



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Catalysis is a key technology for our society with extensive contributions in diverse areas such as energy, chemicals and food production, cosmetics, health, etc. [1, 2]. Heterogeneous catalysis, where the catalyst and the substrate are in two different phases, provides easy catalyst separation and reuse, which often results in extended catalyst lifetime and use of lower amounts of solvents. These features have made heterogeneous catalysts predominant in industry (about 80% of processes). However, heterogeneous catalytic systems typically require harsher reaction conditions than homogeneous analogs and this may lead to lower selectivities. Heterogeneous catalytic systems may also suffer from mass transport limitations, which may decrease the activity. In addition, the exploration of reaction kinetics and mechanistic understanding are often more problematic for heterogeneous catalysts. Conversely, homogeneous catalysts usually provide high activities and selectivities under milder reaction conditions. They also allow easier mechanistic investigations. However, since the catalyst and the substrate coexist in the same physical phase (typically a liquid phase), these systems inherently suffer from difficult catalyst separation and reuse. Hence, down-stream separation procedures such as precipitation, extraction or ultrafiltration are typically required to recover the catalyst. These operations need additional equipment and undesirable large amount of solvent and entail extra costs.
To circumvent these issues, several strategies have been implemented for the recycling of homogeneous catalysts in large-scale processes. These strategies include: (1) employing specially designed membranes capable of selectively percolating catalyst-supporting molecules (such as dendrimers or colloids) through micro-/ultra-/nanofiltration or reverse osmosis processes [3, 4], tailored to the size and geometry of the catalyst; (2) utilizing biphasic catalysis, where the catalyst is immobilized in a separate, immiscible liquid phase from that of the reaction products [3] (for instance, the Rhône-Poulenc/Ruhrchemie hydroformylation of propene and butene involving water as the immiscible phase [5]); and (3) employing catalyst confinement techniques [6], where the catalyst support can be a solid phase with the molecular catalyst either grafted onto the surface or enclosed within the pores of insoluble polymers (such as resins), inorganic oxides (like silica and alumina), metal-organic frameworks (MOFs) or carbon materials (including carbon black, carbon nanotubes, and graphene), known as heterogenized homogeneous catalysis, or a separate liquid phase, referred to as liquid/liquid biphasic catalysis. The latter strategy is an effective and low-cost solution to preserve the catalytic performance and facilitate catalyst recovery. Whatever the catalyst support, solid or liquid, catalyst confinement approaches combine the benefits of homogeneous and heterogeneous catalysis. This is also possible via micro-heterogenization using colloidal assemblies [3], also referred to as nanoreactors.
Polymeric nanoreactors that contain covalently linked ligands in their core architecture offer an interesting approach for anchoring and protecting the catalyst from the surrounding environment, while facilitating its recovery and recycling [4]. One of the prominent techniques for catalyst recovery and recycling is liquid/liquid biphasic catalysis using two immiscible liquid phases, one containing the catalyst and the other one containing the reaction products and any unreacted substrate [5, 7, 8]. In this approach, the two phases can be separated by a simple decantation, thus facilitating the reuse of the catalyst phase without the introduction of any thermal or chemical stress. Four distinct operational modes may be associated to the liquid/liquid biphasic approach, as listed for the biphasic aqueous catalysis [5]: (i) homogeneous in the aqueous catalyst phase, if the substrate is sufficiently water-soluble; (ii) homogeneous in the substrate/product phase, if the catalyst is sufficiently soluble in the substrate phase or can be transported there by a temperature stimulus (thermomorphic approach) [9, 10, 11] or by a phase-transfer agent; (iii) interfacial when neither component is sufficiently soluble in the other component phase and the transformation occurs exclusively at the interface; (iv) homogeneous within the nanoreactors, which form a stable dispersion in a phase different from that of the substrates/products [12, 13]. The latter approach, with the catalyst confinement within a small container, can allow precise control over its interaction with the substrate. This can lead to different reaction pathways compared to the free catalyst, resulting in significantly improved selectivities compared to the non-confined analogs [14, 15]. The catalyst host can either be molecular or macromolecular. This review deals with macromolecular nanoreactors.
Nanostructured materials can be produced for use as nanoreactors [16, 17, 18, 19]. They have a higher surface area relative to their volume compared to complex and bulk materials. This property allows for enhanced interactions with the surrounding environment, leading to increased reactivity and improved performance in various applications. Also, nanostructured materials often exhibit, despite their small size, excellent mechanical properties like high strength and hardness compared to their bulk counterparts [20]. This is mainly due to their unique structural features, including the presence of grain boundaries and interfaces, which can hinder dislocation movement and improve overall material strength. In addition, nanostructured materials can exhibit enhanced electrical conductivity and optical properties [21, 22, 23], making them suitable for various electronic and optoelectronic applications as well as favorable for energy storage and conversion applications. For example, a high surface area improves electrode–electrolyte interactions in batteries and supercapacitors, resulting in enhanced energy storage capabilities [24, 25]. Most importantly, metal nanomaterials with high specific surface area are often used as catalysts [4, 26, 27] due to their ability to provide a large number of active sites for chemical reactions. The increased surface area allows for more efficient adsorption and reaction processes, leading to enhanced catalytic activity and often also selectivity.
A powerful toolbox is available to access a wide array of polymeric nanoreactors [8, 9] through controlled and living polymerization techniques [28] such as reversible addition-fragmentation chain transfer (RAFT) polymerization, ring-opening anionic, -cationic and -metathesis polymerization (AROP, CROP and ROMP), and atom transfer radical polymerization (ATRP). These polymerization methods have successfully been used to synthesize various nanoreactor architectures for application in catalysis, including self-assembled micelles, multifunctional micelles [29], polymersomes [30, 31, 32, 33, 34, 35] and star polymers [36] (Figure 1). In all cases, a common aim is to embed and protect the catalyst while allowing the substrate to easily reach the reactive sites and the products to migrate back toward the bulk phase. This review provides an overview of the currently known polymer-based nanoreactor systems used in biphasic catalysis, designed from macromolecular building blocks. Special attention is given to core-crosslinked micelles (CCMs) obtained by RAFT polymerization. The progress made on the synthesis of this particular type of unimolecular nanoreactors and on their use in micellar-type aqueous biphasic catalysis will be highlighted.
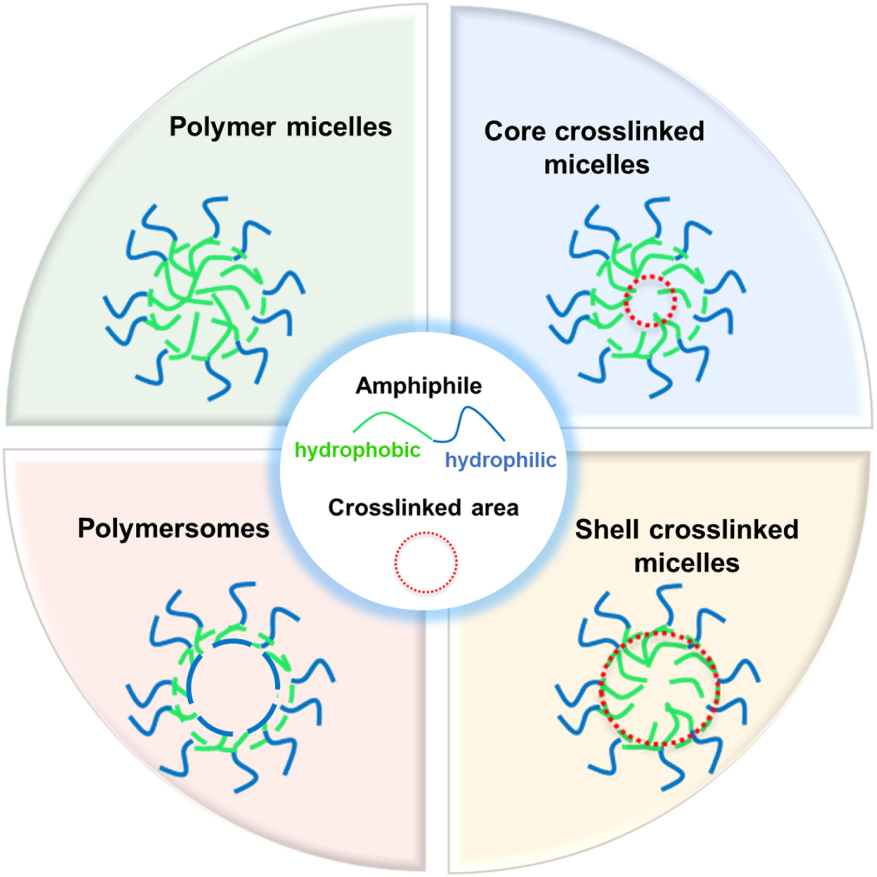
Different polymeric nanoreactors for catalytic applications.
2. Catalysis within confined spaces
One of the captivating approaches in homogeneous biphasic catalysis is micellar catalysis. Each micelle is an independent reaction locus and can thus be described as a catalytic “nanoreactor” [4, 14, 15, 16, 29, 31, 37, 38, 39, 40, 41]. Two fundamentally different implementations of micelles in catalysis can be distinguished [42]. In the initially developed one, the catalyst is dissolved in the aqueous phase, or held close to the micellar surface by coulombic forces (if the catalyst has an opposite charge to that of the micelle surface) or by covalent grafting. The catalytic reaction occurs at the interface between the bulk aqueous phase and the nanoreactor core where the hydrophobic substrate is located. The beneficial effect of the micelles is thus merely the increase of the water/organic interface, thus improving mass transport kinetics in an interfacial catalysis approach. This is what the “micellar catalysis” terminology has typically been referring to, but a more appropriate description should be “micelle-aided catalysis”. This topic has been extensively reviewed and will not be considered here [1, 43, 44, 45, 46, 47, 48]. The second type of micellar catalysis, developed more recently, deals with systems where the catalyst is embedded, by interaction with core-anchored ligands, in the hydrophobic core of the micelles. The catalytic reaction occurs in the homogeneous environment of the hydrophobic micellar core, benefitting from a high local concentration of both substrate and catalyst [14, 42]. The kinetics of the overall transformation can thus favorably compare with that of the analogous homogeneous process.
The first reports on micellar catalysis, based on micelles that are self-assembled from a simple surfactant (e.g. potassium dodecanoate, hexadecyltrimethylammonium bromide (CTAB), sodium dodecyl sulfate, etc.), date back to 1970 [49]. Many additional investigations into the preparation, characterization and catalytical applications have followed [43, 44, 49, 50, 51, 52, 53, 54, 55, 56, 57].
