


 CC-BY 4.0
CC-BY 4.0
1. Introduction
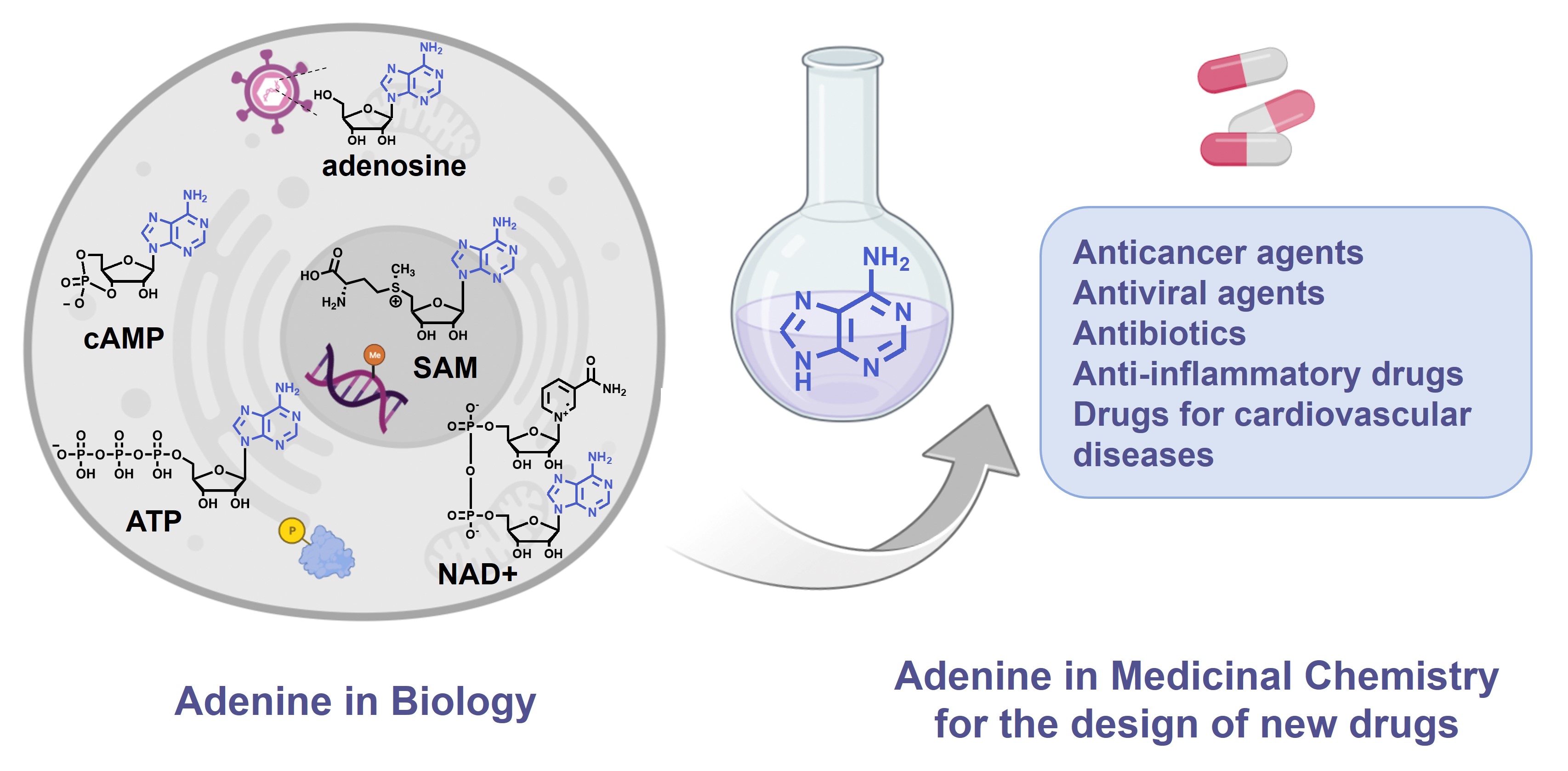
The purine ring system, and especially adenine, is one of the most ubiquitous natural heterocyclic compounds and plays major roles in various biological processes. In addition of being a nucleobase and building block of the genetic material and its transcription and translation in proteins, adenine stores energy in the form of Adenosine TriPhosphate (ATP) and is found in the structure of cofactors of many enzymes such as ATP, S-Adenosyl-L-Methionine (SAM or AdoMet), Nicotinamide Adenine Dinucleotide (NAD), Flavin Adenine Dinucleotide (FAD) and in substrates of metabolic pathways, as acetyl-Coenzyme A (acetyl-CoA) (Figure 1). ATP is both the source of energy at the cellular level, a precursor to DNA and RNA synthesis and a cofactor of protein kinases to phosphorylate their substrates [1]. SAM is used as methyl donor by MethylTransferases (MTases) to methylate histones, DNA, lipids and small biological molecules [2]. NAD+ is a cofactor for redox reactions and has a central role in energy metabolism. It is also a coenzyme for non-redox NAD+-dependent enzymes (CD38, sirtuins and poly(ADP-ribose)) and is a substrate for Nicotinamide Adenine Dinucleotide Kinases (NADK) [3]. Some signaling molecules such as cyclic Adenosine MonoPhosphate (cAMP) contain also an adenine scaffold. cAMP is an ubiquitous second messenger that regulates various biological processes, interacting with protein effectors such as Protein Kinase A (PKA), Exchange Proteins Activated by cAMP (EPACs) and Cyclic Nucleotide-Gated (CNG) channels [4].
The adenine scaffold (in blue) in biological molecules and therapeutic agents.
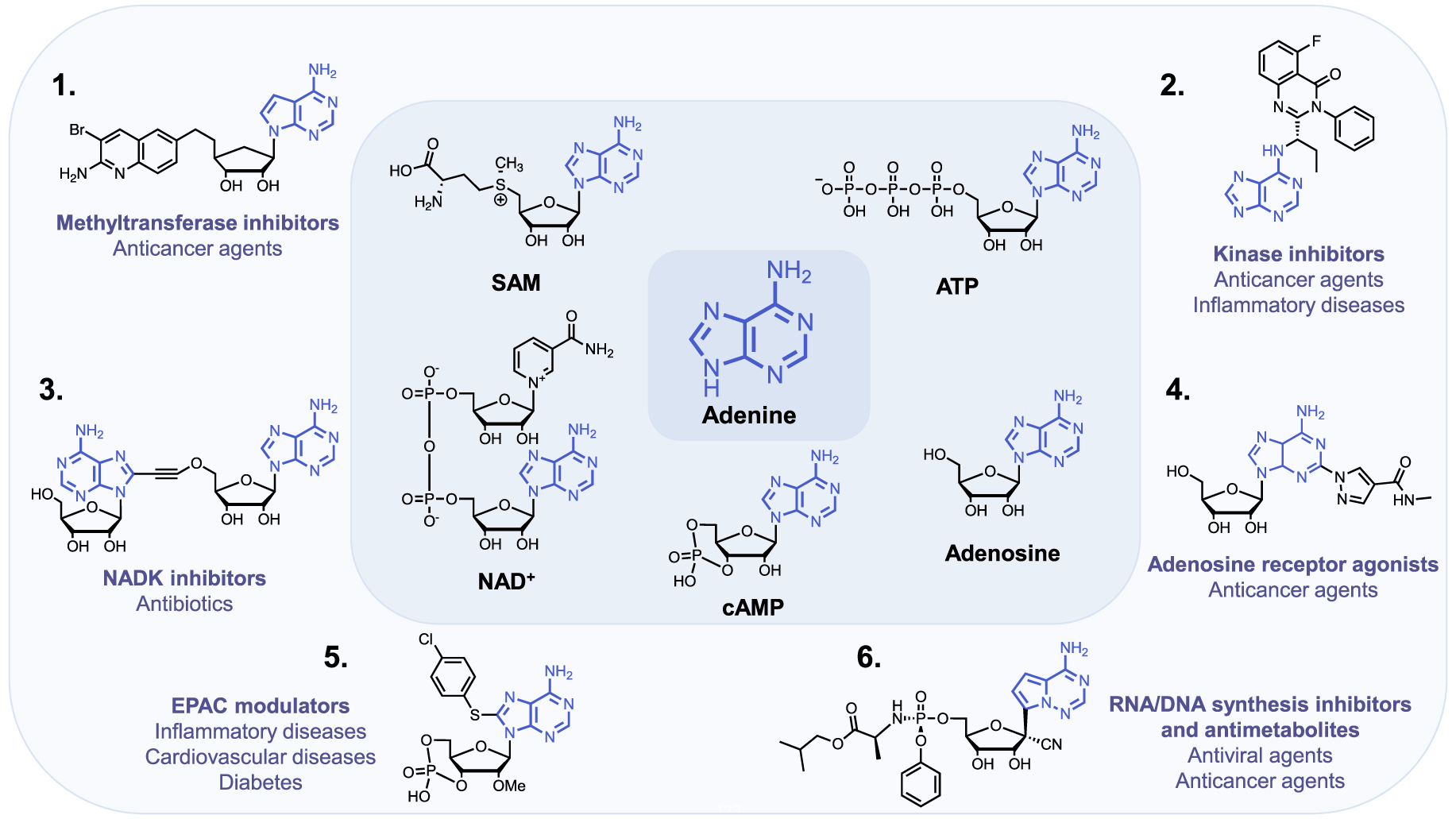
Importantly, these biological pathways are deregulated in diseases and the aforementioned protein families represent major therapeutic targets. Thus, the adenine scaffold has been exploited to design and develop therapeutic agents [5, 6, 7]. Chemical modifications of the adenine scaffold—giving adenine analogues—have been carried out to identify inhibitors of these pathways deregulated in diseases. Kinases and MTases inhibitors, based on the adenine scaffold are used for cancer treatment [8, 9]. Adenosine analogues inhibiting DNA/RNA synthesis are widely utilized as anticancer and antiviral agents [10, 11]. NAD Analogues are investigated to target NADK in the context of bacterial infection [12]. Finally, cAMP analogues are emerging as modulators of EPACs, playing a role in inflammation, diabetes and cardiovascular diseases [13]. Another example is the use of adenosine receptor agonists as anticancer agents [14].
