricarbonyl probe (Mn1-Re, Figure 20) was studied in the cellular model HT29-MD2 (Section 3.4b). Its integrity was probed by analyzing the Re and Mn colocalization by X-ray fluorescence imaging, Re tagging the Mn-ligand. The Re-maps and Mn-maps where not fully co-localized, with spots where Re (ligand) was present and not Mn, revealing some degree of decoordination in cells, as analyzed by IMS/MS (Section 3.6). Interestingly, Mn and Re showed a perinuclear accumulation (see Figure 20). In contrast, Mn1 has been seen homogeneously distributed (Figure 13, distribution of Mn similar to that of K). Interestingly, the two compounds, Mn1 and Mn1-Re, showed a similar bioactivity and similar concentration in mitochondria, despite a higher overall cellular concentration of Mn1-Re (Figure 20). This is probably because the cellular activity of these compounds is governed by the concentration reached at the mitochondria [53].
Up: elemental distribution of P, K, Mn, Re, and Zn in an HT29-MD2 cell incubated with Mn1Re. The phosphorus (P), and zinc (Zn, Kα-lines) maps are used to identify the nucleus area. The overlay (top right) corresponds to the Mn (Kα-line) (magenta) and Re (Lβ-line) (green) maps. The regions corresponding to an overlap of both elements are displayed in white. Intestinal epithelial cells HT29-MD2 were incubated for 2 h with Mn1Re (100 mM, 0.02% DMSO) before cryofixation and freeze-drying. Images were recorded on the 2-ID-D beamline of the APS synchrotron (excitation at 12.0 keV; integration time, 2 s per pixel; pixel size, 200 nm). Down left: C: control HT29-MD2 cells, Mn1/Mn1Re: HT29-MD2 cells incubated with Mn1/Mn1Re (10 and 100 μM, 6 h). IL8 secretion in HT29-MD2 cells. Intestinal epithelial cells HT29-MD2 were incubated for 7 h under different conditions indicated in the figure. LPS (0.1 μg⋅mL−1) was added at the end of the first hour. Data represent means ± SEM for 7–10 independent experiments. (∗) p < 0.05, (∗∗) p < 0.01 versus LPS condition. Down right: C-control cells, Mn1/Mn1Re-HT29-MD2 cells incubated with Mn1/Mn1Re (100 μM, 6 h). The total Mn content in cell lysates was determined in acid-digested cell lysates by titration using EPR. Data represent mean ± SEM for 4 independent experiments. (∗) p < 0.05, (∗∗) p < 0.01 versus C. The total Mn content in mitochondria-enriched fractions was determined by ICP-MS. Mitochondria were isolated using a mitochondria isolation kit (Thermo Fisher Scientific). Data represent mean ± SEM for 7 independent experiments; for details see Ref. [53].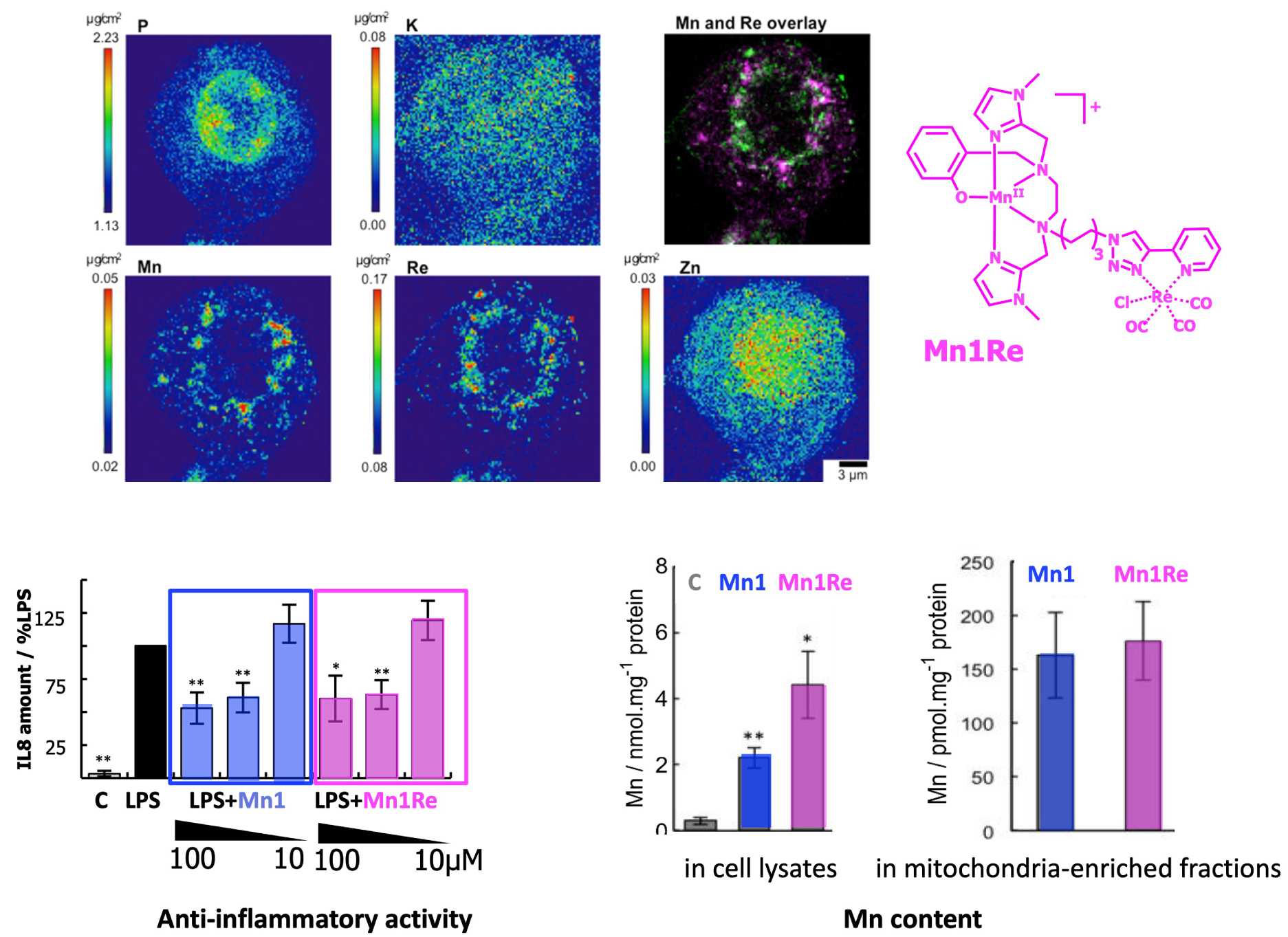
6. Conclusion
“[Naturam] si sequemur ducem, numquam aberrabimus”, Cicero, De officiis, I, 100.