3. Macromolecular nanoreactors
For micelles generated by amphiphilic macromolecules, typically named “polymer micelles”, different morphologies exist depending on the packing parameter p that was defined by Israelachvili [58] through Equation (1), where v is the volume, a0 is the optimal area of the head group and lc is the length of the hydrophobic tail.
| (1) |
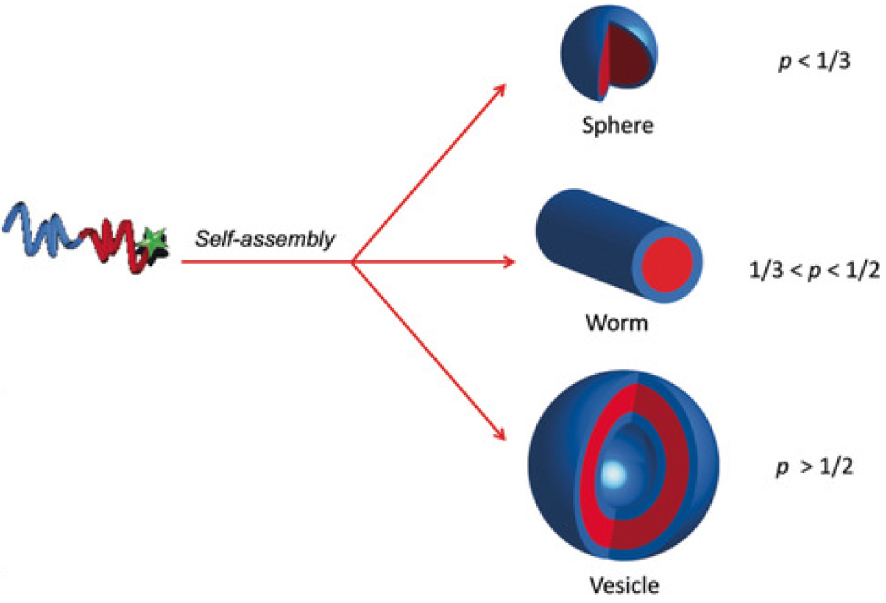
Dependence of the morphology of the self-assembled objects on the packing parameter p. Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [59]. Copyright 2019.
Another factor that also plays a major role is the hydrophobicity of the local pocket [59]. Self-assembled nanoreactors are non-covalent macromolecular entities assembled from their initial building blocks [31, 33, 34, 35, 60]. They form good compartmentalization of catalytic systems, providing advantages on the kinetics (faster reaction rate) and on thermodynamics (lowering the energy of the transitional state). In an aqueous medium, the formation of amphiphilic micelles (Figure 3) that tend to assemble above the so-called “critical aggregation concentration” leads to complex supramolecular architectures depending on the structure of the molecular amphiphiles [61].
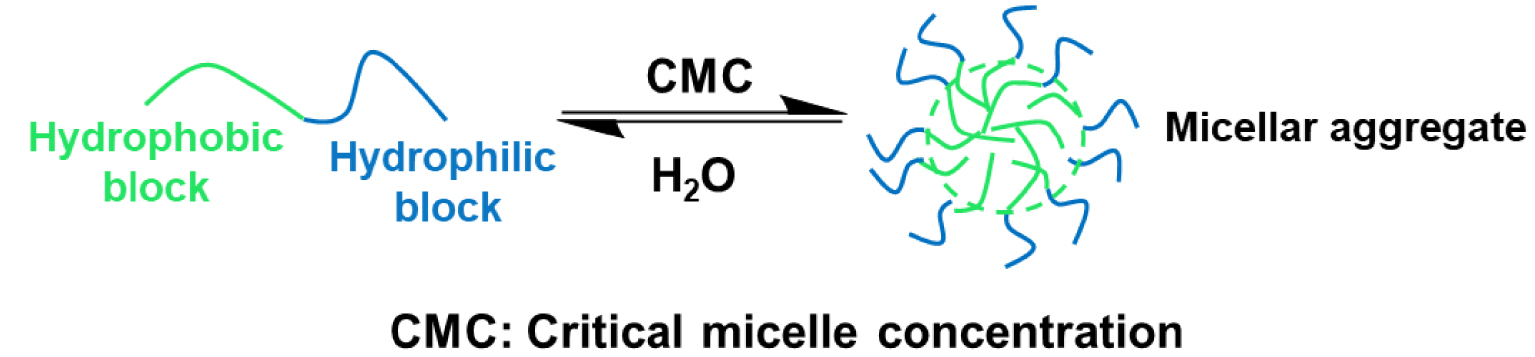
Self-assembly of amphiphilic micelles in water.
As a function of the packing parameter p defined above, three different morphologies can be observed, combining the following terms of Gibbs free energy [37]:
- a favorable entropic contribution resulting from the assembly of the hydrophobic parts of the molecule,
- a tendency of amphiphiles to minimize unfavorable lipophilic–water interaction by closely packing and to spread apart as the result of electrostatic repulsion between the hydrophilic head groups, defined as surface term,
- a limitation to the possible geometry of aggregation, requiring that the hydrophobic cores only assemble in water or polar solvent, defined as the packing parameter explained above.
These aggregates have dimensions in the nanometer regime. The inclusion of catalytic sites in these aggregates, whether by the polymerization or by the coordination of the pre-catalyst, results in the formation of nanoreactors. In such nanoreactors, the reaction selectivity can be modified by tuning the nature of the monomers in the core block.
3.1. Polymer micelles
Polymer micelles [62, 63, 64, 65] (PMs) are described as aggregates resulting from the self-association of amphiphilic polymers owing to hydrophobic interactions between polymer molecules. However, interactions may also be electrostatic or via hydrogen bonds or coordination bonds. The morphology of these block copolymer micelles resembles that of molecular surfactant micelles. They can be spherical or cylindrical micelles, as well as vesicles, where the hydrophobic polymer chains form the core and the hydrophilic polymer chains form the shell when dispersed in aqueous media. The morphology of the PMs depends on the ratio between the hydrophobic and hydrophilic chains, capable of accommodating the catalyst [62]. PMs are efficient systems for the encapsulation of hydrophobic compounds due to the hydrophobic nature of their core and the hydrophilic corona, and these nanoreactors have many applications besides catalysis, especially in the medical field for, e.g. drug delivery [66, 67] and magnetic resonance imaging (MRI) [68].
Chen and coworkers [69] have described the synthesis and application of thermo-responsive polymer micelle-based nanoreactors containing polyoxometalates (POMs) and poly(N-isopropylacrylamide)-b-poly(L-lysine) (PNIPAM-b-PLys-POM) electrostatically linked via the POM and amino groups of poly(L-lysine) for catalytic wet hydrogen peroxide oxidation (CWPO) of phenol. Similarly, Suzuki et al. [70] performed Pd-catalyzed Mizoroki–Heck coupling of iodobenzene and n-butyl acrylate in aqueous solution using thermo-responsive PMs based on poly(N-isopropyl-acrylamide) (PNIPAAm), which is known as an efficient thermo-responsive polymer due to its lower critical solution temperature (LCST) of 32 °C in water [71] (Figure 4).
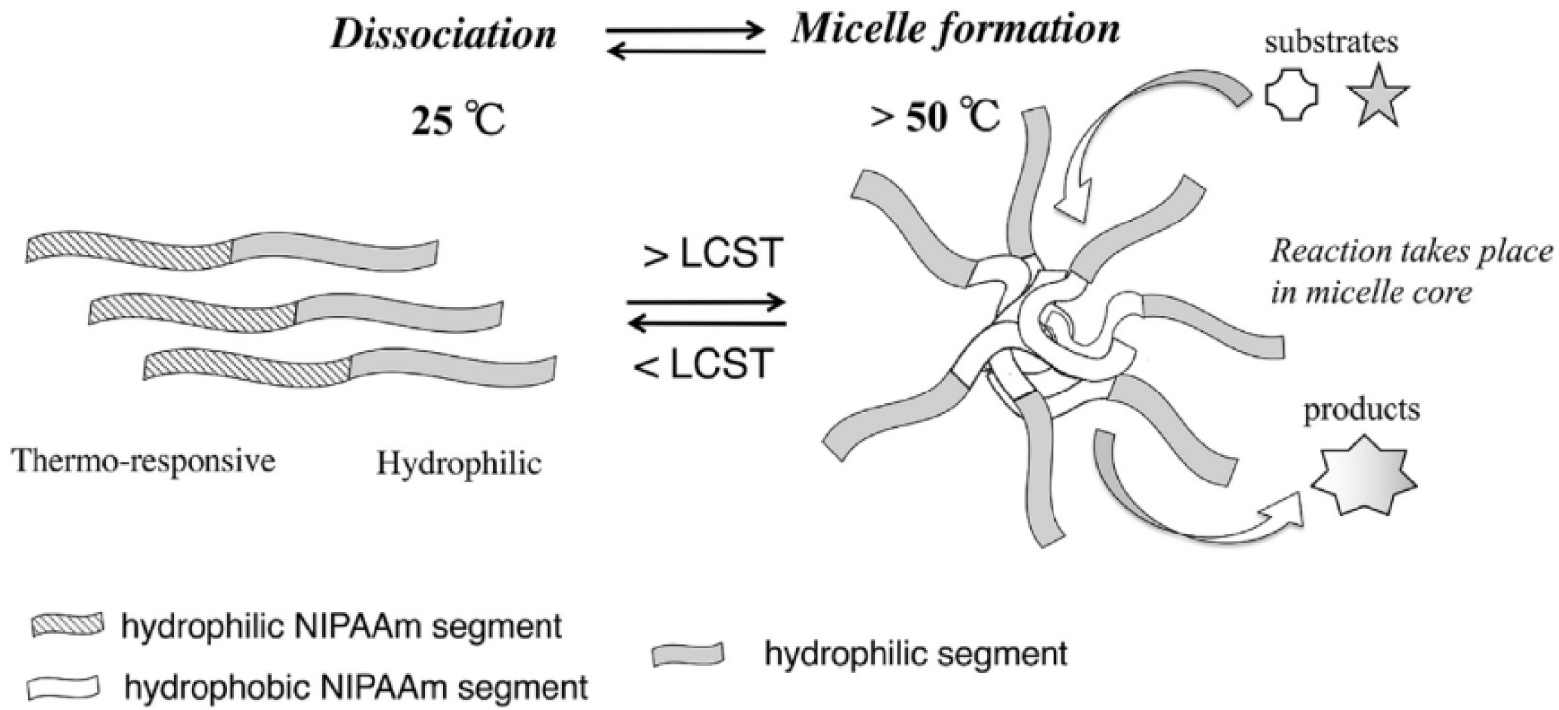
Thermo-responsive micelles switched on/off by temperature. Reprinted with permission of Elsevier from [70]. Copyright 2019.
The block copolymer 2 with a thermo responsive NIPAAm block and a neutral poly(ethylene glycol) (PEG) chain was prepared from 1 via ATRP (Scheme 1). The resulting block copolymer had a narrow size distribution (15–20 nm at 25 °C) and was water-soluble at room temperature but when heated up to 60 °C the polymer was insoluble, and the solution became opaque. Characterization by dynamic light scattering (DLS) demonstrated that the formation of the PMs by 2 was thermo-induced and the switchable nature improved product extraction. The PMs also limited the use of organic solvent for product separation from the aqueous reaction mixture, but 2 mol% of Pd was required to obtain good product yield in the Pd-catalyzed Mizoroki–Heck cross-coupling [70].