Herein, we review the highlights of the use of the adenine scaffold for the design of versatile therapeutic agents and its role in biological processes. This review is not meant to be exhaustive and presents its chemical modulation in Medicinal Chemistry through chosen examples of approved drugs and inhibitors in clinical trials and preclinical studies for selected therapeutic targets.
2. S-Adenosyl-L-Methionine/S-Adenosyl-L-Homocystein (SAM/SAH) analogues to target SAM-dependent methyltransferases
S-Adenosyl-L-Methionine is one of the most ubiquitous cofactors. It is mainly used by the SAM-dependent MethylTransferases (MTases) as methyl donor to methylate their substrates (DNA, RNA, proteins, carbohydrates and metabolites). These methylated substrates contribute for example to the epigenetic regulation, protein function modulation, metabolites synthesis and degradation etc … [15, 16, 17, 18, 19].
SAM is a hybrid compound of adenosine bonded to the sulfur atom of methionine. The sulfonium group that bears the methyl group confers the SAM’s reactivity. Under the catalytic reaction, the nucleophilic substrate (N-, C-, O- or S-nucleophiles) attacks the electrophilic methyl group through an SN2-type mechanism generating S-Adenosyl-L-Homocystein (SAH) as byproduct (Figure 2A) [20, 21]. The methyl group transfer happens in the catalytic site of MTase that is composed of two pockets: a SAM binding site and a substrate binding site [15].
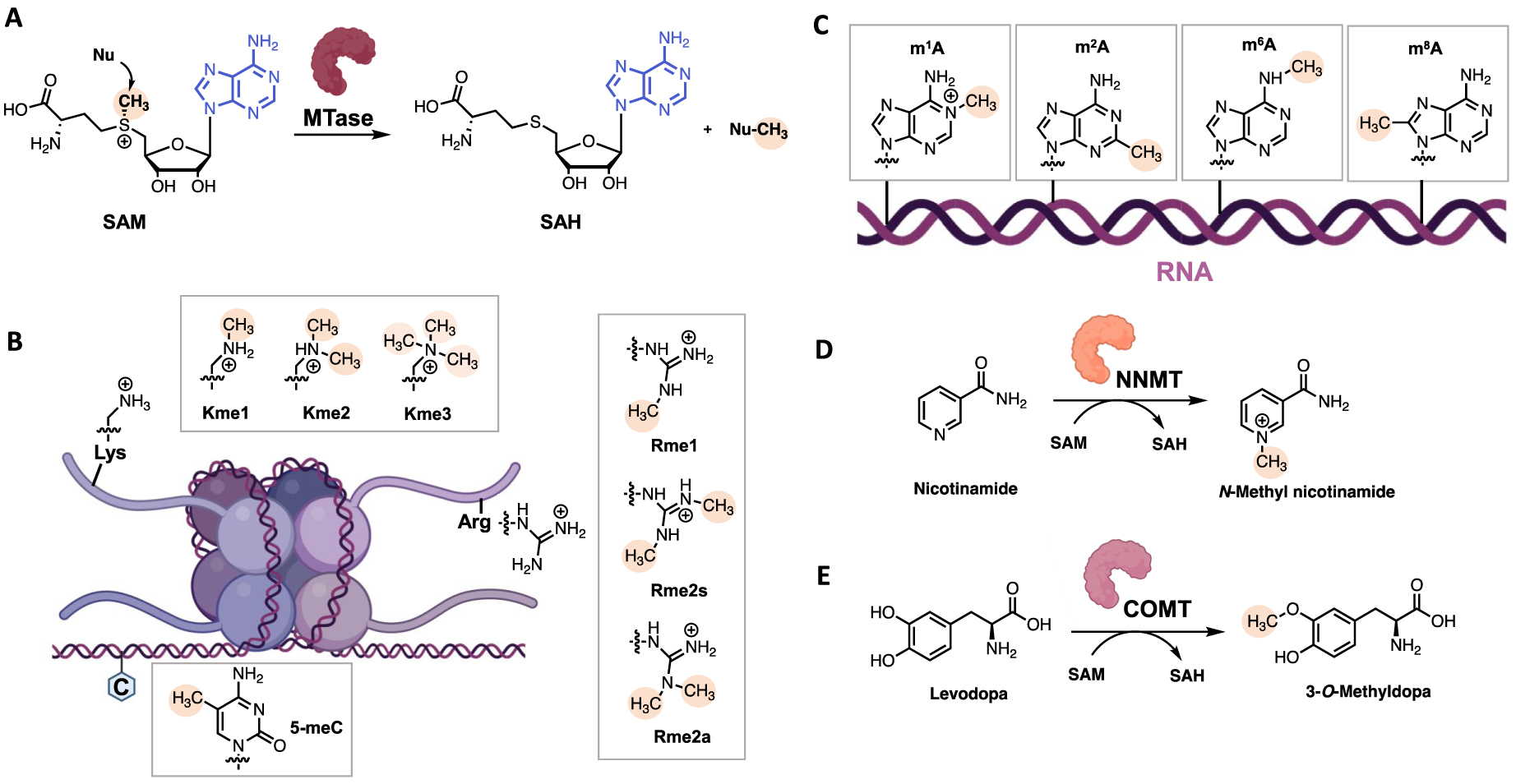
(A) SAM-dependent MTase-catalyzed methylation reaction; (B) examples of methylation products: mammalian histone and DNA methylation by HMTs and DNMTs; (C) examples of adenine methylation on RNA; (D) NNMT-catalyzed methylation of nicotinamide; (E) COMT-catalyzed methylation of Levodopa. (Biorender.com was used to prepare part of the figure.)
SAM-Dependent MTases are a wide and diversified class of enzymes. They can be classified according to their type of substrate. First, Histone MethylTransferases (HMTs) catalyze the transfer of methyl group on lysine and arginine residues of histones and are partitioned into Protein Arginine MethylTransferases (PRMTs) and Lysine MethylTransferases (KMTs) (Figure 2B) [22, 23]. Other protein methyltransferases methylate side chains of non-histone proteins [24], especially N-Terminal MethylTransferases (NTMTs) have recently raised interest recently [25, 26]. Among the DNA MTases, C5 DNA MethylTransferases (DNMTs) catalyze the methylation of C5-position of cytosines (5mC) in DNA (Figure 2B), which plays a major role in human diseases, and for which we and other have developed several inhibitors [27]. Most recently, the biological role of RNA methylation states (as on adenine, Figure 3C) is emergining. Additional MTases are responsible for the methylation of small molecules such as Nicotinamide N-MethylTransferases (NNMTs) that methylates nicotinamide and Catechol O-MethylTransferases (COMTs) that methylate catecholamines for neurotransmitter degradation [28] (Figure 3D, E).
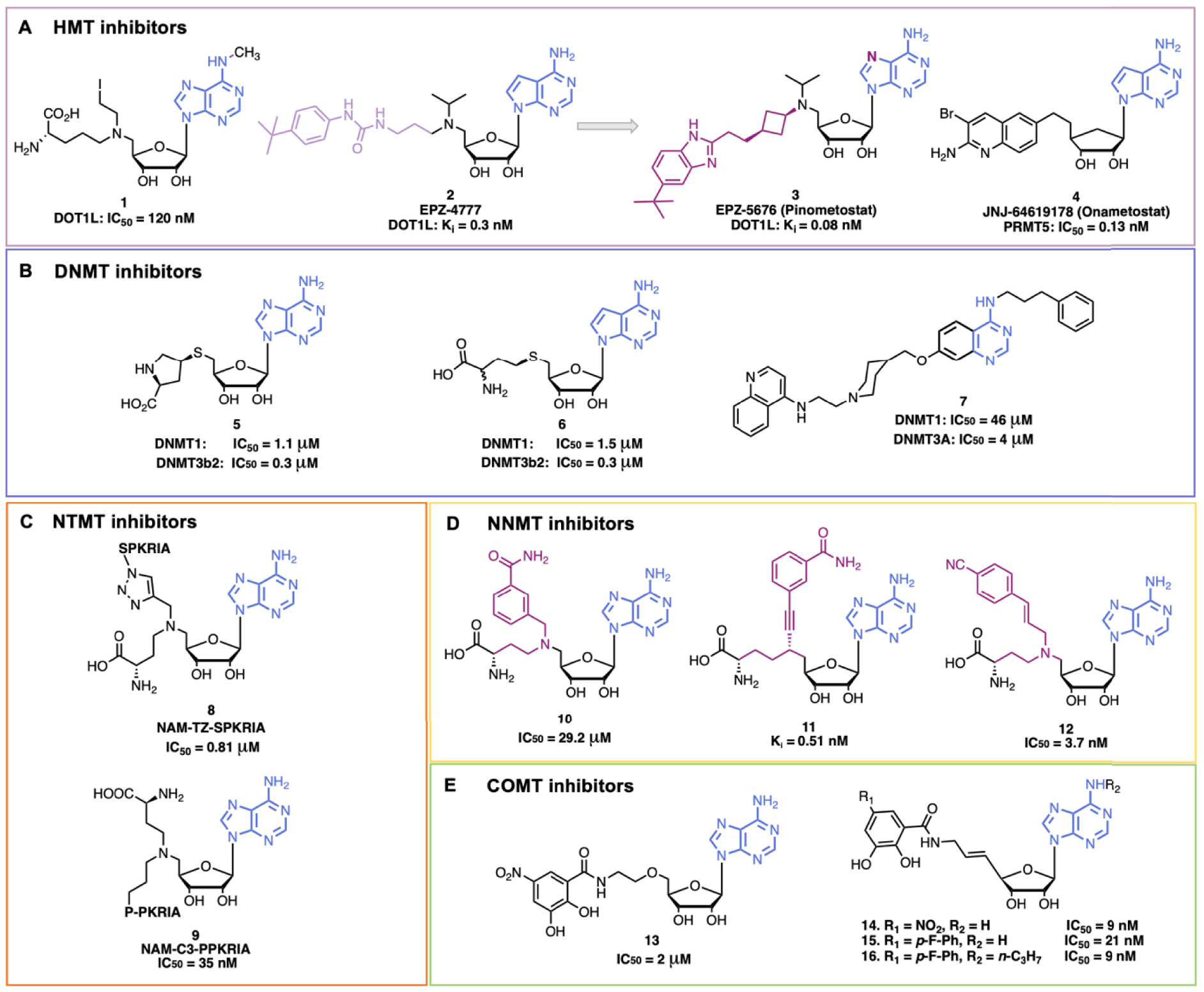
Examples of some SAM/SAH inhibitors. (A) HMT inhibitors; (B) DNMT inhibitors; (C) NTMT inhibitors; (D) NNMT inhibitors; (E) COMT inhibitors.