Let Nature provide us guidance: we shall never get lost.
We would like to end this article on a less scientific note and comment on the research journey we have just described in the fields of bioinorganic chemistry and inorganic chemical biology, going from chemical design of metal complexes to their studies in cells.
Biomimicry has emerged as a highly heuristic approach: why not learn from the strategies selected by Nature during an evolutionary process spanning millions of years, and put them to use [126]? On a macroscopic scale, this might mean reproducing the movement of an insect to create a highly mobile robot or drawing inspiration from animal skeletons to create highly resistant vaults, as Antoni Gaudi, the precursor of biomimicry in architecture, did. This can be translated to the molecular scale: as chemists, we can seek to reproduce the way a biological system works, and move towards the design of bio-inspired catalysts of industrial or therapeutic value. The use of the term “bio-inspiration” instead of “biomimicry” aims to extend the approach beyond simple copying of structures to the creative development of new systems based on fundamental molecular principles deciphered in biology.
We have tried to illustrate this process in the case of highly efficient SODs, with physico-chemical features that could be implemented in low molecular weight complexes, namely the tuning of the redox potential to an optimal value for this redox catalysis, and a positively charged funnel in an overall negatively charged protein, for the electrostatic attraction of the negative substrate, superoxide. Transposing to a scale other than what nature has set up is not always straightforward, as we saw in the case of electrostatic attraction for the transition from the protein scale to those of low-molecular-weight systems, where simply increasing the overall charge was not enough without 3D organization. However, such approaches enable us to better understand Nature’s challenge piece by piece and to learn—or at least make progress in—mastering them at the molecular level.
Came then the need to design dedicated cellular assays to test these catalysts in biological environments. In a way, where biochemistry purifies metalloproteins from biological environments for study in test tubes, cellular and biological inorganic chemistry reverses the process, with the chemical design of metal complexes in the round flask which are then introduced in cellular or biological environments, where they are studied and evaluated.
Immediately, the question of sub-cellular location arose and we embarked on the path of bio-imaging. XFM, suitable for metal imaging, was an obvious technique to explore as we wanted not to modify the low-molecular weight complex and its physico-chemical properties too much.
The idea of developing metal-based IR-probes came about in a different way: this was the story of an encounter, as often in science, with a physicist, Professor A. Dazzi (université Paris-Saclay), who was developing a nano-IR imaging strategy (AFM-IR technique mentioned above). This technique which he had previously used to map biopolymers (DNA, sugars, proteins) was so amazing that we wanted to use it to detect and map non-endogenous molecules. Because of its IR-signature, the M(CO)3 moiety appeared as the ideal molecular core to test that, and their metal-core made them interesting probes to use in XFM.
It also illustrates how science progresses, tackling one question after another, one answer leading to a new more important question or idea to follow. Clearly, Nature is not only a formidable source of inspiration, but also an endless reservoir of interlocking questions and challenges for chemists.
This whole journey was made possible in the context of collaborative work. First, within the research group (https://ens-bic.fr/) where we share a common interest in bioinorganic chemistry and redox aspects, but with complementary expertise (peptide chemistry, organometallic chemistry, ligand synthesis, spectroscopies and now nanoparticles, redox biology and optogenetics). When you do not travel alone, the path is easier and enriched by discussions. But it also means collaborating with other disciplines: physicists, biologists, medical doctors, etc. Working in a multi-disciplinary environment leads to take that step aside that changes everything: it implies being an expert (in one’s own field) and at the same time an ignoramus (in another), with a renewed sense of wonder when you begin to grasp something new. It is also sharing with students, especially PhD students, with whom we live in the laboratory for at least a few years, and who bring a new vision and new questions, making research a place where we are always being challenged.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors want to acknowledge ENS-PSL, PSL University, CNRS, MITI-CNRS, Sorbonne University (SU) and the other bodies that funded this work. ANR-15-CE07-0027 MAGIC, DEI20151234413 (Fondation pour la recherche médicale 2016), and BACTMAN and ANACOMDA (Mission pour les initiatives transverses et interdisciplinaires-CNRS), the Association François Aupetit (AFA, research fellowship), ANR 21-CE18-0053-02 MOBIDIC, ANR 20-CE07-0039-01-CATMAN, ANR 23-CE23 METALINFY; Idex PSL Qlife project Main ANR Q-life ANR-17-CONV-0005; CEFIPRA project no. 6505-1; ANR-16-CE18-0017-01 SATIN; EMERGENCE SU EMRG-24 TOTEM; ANR-22-PEBI-0003 (PEPR).