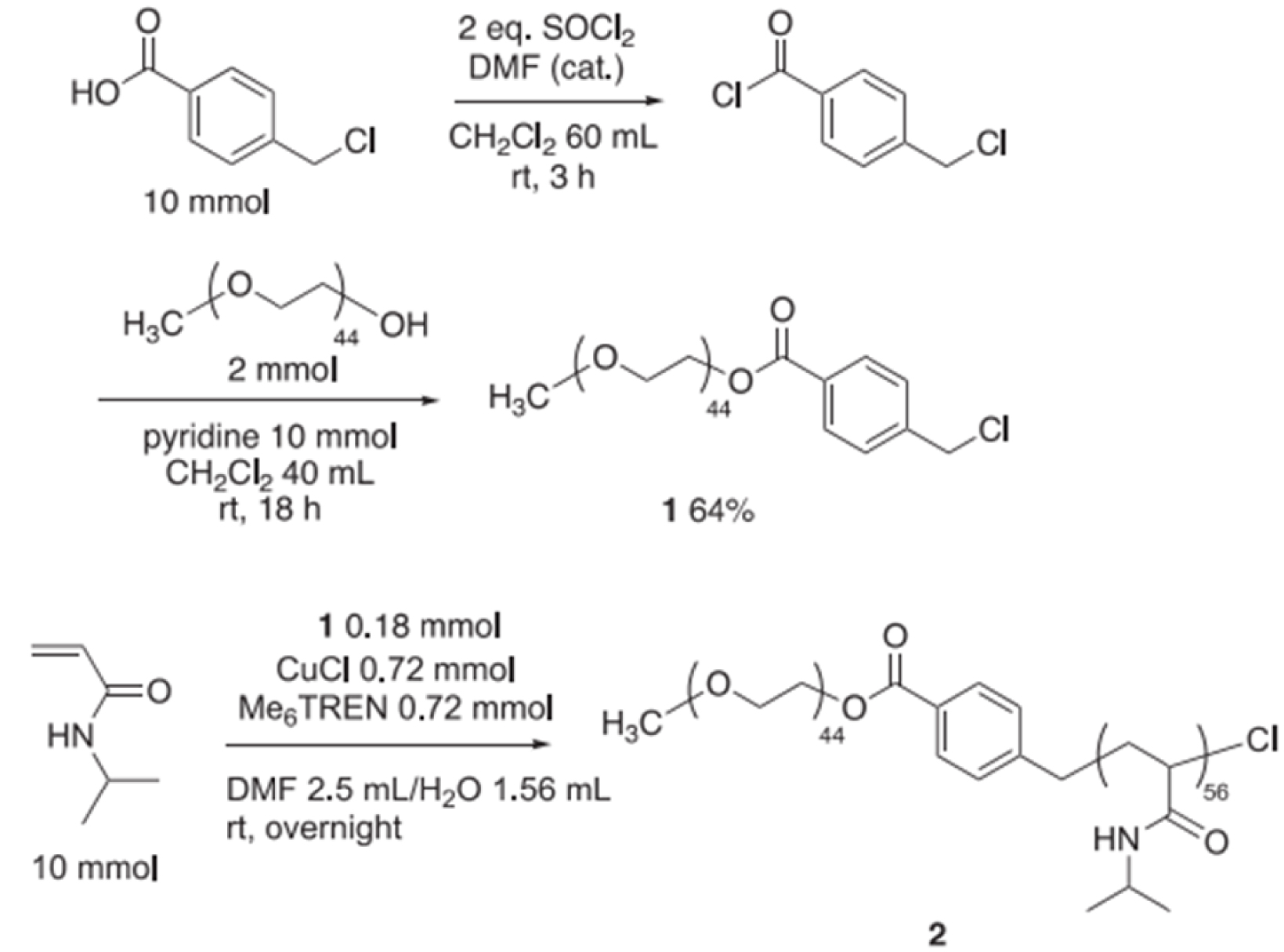
Preparation of NIPAAm-b-PEG 2 with PEGylated ATRP agent. Reprinted with permission of Elsevier from [70]. Copyright 2019.
NaCl was also introduced to the block polymer containing proline to lower the solubility of the organic solute in the aqueous phase and help directing it to the core of the PMs [70]. This “salting-out” protocol led to the design of a new thermo-responsive block copolymer with an anionic sodium sulfate segment, PNIPAAm-b-PSSNa 9, also prepared using RAFT polymerization. In the synthesis protocol, N-isopropylacrylamide and sodium p-styrenesulfonate were polymerized in the presence of the RAFT agent 6 forming 7, followed by depolymerization of p-styrenesulfonate by a radical desulfurization process that led to the removal of trithiocarbonate moiety at the end of the polymer-chain to yield the anionic-type copolymer (Scheme 2). A larger particle size was obtained with the anionic PMs compared to the neutral one, likely due to electrostatic repulsion among the anionic polymer chains. Further analysis also showed that the anionic copolymer exhibited its LCST behavior at 40–55 °C. When the aqueous Pd-catalyzed Mirozoki–Heck coupling of iodobenzene and n-butyl acrylate was performed with 9 using 0.5 mol% Pd, a better yield (though moderate) was obtained compared to the previous reaction with the neutral copolymer and 2 mol% Pd (Scheme 2), thus demonstrating how rational PM design can improve catalytic performance. To recover the catalyst, the authors investigated various solvents for product extraction. In general, using diethyl ether proved less effective than using ethyl acetate. Interestingly, polymer 9 demonstrated higher efficiency than polymer 2, despite the fact that the recovered amount of 5a was only half that obtained with pure water. These findings suggested that thermoresponsive block copolymers PNIPAAm-b-PEG 2 and PNIPAAm-b-PSSNa 9 are not only effective in the Pd-catalyzed Mirozoki–Heck reaction in water, but also in extracting the product from the aqueous reaction mixture.
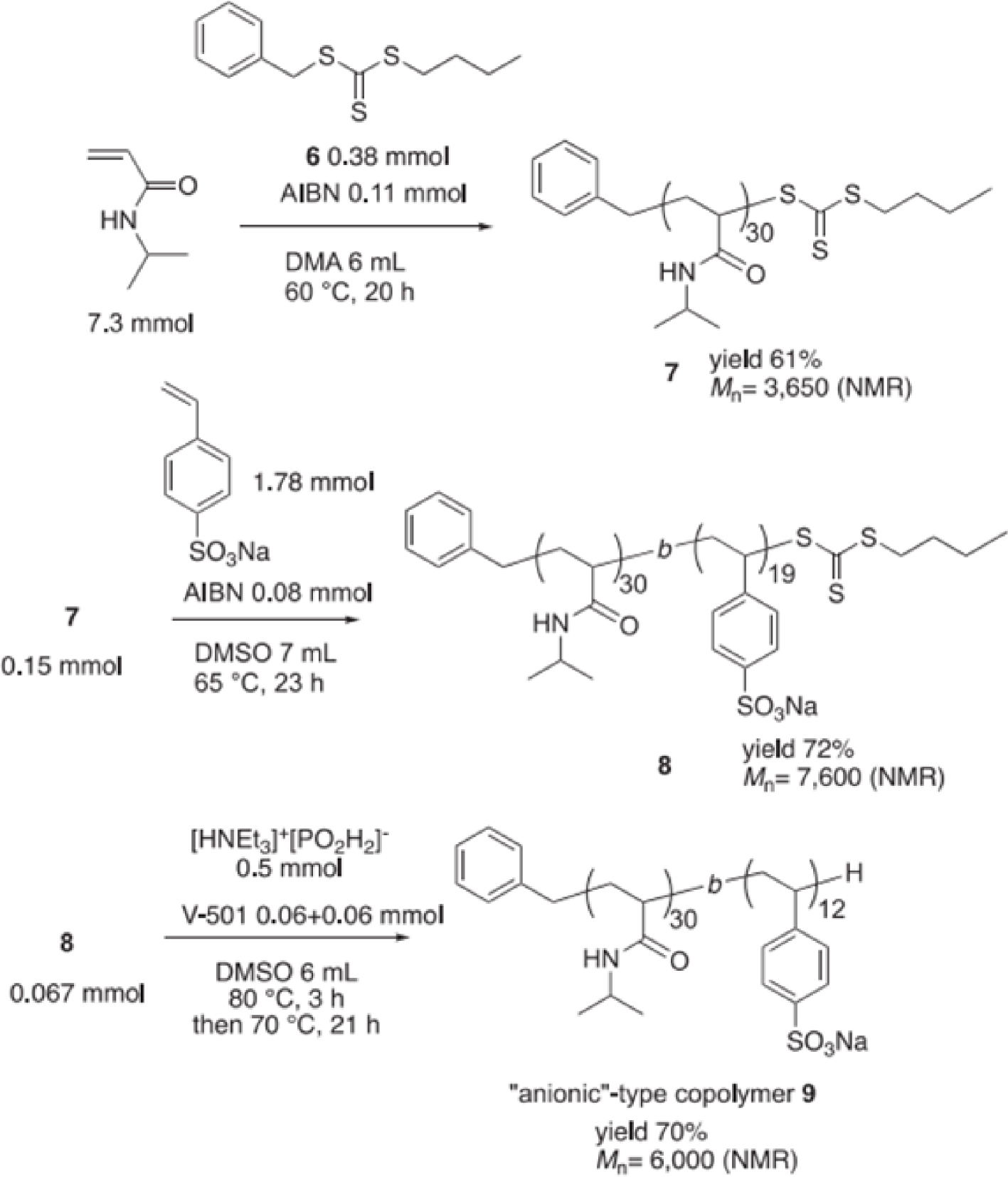
Preparation of PNIPAAm-b-PSSNa 9 by RAFT polymerization. Reprinted with permission of Elsevier from [70]. Copyright 2019; permission conveyed through Copyright Clearance Center, Inc.
The introduction of the catalyst in the core of the micellar nanoreactors can be realized in different ways, either by polymerizing a pre-catalyst-functionalized monomer into the hydrophobic compartment, followed by self-assembly, or by ligand exchange between a molecular pre-catalyst, introduced by mass transport inside the already assembled micelles, to a core-anchored ligand. An example of the former method [72] is the copolymerization of styrene and a functionalized styrene containing a 4-(dimethylamino)pyridine (DMAP) organocatalyst, using the RAFT technique, to yield a hydrophobic macroRAFT agent 2, which was then chain-extended with a hydrophilic poly(N-isopropylacrylamide) (PNIPAM) block (Scheme 3). The catalytic activity of the compartmentalized organocatalyst in the core was found to be high, improving the DMAP-catalyzed acylation reaction of alcohols with anhydrides, with rates up to 100 times compared to those for unsupported DMAP in organic solvents (Table 1). The authors explored catalyst recycling by leveraging the system stimulus-responsive property. Following the extraction of the synthesized product with diethyl ether, the aqueous phase containing the micellar catalyst underwent heating to a temperature over 50 °C. Hereby the polymer transformed into a fine powder, which was subsequently gathered through centrifugation. These catalytic nanoreactors were recycled up to six times without losing catalytic activity.