Due to the plethora of biological functions of MTases but also to their involvement in various diseases, SAM and SAH analogues have been developed and exploited either as MTases inhibitors or as tools to study the functions of MTases[29].
2.1. Histone MethylTransferases (HMT) inhibitors
HMTs are a broad class of enzymes with more than 50 members of KMTs and 9 PRMTs [30]. KMTs catalyze mono- (Kme1), di- (Kme2) or tri- (Kme3) methylation of lysine residues, whereas PRMTs catalyze monomethylation of arginines (Rme1) or dimethylation of arginines (in a symmetrical (Rme2s) or asymmetrical (Rme2a) manner) (Figure 2B). HMTs are involved in various diseases, in particular cancers and infectious diseases, and are thus very interesting therapeutic targets [31, 32, 33, 34, 35]. SAM and SAH scaffolds were exploited to develop numerous HMT inhibitors [36]. We chose few examples among the large literature to show the design and application of adenine-based inhibitors (Figure 3).
SAH is an endogenous inhibitor of many MTases and shows, for example, a similar inhibitory potency for PRMT4, G9a and DOT1L (IC50 ≈ 1.5 μM). Hence, co-crystal structures of SAH complexed with different HMTs were exploited to develop HMT inhibitors. DOT1L, the only HMT that catalyzes H3K79 methylation, attracted the interest notably because it is a major target for acute leukemia with Mix Lineage Leukemia (MLL) translocations. Interestingly, DOT1L is a KMT that does not bear the catalytic SET domain but rather shows a similarity in the 3D structure of the catalytic pocket with the PRMT’s one, and exhibits a unique hydrophobic subpocket in the adenosine binding site [37]. Following this rational, Yao et al. designed a potent and selective compound, 1 (Figure 3A), with a methyl substitution on the N6 position of SAH (to protrude interactions in the hydrophobic pocket specific of DOT1L) and the C–S bond of SAH was replaced by a C–N bond (DOT1L, IC50 = 120 nM) (Figure 3A) [38]. In 2011, Epizyme Inc. developed a new potent DOT1L inhibitor EPZ-4777, 2, where the 7-deaza SAM mimics is linked to a bulky 4-tert-butylphenyl group to occupy also the substrate pocket (DOT1L, Ki = 0.3 nM ± 0.1) [39, 40]. This compound is a SAM competitive inhibitor and the 4-tert-butylphenyl group occupies a newly opened hydrophobic subpocket. EPZ-4777 efficiently inhibits the proliferation of MLL-rearranged leukemia cell lines but the poor pharmacokinetic properties of the compound blocked its progress. To improve the pharmacokinetic parameters, a new compound EPZ-5676, 3 (Pinometostat), was developed. In EPZ-5676 the propyl chain of EPZ-4777 is replaced by a cis-cyclobutyl ring and the benzimidazole moiety replaces the aryl-substituted urea group. Moreover, the adenosine part of SAM is conserved in EPZ-5676 [41]. The compound is very potent (DOT1L, Ki = 0.08 nM ± 0.03) and is selective towards DOT1L. In addition, EPZ-5676 shows better residence time (>24 h versus 1 h) and entered clinical trials (NCT03701295). Interestingly, since EPZ-4777 weakly inhibits PRMT5 (at 30 μM), it served at starting point for the development of new PRMT5 SAM-competitive inhibitors. After extensive SAR exploration, compound JNJ-64619178, 4 (by Johnson and Johnson), was developed (Figure 3A). It is a bisubstrate inhibitor composed of a 7-deaza SAM mimics and a substituted quinoline moiety, occupying the second substrate binding pocket. JNJ-64619178 is potent (PRMT5, IC50 = 0.13 nM) and selective towards PRMT5; it displays favorable pharmacokinetic properties [42] and entered clinical trial notably for the treatment of advanced solid tumors (NCT03573310) [9].
2.2. DNA MethylTransferases (DNMT) inhibitors
DNA methylation, mediated by DNMTs, contributes to maintain normal cell function and to embryonic development. DNMT1 catalyzes methylation of newly synthesized DNA strands, while DNMT3A/B are responsible for de novo DNA methylation [43]. In humans, DNA methylation mainly takes place on position 5 of cytosine in a CpG dinucleotide context and is generally associated with gene repression when in promoters. Abnormal DNA methylation patterns and alterations in DNMTs activity can induce tumor development and maintenance. DNMTs are thus interesting anticancer targets [27]. Azacitidine (Vidaza™) and Decitabine (Dacogen®), are two DNMT inhibitors approved by the FDA (US Food and Drug Administration) [44, 45]. These drugs are cytosine analogues that replace cytosine in DNA, and in RNA for the former. However, they lack selectivity towards CpGs and show poor stability. More recently, a new class of reversible DNMT1-selective inhibitors has been developed by GlaxoSmithKline (GSK), targeting allosteric sites. The compound GSK-3484862 that contains a dicyanopyridine moiety is promising with its low cytotoxicity and great specificity [46, 47]. In addition, adenine analogues have been investigated to mimic SAH. MethylGene Inc. performed structural modifications of SAH and showed that substituting the homocystein with a proline mimics confers potent activity (compound 5 in Figure 3B) [48], while the 7-deaza SAH, 6, showed similar activity as SAH [49]. Finally, DNMT bisubstrate inhibitors with a quinazoline moiety as adenine mimic were developed by our laboratory and others [50, 51, 52, 53]. Compound 7 inhibited DNMT3A and DNMT1 at 4 μM and 46 μM respectively (Figure 3B) [54].
2.3. N-Terminal MethylTransferase (NTMT) inhibitors
Protein N-Terminal MethylTransferase 1 (NTMT1) methylates the N-terminal amine of proteins and recognizes the canonical motif X-P-K (with X = A, P or S) [55]. Dysregulation of NTMT1 is associated with several cancers and aging processes [56]. NTMT inhibitors have been developed notably to study the biological functions of Nα methylation. A first NMT1 bisubstrate inhibitor NAM-TZ-SPKRIA, 8, has been developed by Zhang et al. (Figure 3C) [57]. It is composed of 5′-N SAM analogue coupled to an hexapeptide (SPKRIA) via a triazole linker. The compound is very potent (IC50 = 0.81 ± 0.13 μM) and shows a 60-fold selectivity over other representative MTases. Later, Chen et al., developed a new series of NTMT1 bisubstrate inhibitors [58]. With a new synthetic route, they replaced the first serine residue of the SPKRIA peptide by a proline residue giving the NAM-C3-PPKRIA, 9 (Figure 3C), exhibiting an improved potency (IC50 = 35 ± 2 nM) and more than 100-fold selectivity over NTMT1 against a panel of MTases. However, the compound showed a poor cell permeability preventing cell-based studies. Nonetheless, this work enabled to obtain the first co-crystal structure of inhibitors with NTMT1 that would help for the development of novel NTMT1 inhibitors.
2.4. Nicotinamide N-MethylTransferase (NNMT) inhibitors
NNMT catalyzes methylation of nicotinamide using the SAM cofactor (Figure 2E). NNMT has a major role in cell detoxification and metabolism. Indeed, NNMT can recognize a broad range of substrates (pyridines, quinolines, diverse heterocyclic metabolites) and catalyzes their methylation, which is followed by their excretion [59]. Nicotinamide is also the precursor of NAD+. Moreover, NNMT balances SAM/SAH ratio that impacts indirectly gene expression [60]. Indeed, it has been reported that NNMT is also involved in the development and progression of cancers, neurodegenerative diseases, diabetes and obesity [61, 62, 63]. To better understand the role of NNMT in these diseases, several NNMT inhibitors have been developed, as potent bisubstrate compounds composed of SAM and nicotinamide mimics covalently bound through a linker. In 2017, Van Haren et al. developed the first bisubstrate compound 10 targeting NNMT (IC50 = 29.2 ± 4.0 μM) [64], followed by other derivatives [65, 66]. In particular, Policarpo et al. synthesized a new potent bisubstrate compound 11 with an increased potency in the namomolar range (Ki = 0.51 ± 0.08 nM) [67]. They figured out that an alkyne linker between the NAM-like and SAM-like fragments was optimal to mimic the transition state geometry. Recently, Gao et al. developed a novel potent bisubstrate compound 12 (IC50 = 3.7 ± 0.2 nM) with a para-cyano substituted styrene scaffold in the nicotinamide mimic that replaces the common benzamide moiety [68]. However, the compound was not potent in cell-based assays probably due to a poor cell permeability. All these findings help the further design and development of new NNMT inhibitors.
2.5. Catechol O-Methyltransferases (COMT) inhibitors
COMT catalyzes methylation of catecholamines (e.g. dopamine and epinephrine) but also of Levodopa (L-dopa) producing 3-O-Methyldopa (Figure 2D). L-Dopa is the precursor of dopamine and is used to treat the symptoms of Parkinson Disease (PD) [69]. Administrating COMT inhibitors avoids L-dopa deactivation and prolongs the effects of PD treatment [70]. COMT bisubstrate inhibitors were developed. Based on the crystal structure of COMT with Mg2+, SAM and 3,5-dinitrocatechol, the bisubstrate compound 13 was designed [71]. Compound 13 is a competitive inhibitor of SAM and a noncompetitive inhibitor of catechol. The potency was increased by rigidifying the linker with a double bond giving compound 14 (IC50 = 9 nM) [72]. To avoid hepatotoxicity, the nitro group was replaced by fluoro-substituted phenyl moiety giving the potent compound 15 (IC50 = 21 nM) [73]. Substitution of the OH at position 3 of ribose gave compound 16 (IC50 = 9 nM) [74].
3. Adenine scaffold in kinase inhibitors
Protein kinases catalyze the transfer of a γ-phosphate group from ATP to a substrate as for example tyrosine, serine or threonine residues of proteins (Figure 4A). Approximately 538 kinases have been identified in the human genome so far [75]. Kinases play major roles in numerous cellular processes such as cell proliferation, transcription, apoptosis and cell survival. However, they are also involved in the development and progression of numerous diseases, especially cancers but also inflammatory, neurological and cardiovascular diseases [1]. Kinases are thus validated drug targets. Over 70 kinase inhibitors targeting more than 20 different protein kinases have been approved with most of them used for cancer treatment [76]. Kinase inhibitors can be classified according to their binding mode: Type I inhibitors are ATP-competitive inhibitors and bind to the active conformation of kinases, Type II inhibitors bind to the inactive conformation of kinases, Type III inhibitors bind to an allosteric pocket adjacent to the ATP-binding pocket and Type IV inhibitors bind to an allosteric pocket distant to the ATP-binding pocket [77].
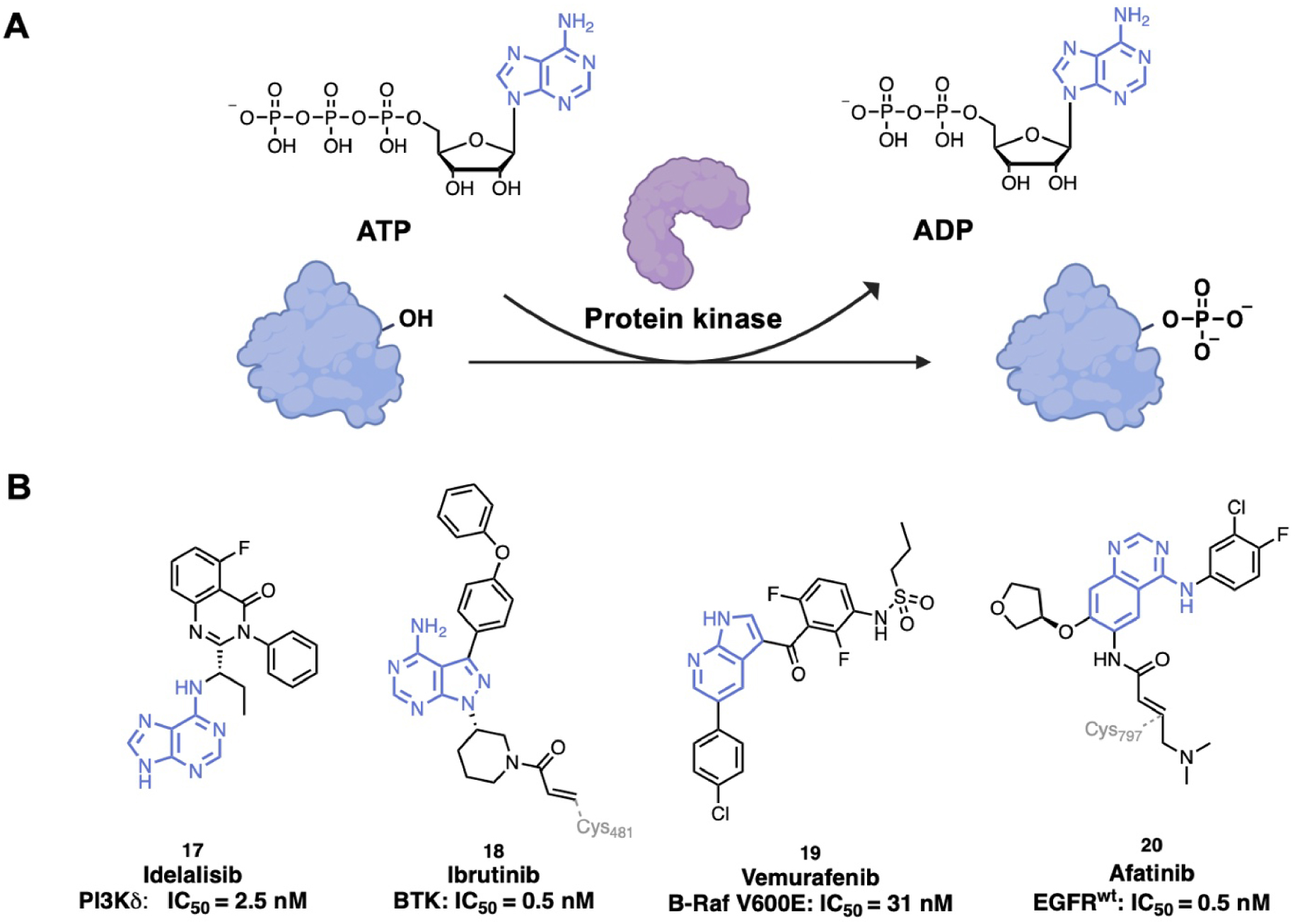
(A) Protein phosphorylation-catalyzed reaction by protein kinases; (B) examples of kinase inhibitors with an adenine analogue scaffold. (Biorender.com was used to prepare part of the figure.)
Most of the kinase inhibitors bind to the ATP-binding site. ATP mimics bind making hydrogen bonds to the kinase hinge backbone similarly to the ATP adenine ring [77]. Different chemical scaffolds of adenine analogues are used (Figure 4B). Some of them present the purine scaffold that occupies the adenine pocket such as Idelalisib, 17, (Zydelig®, by Gilead Sciences), a PhosphoInositide 3-Kinase δ (PI3Kδ) inhibitor approved to treat relapsed Chronic Lymphocytic Leukemia (CLL) [78]. Idelalisib establishes a type II binding [79]. The 5-fluoroquinazolinone part binds to an induced hydrophobic specificity pocket and the purine moiety occupies the adenine pocket by forming a key hydrogen bond between the N6 position of the adenine ring and Val828 [80, 81]. The pyrazolo aminopyrimidine scaffold is also used as adenine mimic in inhibitors such as in Ibrutinib, 18, (Imbruvica®, by Pharmacyclics Inc.), a Non-Receptor protein Tyrosine Kinase (NRTK) targeting Burton’s Tyrosine Kinase (BTK) that was approved to treat mantle cell lymphoma and CLL [82]. Crystallography studies showed that Ibrutinib is an irreversible BTK inhibitor, which covalently binds through Michael addition with Cys481 of BTK [83]. However, some insights on the mechanism of action remain to be understand, as more recently, the determination of the structure of Ibrutinib with SRC (a non-receptor protein tyrosine kinase, PDB ID: 6L8L) showed that Ibrutinib binds via a non-covalent manner to the protein [84]. Pyrrolo pyridine scaffolds are also used, as in Vemurafenib, 19, (Zelboraf®, licensed to Roche), an inhibitor of the serine/threonine B-Raf enzyme (member of the Raf kinase family) approved for late-stage melanoma [85]. The structure of Vemurafenib complexed with V600E B-Raf, determined by X-ray crystallography (PDB ID: 3OG7), showed a type I inhibition mechanism. The pyrrolo pyridine core of Vemurafenib mimics the adenine core of ATP by forming hydrogen bonds with hinge residue Cys532 and Gln530. Moreover, the sulfonamide moiety interacts through hydrogen bonds to the DFG residues and the 4-chlorophenyl moiety is exposed to the solvent [86]. Since resistance has appeared with mutations in the protein, new inhibitors have been designed (Belvarafenib by Hanmi Pharmaceutical/Genetech and Lifirafenib by Novartis for example) [87]. Kinase inhibitors with a fused six-membered ring system such as a quinazoline, quinoline or isoquinoline moiety are the most used ATP mimics (18 FDA approved inhibitors) [77]. For instance, Afatinib, 20, (Gilotrif®, by Boehringer Ingelheim), composed of a quinazoline moiety, is an irreversible inhibitor of Human epidermal growth factor receptor 2 (Her2) and Epidermal Growth Factor Receptor (EGFR) kinase. It was the first irreversible kinase inhibitor clinically approved for treatment of Non-Small Cell Lung Carcinoma (NSCLC) [88]. A type I binding mode was shown with co-crystal structures of EGFR-Afatinib (PDB ID: 4G5J) and mutant T790M EGFR-Afatinib (PDB ID: 4G5P). The quinazoline core binds with a conserved hydrogen bond to the hinge residue Met793 [89].