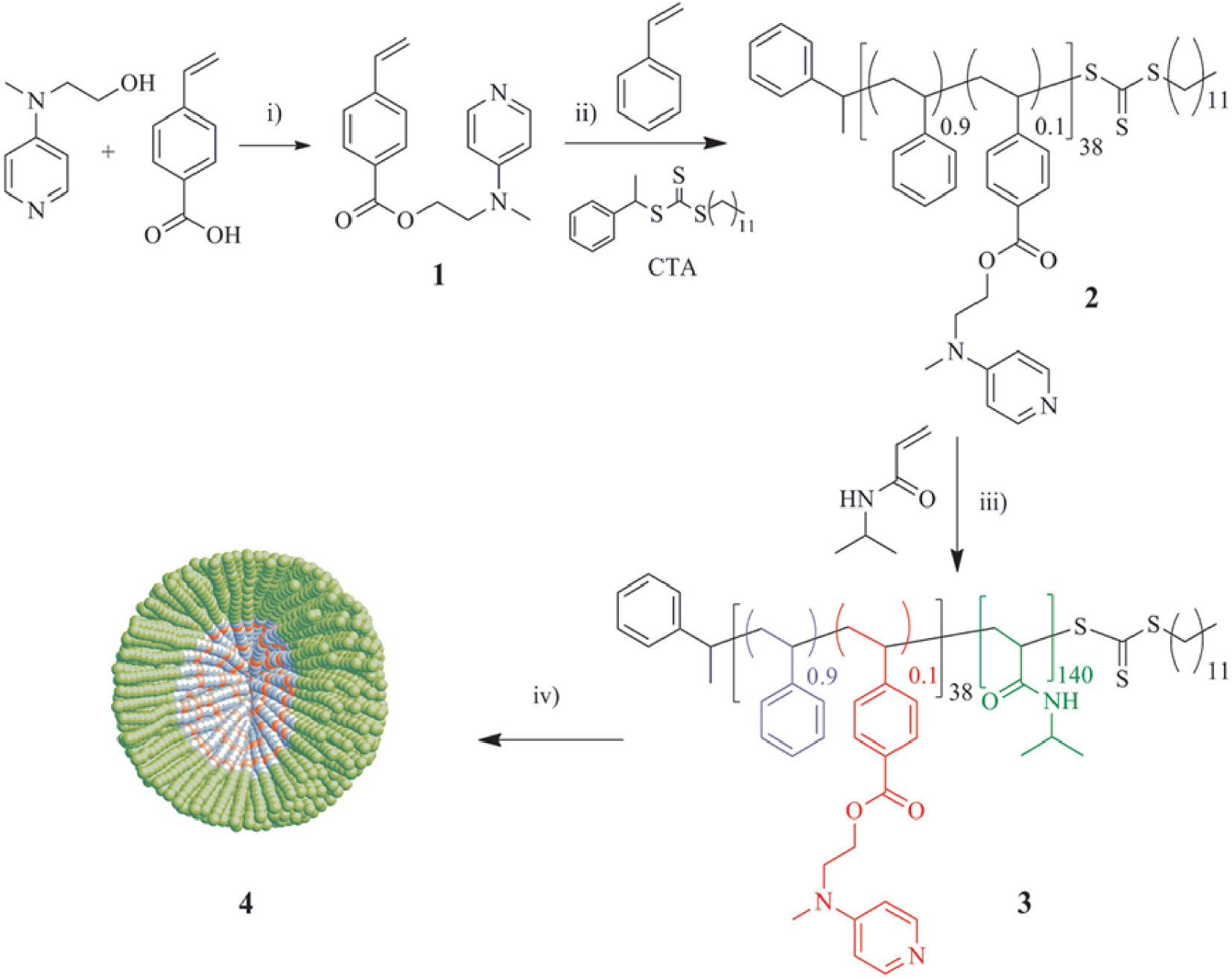
Four-step synthesis of micelles 4 containing the DMAP functionality covalently attached to the hydrophobic core. Reprinted with permission of American Chemical Society from [72]. Copyright 2012.
Acylation reactions in the micelles 4 (adapted from [72])a
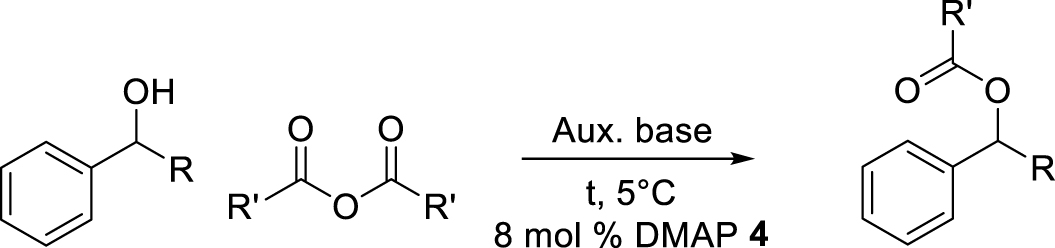
| Entry | R | R′ | Conversion (%) (15 min) | Conversion (%) (24 h) |
|---|---|---|---|---|
| 1 | CH3 | CH3 | 26 | 32 |
| 2 | CH2CH3 | CH3 | 28 | 29 |
| 3 | CH3 | CH2CH2CH3 | 47 | 53 |
| 4b | CH3 | CH2CH2CH3 | 65 | 66 |
| 5b | CH2CH3 | CH2CH2CH3 | 94 | 98 |
a All reactions contained 8 mol% DMAP, [OH] = 0.02 M, 1.5 equiv of auxiliary base (TEA), 1 equiv of alcohol, and 3 equiv of anhydride. Conversions determined by HPLC measurements with mesitylene as the internal standard. b N,N-Diisopropylethylamine (DIPEA) was used as auxiliary base instead of TEA.
Despite successful applications, these and other micellar catalysts have one major drawback: the equilibrium between micelles and free surfactant macromolecules. This equilibrium can lead to undesired formation of stable emulsions, slow micelle/product separation caused by excessive swelling, and catalyst loss through the formation of Langmuir–Blodgett films at the liquid/liquid interface and/or inverse micelles in the bulk organic phase [73]. Another major drawback related to the thermo-responsive PMs is their dynamic nature that renders them sensitive to their environment. To circumvent these issues, the micelles can be turned into unimolecular persistent objects by crosslinking, which can be accomplished by various strategies as described below. These cross-linked PMs constitute an attractive alternative with improved characteristic and catalytical behavior.
3.2. Polymersomes
The stability of micelle- and vesicle-based systems depends on the environment, such as the temperature of the solution and the concentration of the surfactant. To obtain better defined and more stable systems, macromolecular polymers have been considered as alternative nanoreactors, with a higher kinetic stability compared to the self-assembled molecular surfactant (vide supra). When vesicles are self-assembled from macromolecular amphiphiles (or block copolymers), they are referred to as polymersomes. Polymersomes [74, 75, 76, 77, 78] have increased rigidity and stability compared to micelles and vesicles, and their structure can vary from simple coil-like diblock copolymers to rod-like, rod-coil diblock or multiblock polymers with or without additional cross-linker groups [79]. The variation in structuring can be tuned by varying the relative block lengths, which results in modified size, structure, polarity, and permeability of the polymersomes.
Eisenberg and coworkers [80] were the first to generate a block copolymer system with six different morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Shortly after, Meijer and coworkers [81] reported the synthesis of polystyrene-poly(propylene)imine, thus starting a new research line based on the relation between amphiphilic properties and molecular structure towards the preparation of many other polymersomes by various approaches [82, 83, 84] and with different application scope. For enzymatic ring-opening polymerization, Nolte and coworkers [85] developed polystyrene-polyisocyanopeptide (PS-PIAT) based polymersomes, while van Hest and coworkers [86] reported a polymersome-stabilized Pickering emulsion at the water/oil interface that was later applied in biphasic enzymatic catalysis. Also, Lecommandoux, van Hest and coworkers [87] reported a cascade reaction in which small polymersomes were loaded into larger ones, resulting in a multi-compartmentalized polymersome-in-polymersome system (Figure 5).
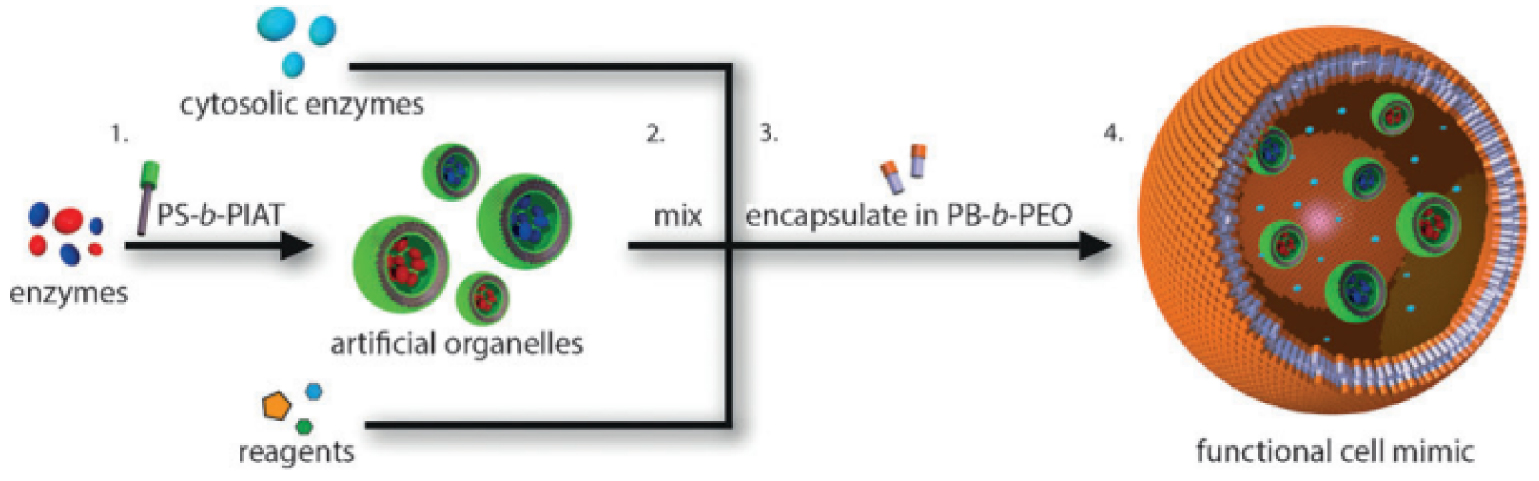
Multi-compartmentalized polymersome-in-polymersome system. Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [87]. Copyright 2014.
To demonstrate the functional and structural control of the system, they investigated a model reaction with isolated enzymes in the different compartments. The first step of the reaction was a Baeyer-Villiger oxidation of a substituted hydroxyphenoxazinone ketone 1. The resulting ester 2 was then hydrolyzed with a lipase (enzyme) to form the corresponding primary alcohol 3, which was subsequently oxidized by alcohol dehydrogenase and a cofactor nicotinamide adenine dinucleotide (NAD) to yield the corresponding aldehyde product 4. The final step of the catalytic transformation was a spontaneous 𝛽-elimination leading to 7-hydroxy-3H-phenoxazin-3-one-10-oxide 5 (Scheme 4). The concept of enzyme compartmentalization in the cascade reaction spatially separated incompatible enzymes to retain their functionality, thus resembling the intracellular organization of eukaryotic cells. This concept allows to separate and conduct incompatible reactions simultaneously in a confined environment with increased efficiency, and is a very powerful tool in material chemistry. For the catalyst recovery, the sample was promptly subjected to centrifugation (25 °C, 500 g force, 4 min). Subsequently, the suspension of polymersomes-in-polymersomes in the aqueous phase was retrieved from the lower layer.