It is worth mentioning that kinase inhibitors are also used to treat non-oncologic diseases such as inflammatory diseases, autoimmune diseases, cardiovascular diseases, diabetes and some neurodegenerative disorders [90]. Noteworthy, some promising kinase inhibitors with as adenine scaffold are currently in preclinical trials for Amyotrophic Lateral Sclerosis (ALS) [91].
4. Nicotinamide Adenine Dinucleotide (NAD) analogues to target Nicotinamide Adenine Dinucleotide Kinases (NADK)
Nicotinamide adenine dinucleotide is an essential cofactor in energy metabolism. NAD is a dinucleotide containing an adenine and nicotinamide linked together through a ribose-phosphodiester backbone (Figure 5A). NAD can be in two forms: in an oxidized form (NAD+) or in a reduced form (NADH). NADK catalyze the phosphorylation of NAD on the 3′-OH of the adenosine to NADP, which is further reduced to NADP(H) (Figure 5B–C) [92, 93]. NADK is the unique enzyme producing NADP de novo and is essential to control the balance of NADH and NADP(H) in various metabolic pathways. NADK has crucial roles in pathogenic bacteria such as Mycobacterium tuberculosis, Staphylococcus aureus, Bacillus subtilis and Listeria monocytogenes [12, 94, 95, 96, 97, 98]. Thus, NADK is a very promising therapeutic target for the development of novel antibiotics [99, 100].
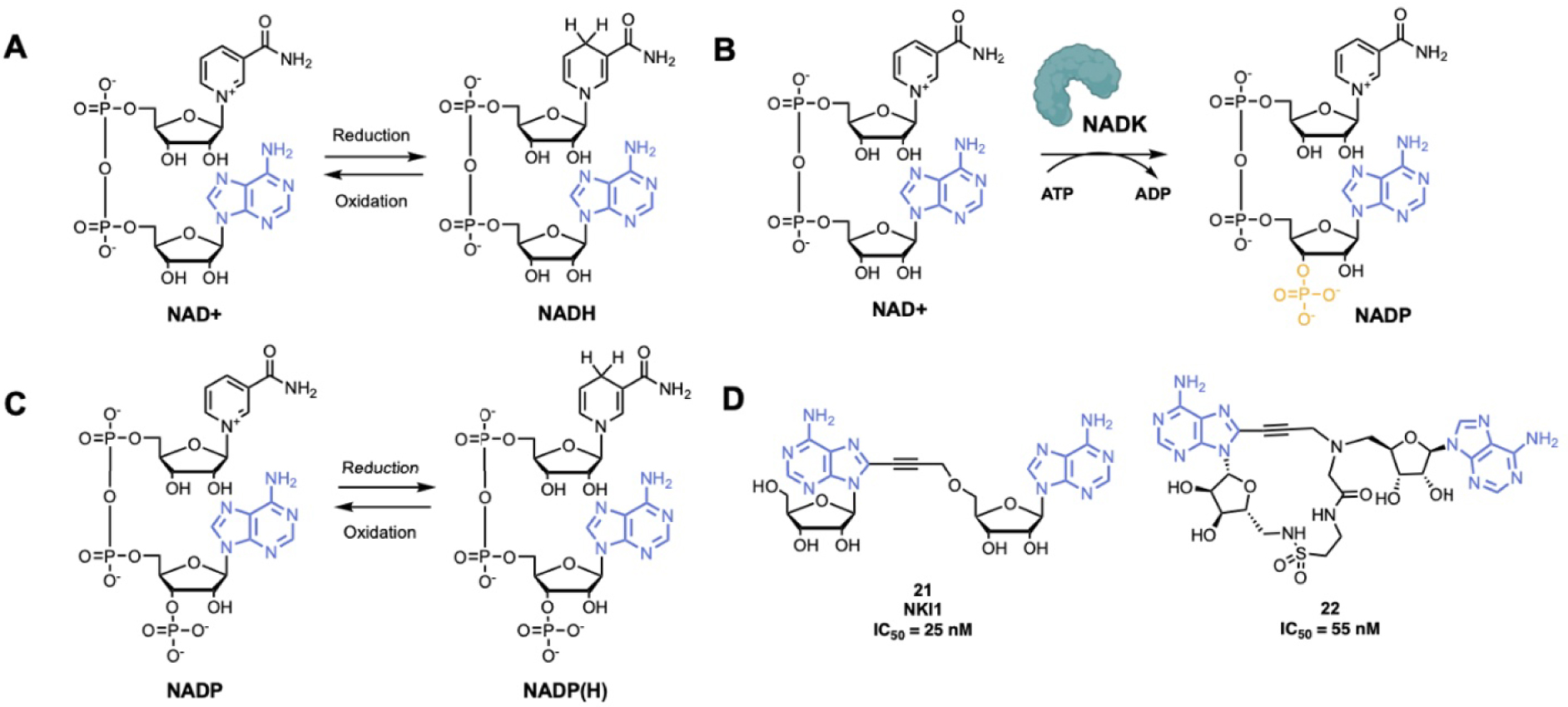
(A) Reduction–oxidation reaction of NAD+/NADH; (B) NAD+ phosphorylation-catalyzed reaction by NADK; (C) reduction–oxidation reaction of NADP/NADP(H); (D) examples of NADK inhibitors. (Biorender.com was used to prepare part of the figure.)
A series of adenosine derivatives targeting recombinant L. monocytogenes and S. aureus NADKs has been recently developed by the group of Pochet [12, 101, 102, 103]. This work led to the synthesis of the first NADK inhibitor NKI1, 21 (Figure 5D), showing activity in mice infected with S. aureus [12]. NKI1 is a dinucleoside of adenosine coupled through a propargyl linker. This linker confers a restricted conformation that is essential for the binding to the NAD-binding site of bacterial NADK. It was suggested that even more constrained compounds would have better affinity and specificity for NADKs [104, 105]. Thus, macrocycle compounds were investigated. The potent compound 22 was developed and the macrocyclization was performed by intramolecular amide bond (Figure 5D). Compound 22 is active at 55 nM on recombinant NADK from L. monocytogenes (LmNADK1) and represents the best NADK-binder described to date [106].
5. Adenosine analogues as adenosine receptor agonists
Adenosine is a major cell regulator that is involved in numerous physiological processes such as the nervous system, vascular function, immune system, and platelet aggregation. More precisely, adenosine interacts with four G Protein-Coupled Receptors (GPCR) Adenosine Receptors (ARs): A1, A2A, A2B and A3 (Figure 6A) [107, 108]. ARs exhibit seven-passed transmembrane α-helix structures, the amino terminus being extracellular and the carboxyl terminus intracellular. Receptors A1 and A3 activate Gi proteins that inhibit the activity of the Adenylyl Cyclase (AC) and consequently reduce the intracellular level of cAMP. On the contrary, receptors A2A and A2B are coupled to Gs proteins that activate AC and induce an increase in the cAMP level. ARs are distributed throughout the body, for example receptors A1 are distributed in the brain, the spinal cord, heart muscles, kidney. Receptors A2A are mainly found in the immune system and the stratum, while receptors A2B are at high concentration in the respiratory pathway. Receptors A3 are located into hippocampus, thalamus, astrocytes, epithelial cells, inflammatory cells etc … [109].
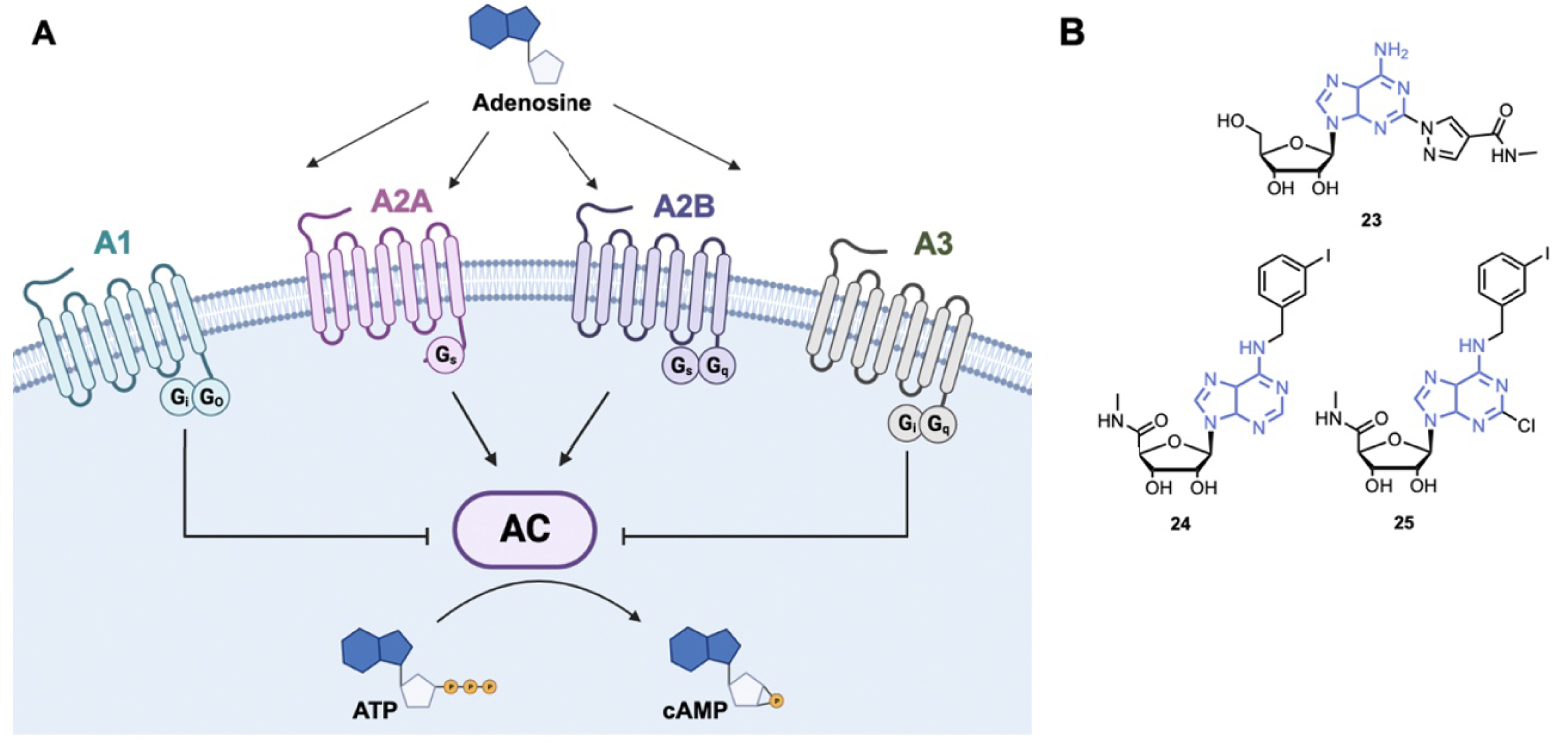
(A) Schematic representation of adenosine signaling; (B) examples of adenosine receptor agonists. AC: Adenylyl Cyclase. (Biorender.com was used to prepare panel A.)