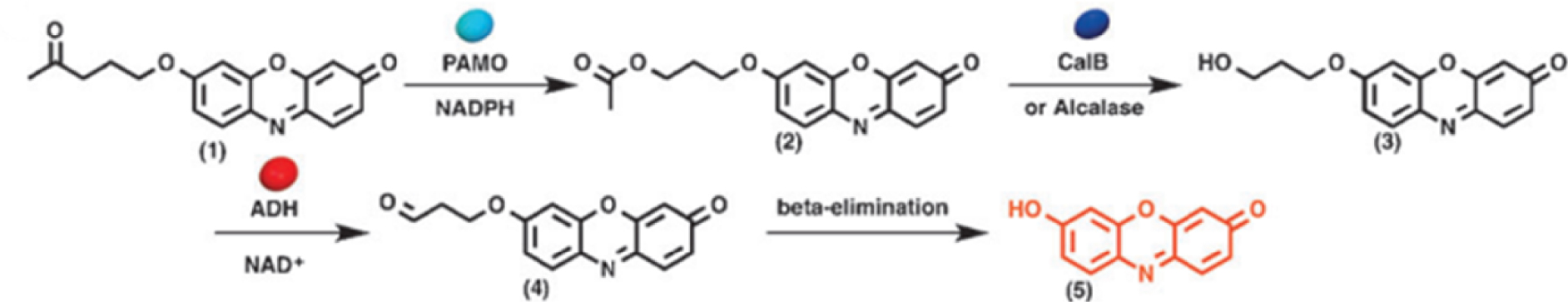
Enzymatic cascade reactions in multi-compartmentalized polymersomes. Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [87]. Copyright 2014.
Using the micellar or vesicular approaches, the formation of stable emulsions may occur due to the excessive swelling of the micellar core [88]. Parameters such as temperature, dilution, and the composition of the mixed solvent [89, 90, 91, 92] determine the equilibrium between micelles/vesicles and single amphiphilic chains. An alternative and promising method to overcome these disadvantages is to cross-link all amphiphilic polymer chains to form unimolecular nanoobjects, thus removing the dynamic micelle/single chain equilibrium [42, 60, 93].
3.3. Unimolecular nanoreactors
3.3.1. Shell cross-linked micelles
Wooley and coworkers [94] developed a method to prepare functionalized micelles from low polydispersity macromolecules with a defined structure and an immobilized permeable cross-linked shell and labile core. Unlike dendrimers [95] these shell-cross-linked knedel-like (SCK) particles have greater peripheral functions and nanometer-size diameters, and are therefore more considered as hybrids between dendrimers, hollow spheres, latex particles, and block copolymer micelles (Figure 6). The SCKs were prepared by a two-step synthesis procedure using a block copolymer of polystyrene and poly(4-vinyl pyridine), PSt-b-PVP, obtained by anionic polymerization. The PSt served as the hydrophobic block and the quaternized PVP generated the hydrophilic block and introduced the cross-linkable group. Besides micellar catalysis [94], the SCKs find many applications in other fields such as, e.g. recording materials, hydraulic fluids, delivery processes, phase transfer reactions, solvation, coatings and fillers.
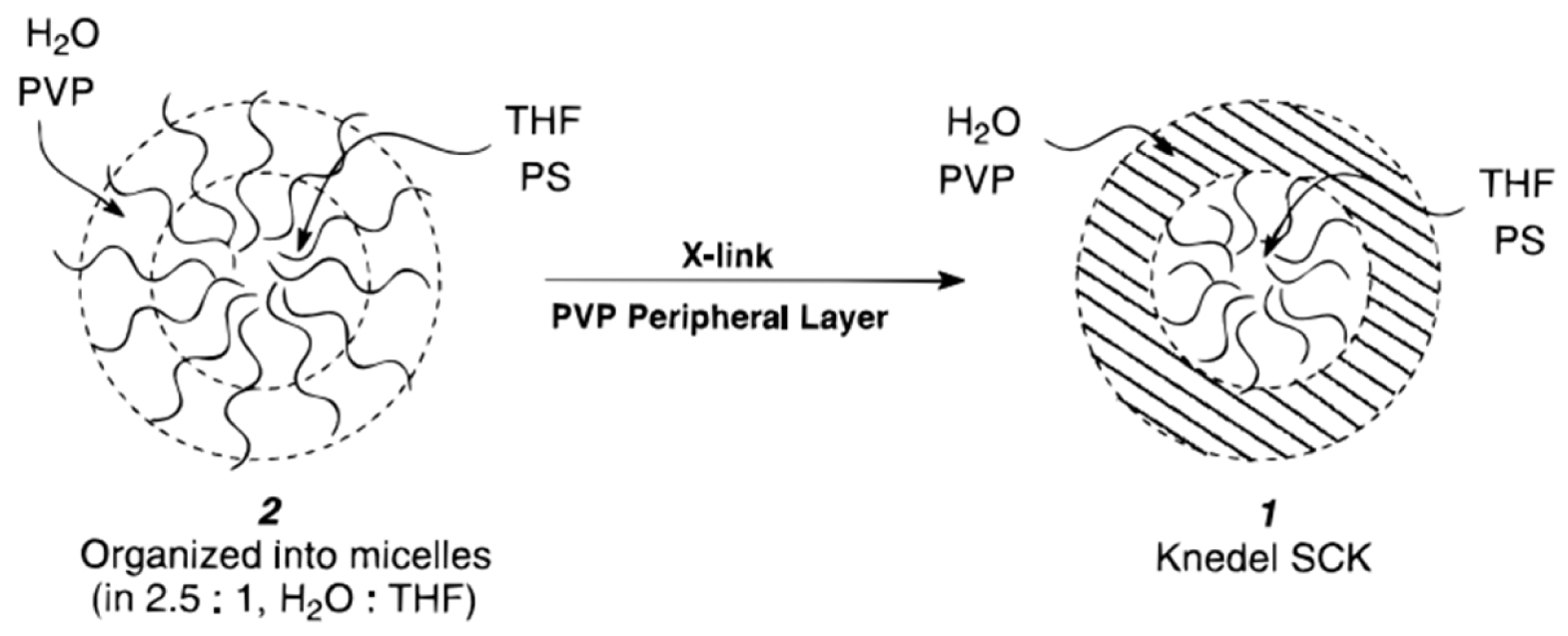
Schematic representation of the basic approach for the formation of SCK’s. Micellization of amphiphilic 2 is followed by cross-linking through the styrene side chains located in the peripheral water to yield 1. Reprinted with permission of American Chemical Society from [94]. Copyright 1996.
O’Reilly and coworkers [96] reported the co-polymerization of an amphiphilic copolymer in which the hydrophobic domain was selectively functionalized with terpyridine groups using nitroxide mediated polymerization (NMP) techniques, that has been extensively studied and optimized by Schubert and coworkers [97, 98] (Scheme 5). The resulting shell cross-linked micelles (SCMs) (Figure 7) were then modified by metal ion complexation, notably Cu+, and used as catalysts for 1,3-dipolar cycloaddition reactions (“click reactions”) of azido- and alkynyl-functionalized small molecules [96].
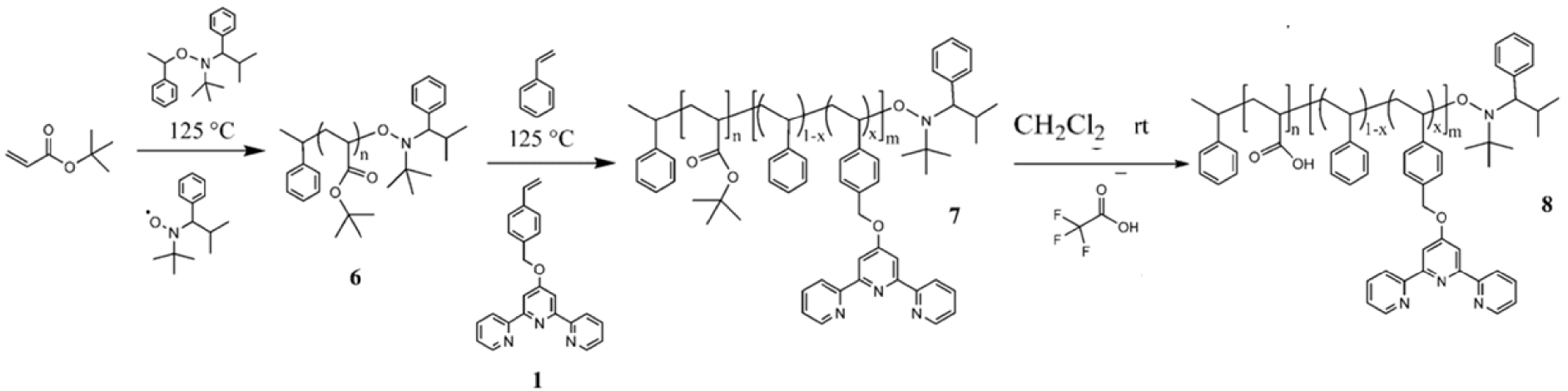
Synthesis of a tert-butyl acrylate block 6 using NMP, followed by chain extension to block copolymer 7 that incorporates terpyridine functionality, and then formation of an amphiphilic diblock copolymer 8 with terpyridine functionality embedded within the hydrophobic segment. Reprinted with permission of American Chemical Society from [96]. Copyright 2008.

Representative transmission electron microscopy (TEM) images of SCM. Reprinted with permission of American Chemical Society from [96]. Copyright 2008.
Weck and coworkers [99] also reported the synthesis of various SCMs and their catalytic applications [100]. Initially, they reported the synthesis of poly(norbornene) block copolymer-based amphiphilic ABC triblock copolymers via ROMP [99]. The middle block (B block) was functionalized with a photochemically active cinnamate group for crosslinking, and the terminal hydrophobic block (C block) contained a Co-salen ligand. The Co-salen functionalized SCMs catalysts were then used for hydrolytic kinetic resolution (HKR) of epichlorohydrin. They showed that the structure of the SCM catalysts was stabilized by the cross-linked shell, which assisted recyclability of the catalysts. Then they also prepared SCMs containing Co(III)-salen cores from amphiphilic poly(2-oxazoline) triblock copolymers [100] and studied substrate selectivity in HKR with various terminal epoxides. More recently, they also developed trifunctional SCMs for enantioselective three-step tandem catalysis [101], which were based on poly(2-oxazoline) synthesized through living CROP [102, 103] that permitted compartmentalization of three incompatible catalysts. The SCM consisted of carboxylic acids in the hydrophilic outer shell, Rh-based N-tosylated 1,2-diphenyl-1,2-ethylenediamine (Rh-TsDPEN) in the intermediate cross-linked shell, and 4-dimethylaminopyridine (DMAP) in the hydrophobic core. The spatial architecture of each catalyst was designed to selectively exploit the path of the substrate during each step of the reaction; the first step involved ketal hydrolysis to the corresponding prochiral ketone, the second step asymmetric transfer hydrogenation (ATH) to yield an enantio-enriched secondary alcohol, and the final step selective acylation to the desired ester product (Scheme 6). Higher conversions were observed with more hydrophobic ketals and anhydrides, and the role of the intermediate shell cross-linked layer in preventing deactivation of the DMAP catalyst supported inside the micellar core was also shown. The conversion was determined from aliquots taken at certain time intervals, extracted with ethyl acetate. No recyclability tests were reported.
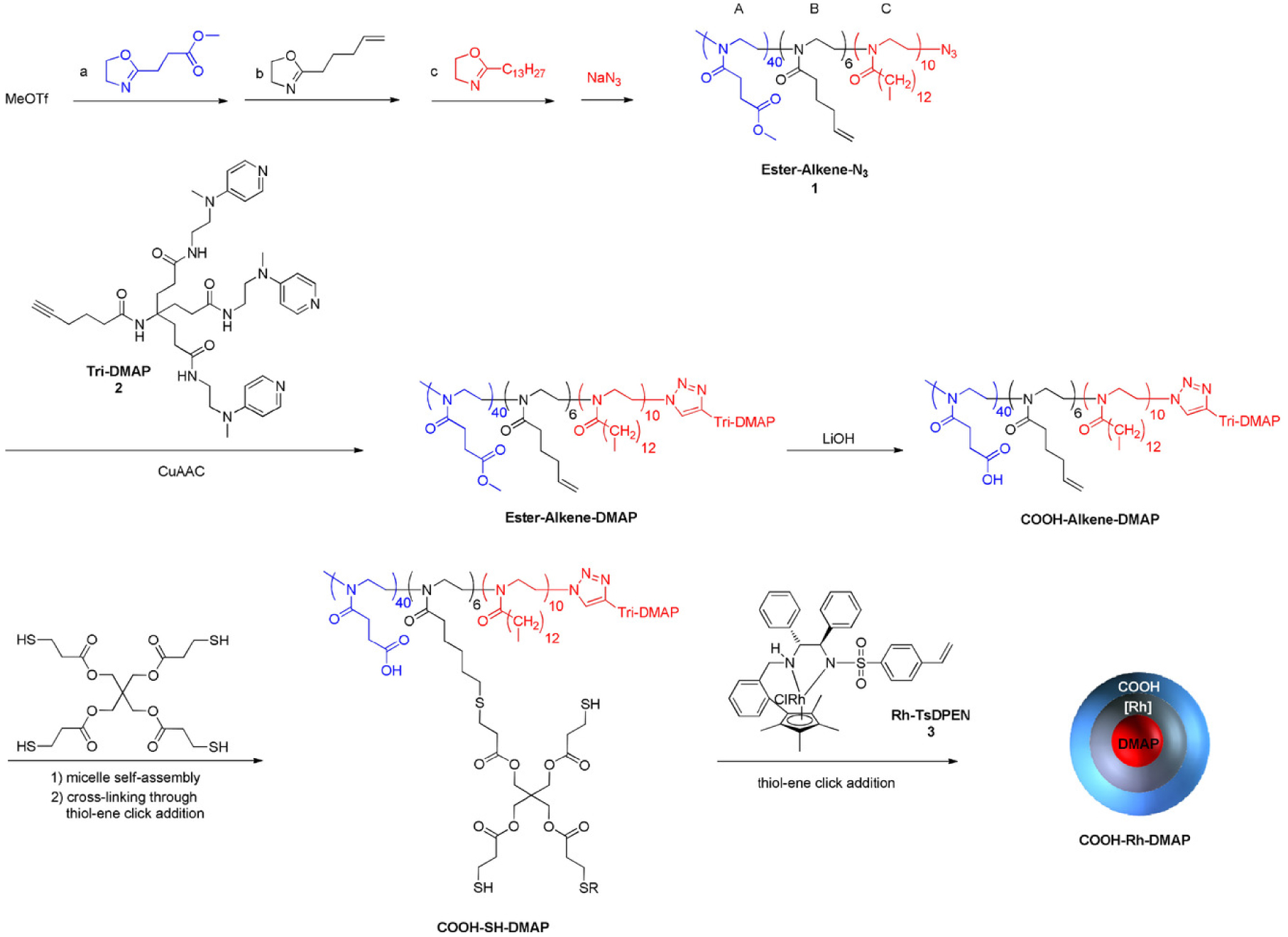
Synthesis of the trifunctional SCM nanoreactor COOH-Rh-DMAP. Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [101]. Copyright 2018.
Optimally, SCMs act as nano-incubators for loaded catalysts and protect the catalyst from passivation or deactivation. However, the cross-linked shell also introduces a physical barrier between the hydrophobic core and the hydrophilic shell, which can lower the mass transport of certain chemicals and limit SCMs applicability for certain catalytical transformations.
3.3.2. Core cross-linked micelles
To circumvent the mass transport restrictions of SCMs, other polymeric structures have been developed. The first structure, called core-crosslinked micelle (CCM), consists of ligand-functionalized amphiphilic diblock copolymers that are cross-linked at the end of the hydrophobic chain. This provides star-block unimolecular nano-objects where the ligands are located on flexible arms outside of the cross-linked area. The second structure, known as nanogel (NG), features a fully cross-linked core, with the catalyst binding sites being situated within the cross-linked core [88]. Various strategies have been reported in the literature for the synthesis of CCMs, including ATRP [104], as used by Sawamoto and Terashima. They were the first to use NG-type nanoreactors in catalysis and the Ru-containing NG polymers made by ATRP [42, 105, 106, 107, 108], and RAFT-mediated polymerization-induced self-assembly (PISA) [109]. The latter strategy is a particularly attractive approach as it allows to produce block copolymer nano-objects with a full synthesis protocol in one-pot. A survey of the different synthetic approaches is beyond the scope of this review and the reader is referred to the literature [110, 111, 112, 113, 114, 115] for further details.
Rieger and coworkers [116] reported the synthesis of a core–shell NG using a previously established method involving RAFT radical polymerization in an aqueous dispersion that provided nanometer-sized, uniform core–shell particles with a cross-linkable core. Poly(N,N-dimethylacrylamide) served as the stabilizing shell, and (N,N-diethylacrylamide) (PDEAAm) was used for the core matrix through aqueous heterogeneous polymerization at a moderate temperature. The NG was formed in water at 70 °C through RAFT dispersion polymerization of N,N-diethylacrylamide (DEAAm; 85 mol%), DMAEA (10 mol%), and N,N-methylenebis(acrylamide) (MBA; 5 mol%), with a stabilizing macromolecular RAFT agent based on poly(N,N-dimethylacrylamide) (PDMAAmmacroRAFT). The resulting nanostructured core–shell NG demonstrated the ability to stabilize Pd0 nanoparticles (NPs) in its core. Pd NPs were incorporated into the NG by adding a PdII salt, expected to coordinate with the nitrogen functions of DMAEA units, followed by metal reduction with ethanol. The hybrid PdNP@NG exhibited stability in both solid and solution states, making it an effective catalyst for the Mizoroki–Heck reaction between n-butylacrylate and activated bromo- and iodoarenes. After catalysis, the hybrid NG could be recovered, dried, and reused for three consecutive runs without significant loss of the catalytic activity. However, after the fourth reuse, a decline in catalytic activity was observed. Analysis of the recovered hybrid PdNP@NG revealed the presence of large Pd aggregates and significant oxidation of Pd0 to PdII (Figure 8). Additionally, analysis by inductively coupled plasma mass spectrometry (ICP-MS) showed a substantial loss of Pd from the polymeric NG support. This suggested that part of the catalysis occurred outside the NG, explaining the observed limited recyclability.
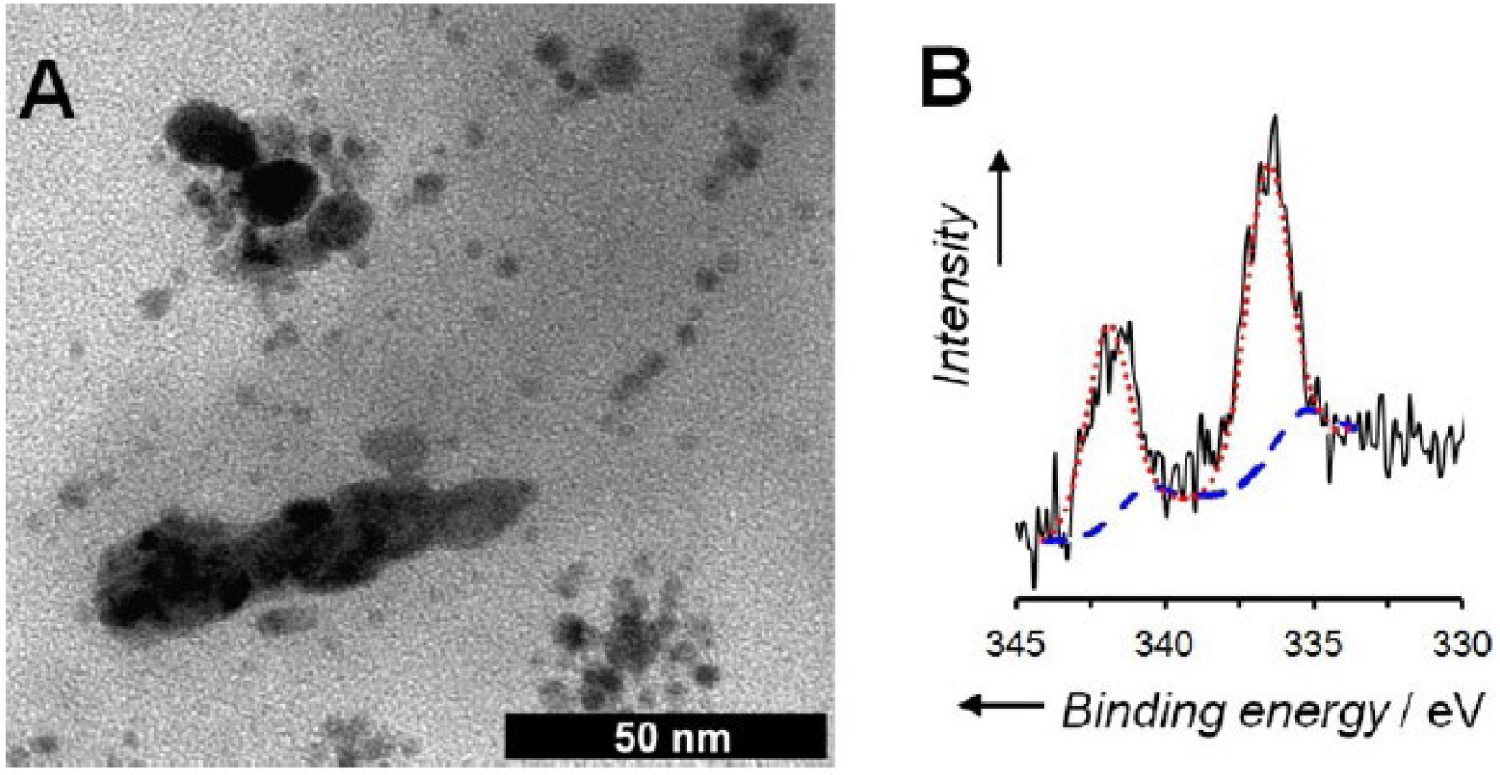
(A) TEM image and (B) Pd 3d X-ray photoelectron spectrum (XPS) of the hybrid PdNP@NG after four catalytic cycles (solid black line) and its deconvolution into Pd0 (dashed blue line) and PdII (dotted red line). Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [116]. Copyright 2017.
Stenzel and coworkers [117] used RAFT polymerization technique to synthesize poly(2-hydroxyethyl acrylate)-poly(n-butyl acrylate) block copolymers using either poly(2-hydroxyethylacrylate) or poly(n-butyl acrylate) macromolecular RAFT (macroRAFT) agents with narrow molecular weight distributions. These macroRAFT agents were further extended by polymerization of a diacrylate monomer, yielding CCMs. No catalytic application was described for these CCMs.