By their role, ARs are involved in various pathological functions in several human diseases as for example in central nervous system disorders, cardiovascular diseases, asthma, and cancer. ARs are thus interesting therapeutic targets and several drugs including agonists, antagonists and allosteric enhancers have been developed and approved. The drugs targeting adenosine signaling pathways are described in a recent review by Kutryb-Zając et al. [14]. A major strategy for the design of agonists of ARs consists in modifying the N-6 position of adenine and the 5′-position of ribose.
5.1. Specific A2A receptor agonist
Regadenoson, 23 (Lexiscan®), is the only A2A receptor agonist that has been approved by the FDA (Figure 6B). Regadenoson is a pyrazole derivative of adenosine that induces coronaries dilatation by binding to A2A receptors of the coronary arteries [110, 111]. Regadenoson is more than 100 times more potent than adenosine as an agonist of A2A receptor. It is used as a stress agent in conjunction with Myocardial Potential Imaging (MPI) for diagnosis of Coronary Artery Disease (CAD). Regarding its therapeutic use, Regadenoson is currently in phase I/II study for the treatment of COVID-19. Indeed, by inhibiting hyperinflammation, Regadenoson could reduce COVID-19 induced lung injury and ameliorate recovery of patients [112].
5.2. Specific A3 receptor agonists
The A3 receptor is a promising therapeutic target because it is highly expressed in the skin of psoriasis patients, in inflamed tissues of arthritic patients and in several solid tumor cells (breast, pancreatic, melanoma …) [113, 114, 115]. Importantly, A3 agonists inhibit cytokine production and downregulate the NF-κB pathway in cancer and inflamed tissues. Piclidenoson, 24 (Figure 6B), is an adenosine A3 agonist showing anti-inflammatory effects in arthritis and inflammatory bowel disease [116, 117]. A phase II study showed that the drug was safe and well-tolerated for patients with rheumatoid arthritis [118]. Piclidenoson was tested in phase III to treat patients with moderate to severe plaque psoriasis (NCT03168256) [119].
The A3 receptor is also overexpressed in HepatoCellular Carcinoma (HCC) tissues and it has been shown that the A3 agonist Namodenoson, 25 (Figure 6B), inhibits tumor cell growth in vitro and in vivo [120, 121]. Namodenoson is currently in phase III to treat advanced HCC (NCT05201404) [119].
Targeting adenosine receptors has shown promising results, however due to the complexity of the AR signaling and the wide distribution in the body of ARs, only a small amount of adenosinergic drugs have been approved. Development of new drugs is currently explored.
6. cyclic Adenosine MonoPhosphate (cAMP) analogues as Exchange Proteins Activated by cAMP (EPAC) agonist modulators
cAMP is a versatile chemical second messenger that mediates extracellular signals to intracellular responses in most types of cells. Many biological processes are regulated by cAMP such as cell activation, proliferation, migration or apoptosis regulation [122]. The cAMP response is first initiated by primary signaling molecules (hormones, neurotransmitters or prostaglandins) that activate mainly G-Protein-Coupled Receptors (GPCR), which in turn activate adenylyl cyclases (Figure 7A). ACs catalyze ATP cyclization to produce cAMP [123]. There are several intracellular receptors for cAMP, as the Protein Kinase A (PKA) family, EPACs and CNG channels [124, 125, 126]. Deregulations in local cAMP signaling has been correlated to various pathophysiological disorders such as cancer, diabetes, cardiac dysfunction and immunological diseases [127, 128, 129].
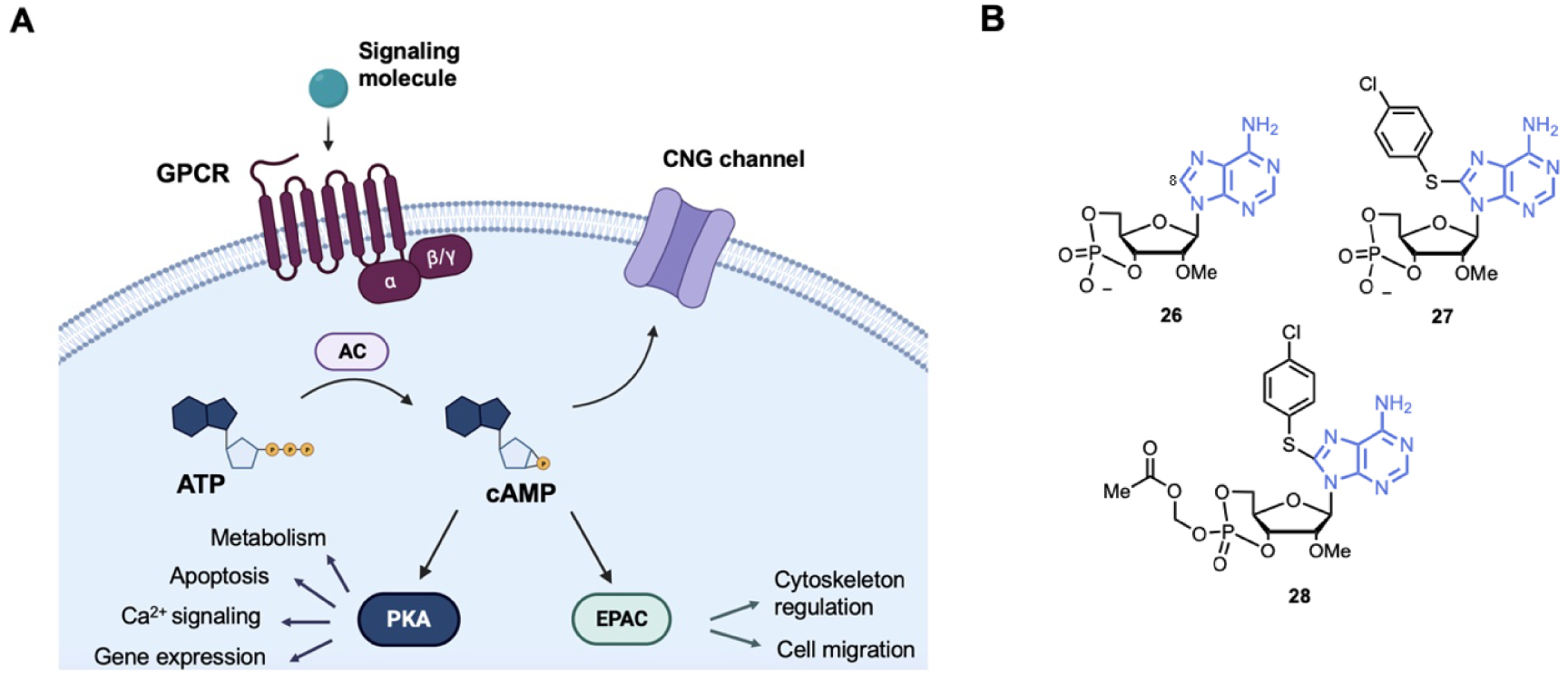
(A) Schematic representation of the intracellular cAMP signaling cascade; (B) examples of cAMP analogues used to target EPACs (Exchange Proteins Activated by cAMP). AC = Adenylyl Cyclase; CNG = Cyclic Nucleotide Gated; PKA = Protein Kinase A. (Biorender.com was used to prepare panel A.)