Furthermore, three generations of CCMs with amphiphilic unimolecular polymer-based nanoreactors [88] having neutral (CCM-N), cationic (CCM-C) and anionic (CCM-A) shells, respectively, have been prepared by RAFT PISA and used for aqueous biphasic catalysis (Figure 9) [118]. In all cases, special attention was devoted to the catalyst recovery using an optimized procedure. Briefly, after phase separation, the latex was extracted with diethyl ether or toluene, and the combined organic phases subjected to analysis by gas chromatography (GC). For the recycling experiments, a fresh substrate solution (same amount as in the initial run) was added to the same vial, followed by reaction and product separation according to the same protocol.
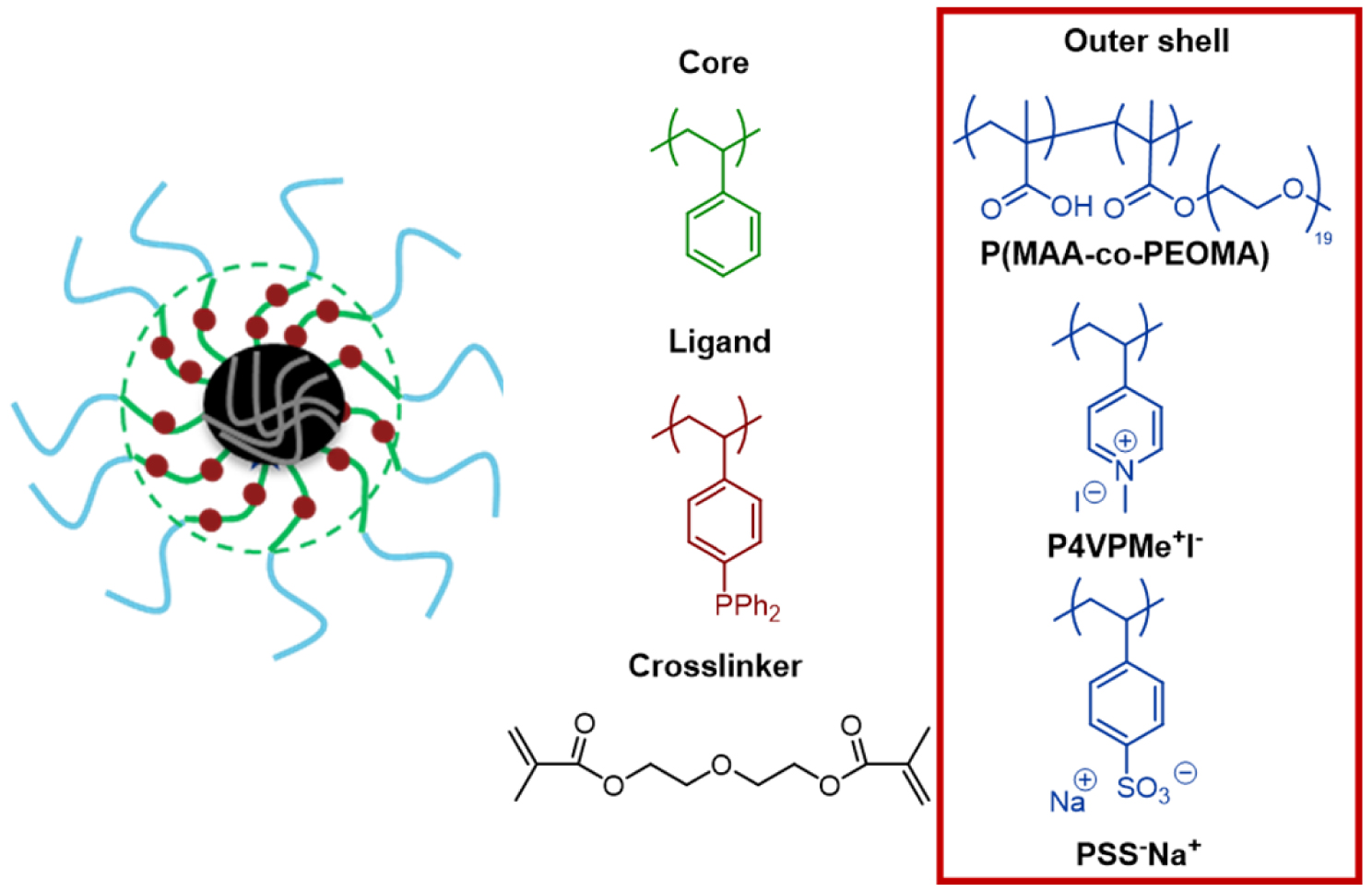
Three generations of CCMs with triphenylphosphine (TPP) core and different outer shell.
The first generation CCM-N [119] was designed with an uncharged water-soluble shell by copolymerization of methacrylic acid (MAA) and poly(ethylene oxide) methyl ether methacrylate (PEOMA) to generate a P(MAA-co-PEOMA) macroRAFT agent (Scheme 7). Addition of styrene (St) and DPPS (diphenylphosphinostyrene) as core ligand generated P(MAA-co-PEOMA)-b-P(St-co-DPPS) amphiphilic block copolymers that self-assembled into well-defined micellar particles (d = 72 nm), which was crosslinked using diethylene glycol dimethacrylate (DEGDMA) forming CCM-N (d = 79 nm). The average formula of the single polymer chains is R0-(MAA0.5-co-PEOMA0.5)30 b-(St1−n-co-DPPSn)300-b-(St0.9-co-DEGDMA0.1)100-SC(S)SPr, with the chain ends (R0 = C(CH3)(CN)CH2CH2COOH and SC(S)SPr) provided by the RAFT chain transfer agent (CTA). The uniformity of the CCM-Ns was demonstrated by size exclusion chromatography (SEC), TEM and DLS analyses, which also evidenced the latex stability. Moreover, nuclear magnetic resonance (NMR) spectroscopy and DLS confirmed the transport of hydrophobic molecules across the hydrophilic shell into the polymer core to be fast and leading to increased particle size owing to the particle swelling (d = 117 nm).
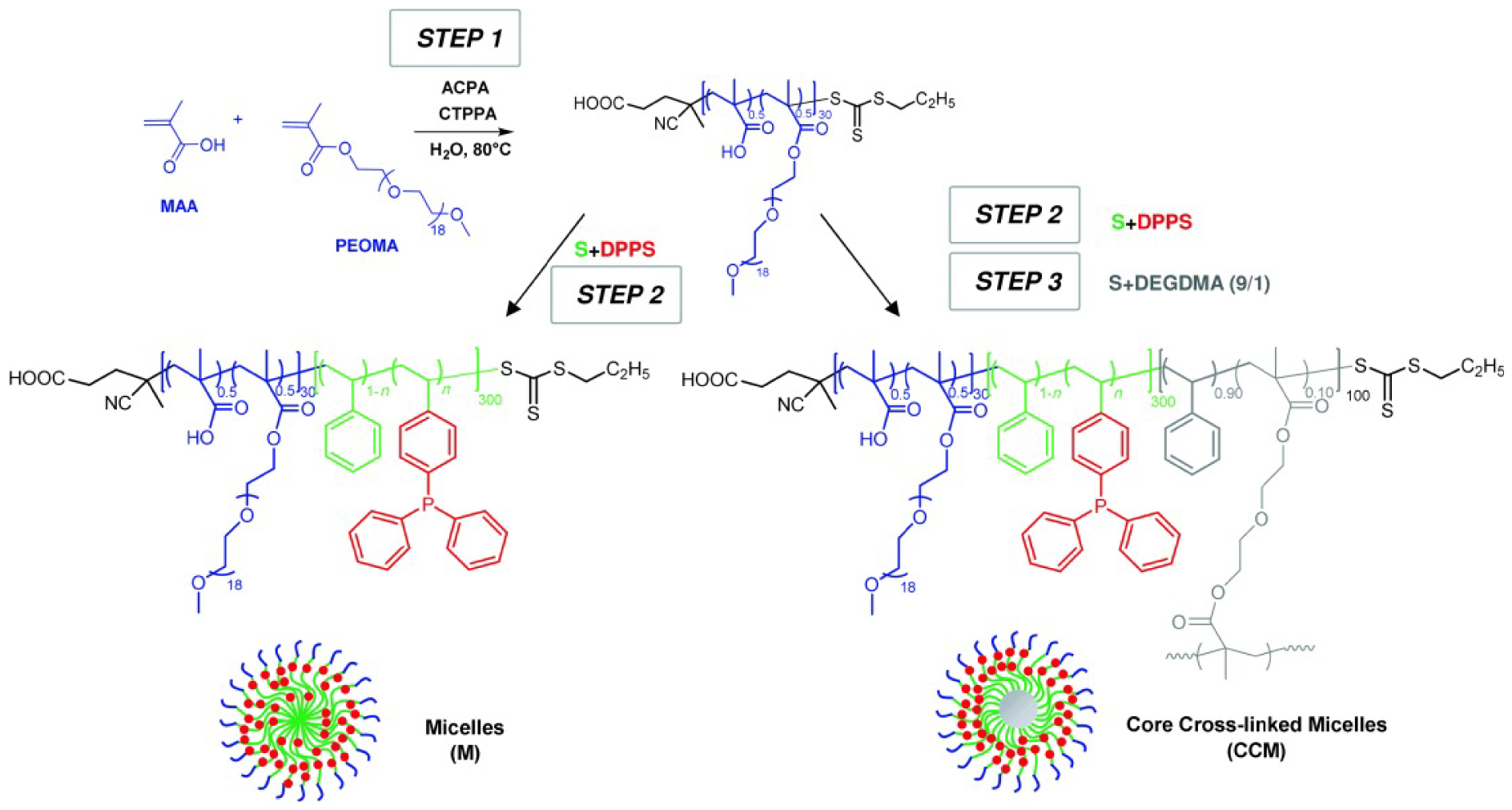
General strategy of RAFT-mediated emulsion polymerization for the synthesis of various types of core-shell nanoreactors. Reprinted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [119]. Copyright 2014.
As a proof-of-principle application, the CCM-Ns were loaded with [Rh(acac)(CO)2] (acac = acetylacetonate) and evaluated for aqueous biphasic hydroformylation of 1-octene [120]. High activity, low isomerization as well as good stability and recyclability (Rh leaching of a few ppm) were found, but the system was moderately mass-transfer limited at high Rh concentrations and non-negligeable leaching appeared to result from particle aggregation. Subsequent follow-up studies addressed the metal leaching mechanism [121], metal migration and cross-exchange in amphiphilic core-shell polymer latexes [122] and mass-transport limitations [123]. Later, Poli, Manoury and coworkers [124] extended the family of ligand-functionalized CCMs using the same synthesis protocol to include the bidentate Nixantphos ligand in the CCM-N. After loading with [Rh(acac)(CO)2] the system was also applied to the aqueous biphasic hydroformylation of 1-octene, where only moderate activity (mass-transfer limitation) but excellent regioselectivity was found for the formation of n-nonanal.
A convergent synthesis of polymeric nanoreactors containing polymerizable RhI-NHCR complexes [53] was then developed (Scheme 8). TEM images of the resulting Rh-NHCR@CCM nanoreactors with R = Me (5a) and R = Mes (5b) (Figure 10) revealed formation of particles with a broad size distribution (diameter of 123.3 ± 19.2 nm). Under optimized reaction conditions, the 5b nanoreactors yielded good activity and excellent recyclability for styrene hydrogenation with Rh-leaching <0.6 ppm per reaction cycle (measured by ICP-MS) after some initial decomposition of the Rh-complex by cleavage of the Rh-NHC bond in the initial two runs (Table 2, entries 1 and 2). Despite the encouraging recyclability results, mass-transfer limitations and impractical slow separation of the catalyst and reaction mixture remained for this catalyst, like for the other CCM-N catalysts.