EPACs activate Ras-like small GTPases (Rap1 and Rap2) that are involved in cell-cell adhesion [122, 125, 130]. EPAC1 and EPAC2 have been identified as potent targets for several human diseases, as in cancer and cardiovascular diseases, thus small molecules modulating EPAC have been developed. EPAC modulators can be classified into cAMP analogues and non-cyclic nucleotide molecules (for a specific review see Wang et al.) [13]. Here we focused on the cAMP analogues (Figure 7B). cAMP analogues display EPAC agonist activity. Bos et al. first showed that 2′-OH modification increases selectivity EPAC/PKA selectivity. For example, substitution of the 2′-OH by a methoxy group led to compound 26 with about 10- to 60-fold selectivity EPAC/PKA [131]. Then, bulky substitution on adenine at the C8-position gave EPAC agonist 27 with an half-maximal EPAC1 activation at 2.2 μM and about 100-fold EPAC/PKA selectivity [132]. However, compound 27 showed poor membrane penetrating capability. Then, a prodrug, compound 28 (Figure 7B) was developed with esterification of the 4-phosphate group. Compound 28 was over 100-fold more potent than compound 27 and did not affect EPAC/PKA selectivity but acted also as a PhosphoDiEsterases (PDE) inhibitor [133].
7. Adenosine analogues as antiviral and anticancer agents
Nucleoside analogues are widely used for antiviral and anticancer therapy by targeting DNA or RNA replication of the virus or the rapidly replicating cancer cells. Nucleoside analogues are mainly “chain terminators” and belong to the inhibitors described as anti-metabolites (Figure 8A). After penetration in the cell, the nucleoside analogues are tri-phosphorylated by viral or human cells. The triphosphorylated compounds are then recognized by polymerases and incorporated into the growing DNA/RNA chain. During DNA/RNA chain elongation, the (n + 1) nucleoside triphosphate is attached to the 3′-OH of the n terminal nucleotide of the growing chain. The 3′-OH group is thus essential for chain elongation and “obligate chain terminators” nucleoside analogues lack the 3′-OH group so that their incorporation consequently results in chain termination [134]. Either the 3′-OH group is substituted with a non-reactive group (–H, –Cl, –F, –Br and –N3 for example) or the deoxyribose is replaced by arabinose derivatives. In general, only few modifications are performed on the nucleobase in order to facilitate the incorporation of the nucleoside analogues as its natural counterpart [135]. Several adenosine analogues are used as “chain terminators” for the treatment of cancer or viral infection.
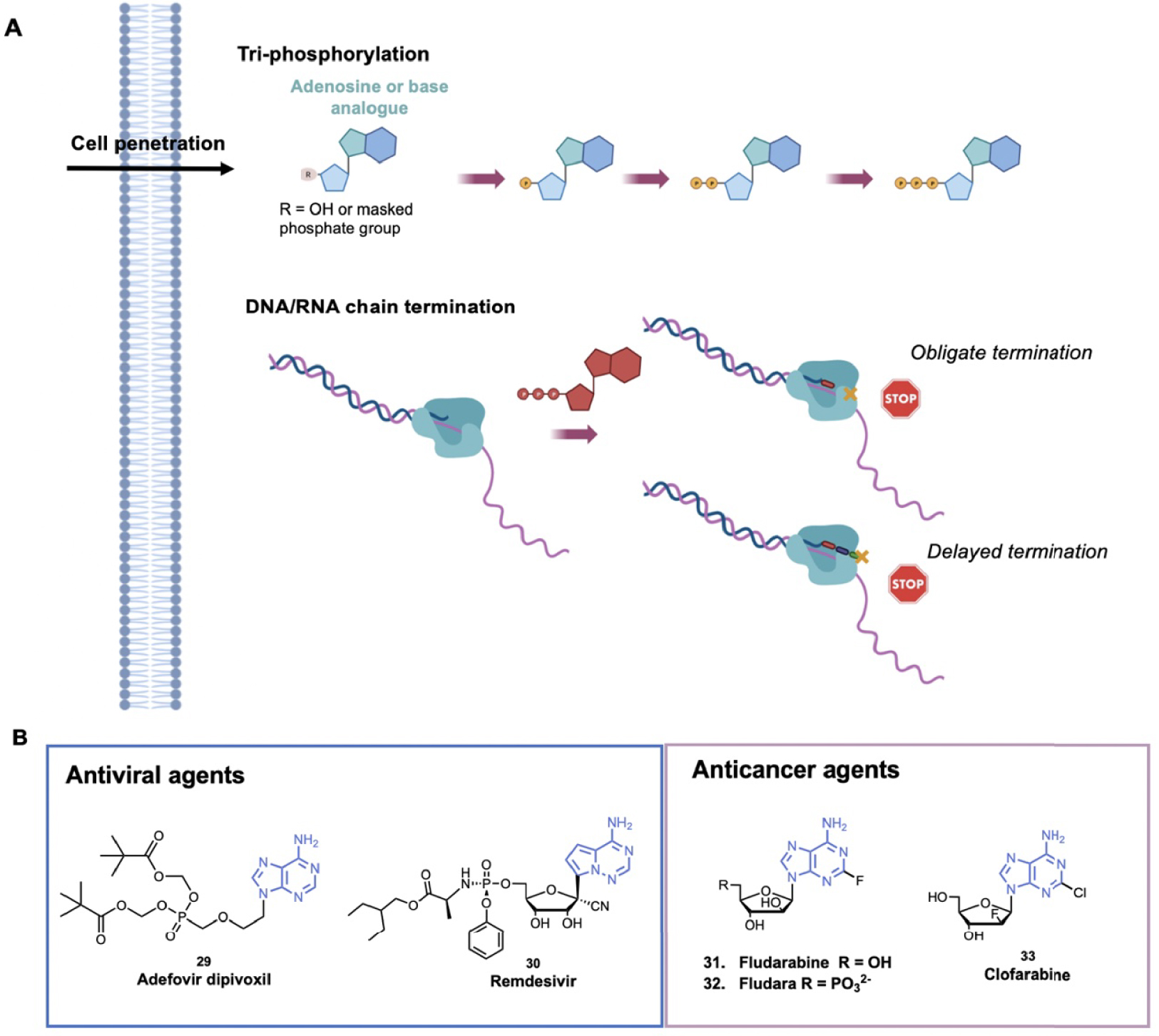
(A) Schematic representation of the main mechanisms of nucleoside analogues to inhibit DNA/RNA synthesis; (B) examples of adenosine analogues as antiviral and anticancer agents. (Biorender.com was used to prepare panel A).
7.1. Antiviral agents
One example of chain terminator antiviral agent is Adefovir dipivoxil, 29 (Hepsera®), that was approved by the FDA in 2002 for the treatment of Hepatis B Virus (HBV) (Figure 8B) [136]. It is a ester prodrug of a viral replication terminator Adefovir that blocks the HBV reverse transcriptase and is the prototype of acyclic adenosine phosphonate analogues. The phosphonate bond (–PC–) mimics the 5′-monophosphate of natural nucleotides and enables Adefovir to be phosphorylated by kinases to phosphono-diphosphate form. Adefovir diphosphate competes with the natural substrate dATP and its incorporation results in blockage of the viral polymerase causing chain termination. The phosphonate bond is more stable than the phosphate bond and cannot be cleaved by phosphodiesterases [137]. Based on this efficient strategy, several similar acyclic nucleoside phosphonate compounds have been optimized. In parallel, some nucleoside analogues display a “delayed chain termination” mechanism [138]. These compounds exhibit a locked sugar conformation, and after their incorporation in the growing chain, two or three other nucleotides can be further incorporated. Then, due to steric hindrance and displacement of the growing chain in the catalytic site, strand elongation is impeded [134]. This mechanism was developed to avoid exonucleolytic removal of nucleoside analogues. For example, Remdesivir, 30 (Veklury®), is a delayed chain terminator that has been recently approved by the FDA for the treatment of COVID-19 [139, 140]. More precisely, Remdesivir is a prodrug of adenosine, the 1′-cyano group avoids the translocation of coronavirus polymerases. According to molecular modeling data, repulsion between the cyano group and S861 in the protein pocket would inhibit the polymerase translocation to position (n + 4) [141].
7.2. Anticancer agents
Fludarabine, 31 (F-ara-A), was approved by the FDA for the treatment of B-cell chronic lymphotic leukemia (Figure 8B). After cell penetration in its nucleoside form, Fludarabine is triphosphorylated (forming F-ara-ATP) and competes with the natural substrate. Incorporation of F-ara-ATP into DNA by DNA polymerase triggers chain termination [142]. Then, to improve solubility, the 5′-monophosphate derivative, 32 (Fludara®), was designed. Fludara® is then converted into fludarabine by phosphatase (Figure 8B). Clofarabine, 33 (Clolar™, Genzyme), a next-generation deoxyadenosine analogue, was approved by the FDA in 2004 to treat relapsed or refractory Acute Lymphoblastic Leukemia (ALL) (Figure 8B). Clofarabine showed an improved stability in the acidic gastric environment and in plasma, notably thanks to the fluorine group at the 2′-position that increases its stability at acidic pH. After its triphosphorylation by kinases, Clofarabine competes with the natural dATP for binding to DNA polymerase −α and −𝜀. At high ratios of Clofarabine/dATP, Clofarabine is inserted into internal and terminal sites of DNA and inhibits DNA elongation [143]. The anticancer activity of Clofarabine is also due to the inhibition of ribonucleotide reductase and induction of apoptosis [144]. These compounds belong to the antimetabolites that are used in the treatment of cancer based on the higher proliferation of the cancer cells, however they target all proliferating cells showing the well-known side effects (e.g. hair loss).
8. Conclusion and perspectives
This review highlights the role of the adenine in the biology of cells and shows through examples the design of adenine-based drugs and their use to treat human diseases, such as cancer, neurological or infectious diseases. The adenine scaffold is involved in many essential biological processes and participate to a broad range of mechanisms, as energy source, cofactor, …. As these mechanisms are aberrant in diseases, they constitute potential targets for treatment and thus the adenosine scaffold is a chemical starting point for the design of drugs in medicinal chemistry. Many chemical modifications have been carried out on the heterocycle moiety and on the side chain giving extensive chemical libraries of adenine derivatives with comprehensive SAR studies. Moreover, it enabled to develop and investigate novel methods in purine synthesis [145, 146, 147].
Importantly, membrane permeability and bioavailability remain an issue and many efforts are carried out to improve the pharmacological properties and extend the adenine analogues repertoire and explore adenine prodrugs. For example, adenosine fleximers, in which the purine base is split into two heterocycles connected by a bond allowing rotation, have been developed (Figure 9A) [148]. Thanks to their flexible structure, these derivatives have shown advantages compared to the corresponding purine-base nucleosides, as they can exert new interactions with the protein partner that are not possible with the corresponding nucleoside or their extended structure can give access to new space in the protein pocket [149]. Indeed, the replacement of a guanine by fleximer in Acyclovir greatly improved its biological activity and the ability of the fleximer analogue to adopt the syn conformation was able to inhibit efficiently a SAH hydrolase [150]. These compounds have many promising applications in particular for their antiviral activity but also as molecular probes and antitumor agents [150].
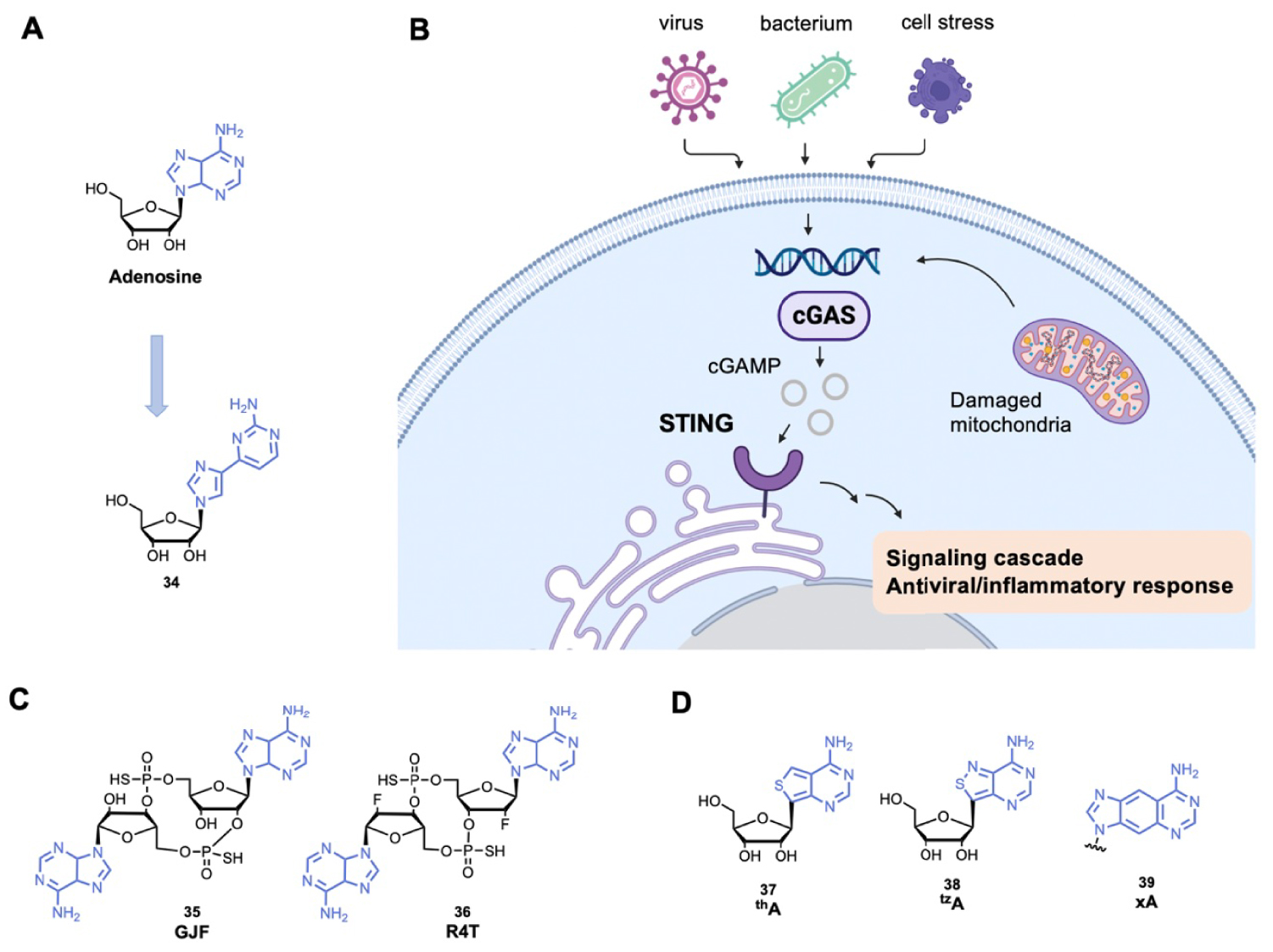
(A) Example of fleximer of adenosine; (B) schematic representation of the STING pathway; (C) examples of cGAMP analogues and (D) examples of adenosine derivatives used as fluorescent probes. (Biorender.com was used to prepare panel B.)
Another emerging and promising topic consists of the development of new modified cyclic dinucleotides targeting STimulator of INterferon Genes protein (STING). STING recognizes 2′3′ cyclic GMP-AMP (cGAMP), which is produced by the cyclic GMP-AMP synthase (cGAS) after recognition of cytosolic endogenous or exogenous DNA (Figure 9B) [151]. The STING activation induces a signaling cascade and transcription of antiviral and proinflammatory genes [152]. Several cyclic dinucleotides with adenosine derivatives are developed to activate STING (Figure 9C), by mimicking cGAMP [153]. Such compounds should be further explored and could be used to improve vaccination, control autoimmune disease or treat cancer.
It is worth mentioning that the quinazoline core is also a common mimic of adenine and used more broadly in the design of drugs as we can see in many kinase and DNA methyltransferase inhibitors for example.
Finally, the adenine scaffold is also used to develop fluorescent probes. To confer and modulate fluorescence properties, modifications are mainly carried out on the C8-position of adenine (and few examples with modifications on C2-position) (Figure 9D). By mimicking the natural non-fluorescent adenine, these compounds have many biological applications such as probing the structure of nucleic acids and their interactions with biological molecules [154, 155, 156]. Finally, purine chemical libraries are extended with the guanine scaffold that is also used in many biological applications including anticancer and antiviral therapy [5, 157].
The interest in the design of analogues of cofactors (SAM, NAD, …) is increasing for both anticancer, antiviral and antibiotics applications. Interesting, the advances in the technologies to detect RNA modifications (epitranscriptomics) pointed out their role in diseases, increasing the research on the design of analogues to target SAM-based enzymes, ribozymes, deoxyribozymes [158, 159, 160, 161, 162].
To conclude, adenine plays a major role in various biological processes but is also involved in processes that are aberrant in diseases. Therefore, it is worth continuing to develop new inhibitors based on this scaffold.
Abbreviations
| AC | Adenylyl Cyclase |
| Acetyl-CoA | Acetyl-CoenzymeA |
| ALS | Amyotrophic Lateral Sclerosis |
| AR | Adenosine Receptors |
| ATP | Adenosine TriPhosphate |
| cAMP | cyclic Adenosine MonoPhosphate |
| BTK | Burton’s Tyrosine Kinase |
| 5mC | 5-methylCytosine |
| CAD | Coronary Artery Disease |
| CLL | Chronic Lymphocytic Leukemia |
| CNG | Cyclic Nucleotide-Gated channels |
| COMT | Catechol O-MethylTransferases |
| DNMT | C5 DNA MethylTransferases |
| EPAC | Exchange Protein Activated by cAMP |
| EGFR | Epidermal Growth Factor Receptor |
| FAD | Flavin Adenine Dinucleotide |
| FDA | U.S Food and Drug Administration |
| cGAS | cyclic GMP-AMP Synthase |
| cGMP | 2′3′ cyclic GMP-AMP |
| GPCR | G-Protein-Coupled Receptors |
| HBV | Hepatis B Virus |
| HCC | HepatoCellular Carcinoma |
| Her2 | Human epidermal growth factor receptor 2 |
| HMT | Histone MethylTransferase |
| KMT | Lysine MethylTransferase |
| MLL | Mix Lineage Leukemia |
| MPI | Myocardial Potential Imaging |
| MTase | MethylTransferase |
| NAD | Nicotinamide Adenine Dinucleotide |
| NADK | Nicotinamide Adenine Dinucleotide Kinase |
| NNMT | Nicotinamide N-MethylTransferase |
| NRTK | Non-Receptor protein Tyrosine Kinase |
| NSCLC | Non-Small Cell Lung Carcinoma |
| NTMT | N-Terminal MethylTransferases |
| PDE | PhosphoDiEsterases |
| PI3Kδ | PhosphoInositide 3-Kinase δ |
| PKA | Protein Kinase A |
| PRMT | Protein arginine MethylTransferase |
| SAH | S-Adenosyl-L-Homocystein |
| SAM | S-Adenosyl-L-Methionine (or AdoMet) |
| sSAR | Structure-Activity Relationships |
| STING | STimulator of INterferon Genes protein. |
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Fundings and acknoweldgements
Authors acknowledge the French National Research Priority Plan PPR Antibioresistance for grant AMI-AMR-2020_ANR-20-PAMR-0011 TheraEPI (to PBA) and The French Minister of Higher Education, Research and Innovation for the ENS Paris Ulm CDSN PhD fellowship to AF.
Biorender.com was used to make parts of Figures graphical abstract, 2, 4, 5, 6, 7, 8 and 9.