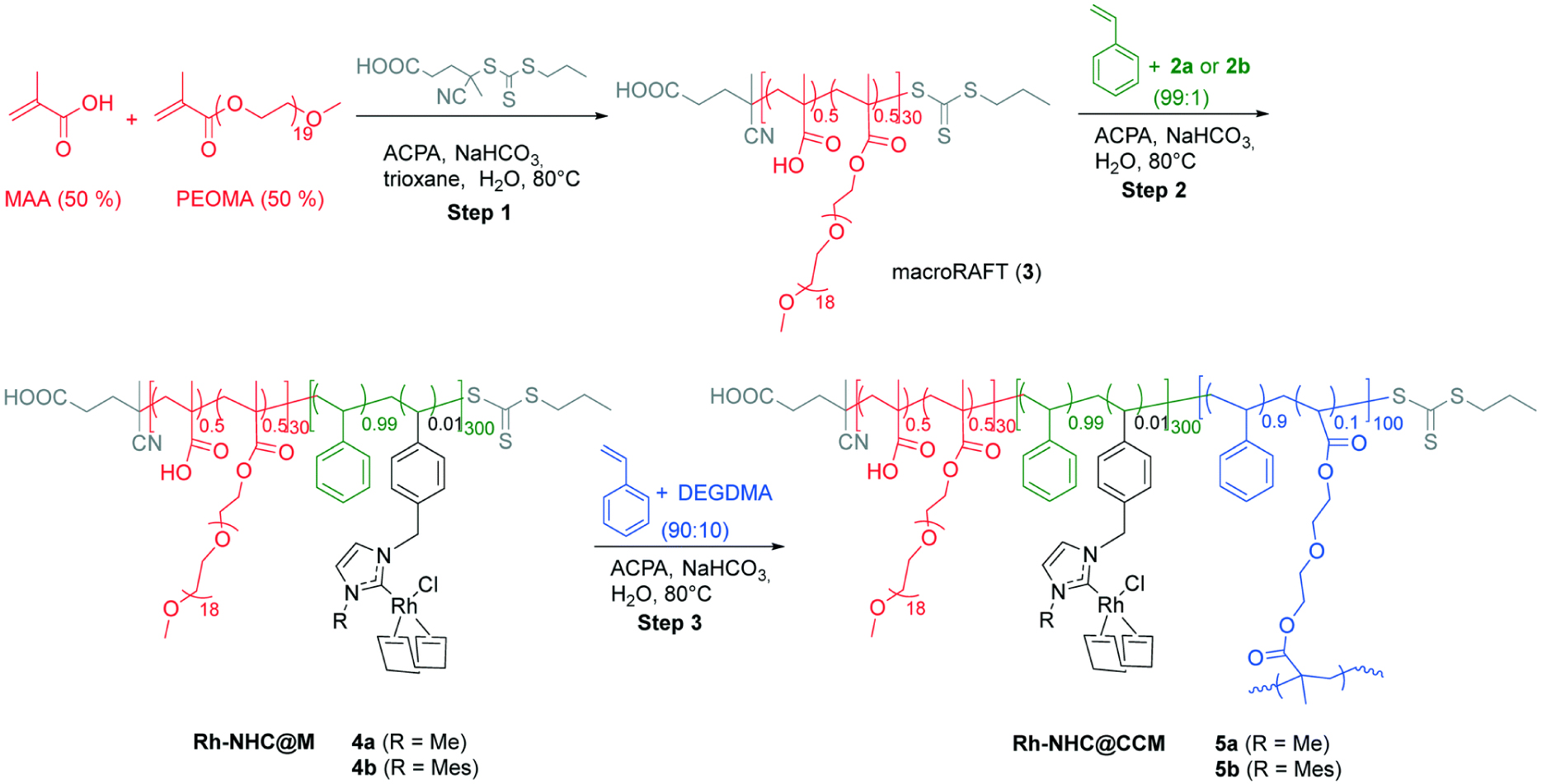
Synthesis of Rh-NHCR@CCMs 5a (R = N-methylimidazole (Me)) and 5b (R = N-mesitylimidazole (Mes)) by RAFT-PISA polymerization. Reprinted with permission of The Royal Society of Chemistry from [53]. Copyright 2021.

TEM images of Rh-NHCR@CCMs 5a (left) and 5b (right) (numbers as in Scheme 8). Reprinted with permission of The Royal Society of Chemistry from [53]. Copyright 2021.
Recycling study of Rh-NHCMes@CCM 5b (number as in Scheme 8) catalyst in styrene hydrogenation (adapted from [53])

| Entry | Run | St/Rh | St Conversiona (%) | EB Selectivitya (%) | Rh leachingb (ppm) |
|---|---|---|---|---|---|
| 1 | 1st | 1000/1 | >99.5 | >99.5 | 0.87 |
| 2 | 2nd | >99.5 | >99.5 | 1.47 | |
| 3 | 3rd | >99.5 | >99.5 | 0.24 | |
| 4 | 4th | >99.5 | >99.5 | 0.34 | |
| 5 | 5th | >99.5 | >99.5 | 0.21 | |
| 6 | 1st | 10,000/1 | 73 | >99.5 | 0.39 |
| 7 | 2nd | 77 | >99.5 | 0.61 | |
| 8 | 3rd | >99.5 | >99.5 | 0.54 | |
| 9 | 4th | >99.5 | >99.5 | 0.31 | |
| 10 | 5th | 98 | >99.5 | 0.13 |
Conditions: styrene (79.3 mg, 0.75 mmol), CCM 5b (85 mg, 7.9 × 10−7 mol of Rh for St/Rh = 1000/1 or 8.5 mg, 7.9 × 10−8 mol of Rh for St/Rh = 10,000/1), decane (31.5 mg, 0.225 mmol), toluene (1 mL)/water (0.5 mL). a Measured by GC. b Measured by ICP-MS.
To circumvent the mass-transfer and separation issues with CCM-N, a second generation of CCM nanoreactors with an outer polycationic shell (CCM-C) based on poly(1-methyl-4-vinylpyridinium) (CCM-Cs) was developed via RAFT polymerization [125]. The macroRAFT agent was synthesized by RAFT polymerization of 4-vinylpyridine (4VP) in aqueous ethanol, followed by a chain extension with a PSt block and quaternization of the P4VP block. A core-anchored triphenylphosphine (TPP) ligand functionality, diluted in styrene was introduced by chain extension in a fourth synthetic step and the CCM-C was finally formed in a fifth step by core cross-linking by a DEGDMA:St (10:90) mixture [126] (Scheme 9). DLS and TEM analyses showed that all obtained polymers (x = 5, 10 or 20%) have spherical morphology, a narrow size distribution (d = 130–150 nm) and a positive zeta potential.
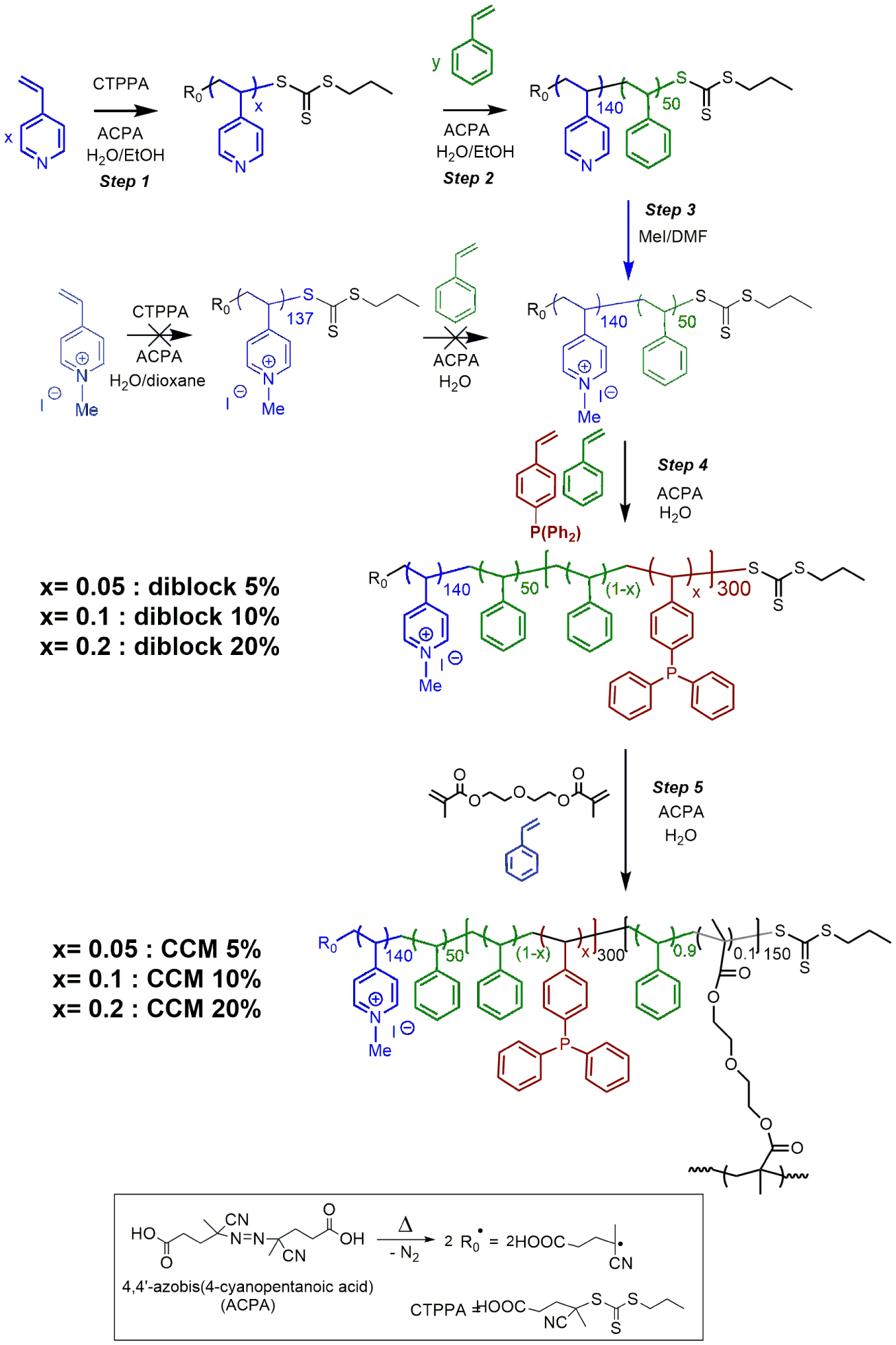
Synthesis of CCM-C. Adapted with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim from [126]. Copyright 2019.
After loading with [RhCl(COD)]2 (COD = 1,5-cyclooctadiene), the aqueous biphasic hydrogenation of 1-octene and styrene revealed improved mass transport properties and superior performance of the CCM-Cs, in terms of both catalytic activity, stability, and recovery, relative to the neutral-shell analogues [126]. Importantly, also less catalyst leaching occurred due to an enhanced ability of the polycationic shell to confine the nanoreactors in the aqueous phase. Moreover, in substrate scope investigations where the catalytic hydrogenation of acetophenone was attempted, the [RhCl(COD)(TPP@CCM-C)] latex turned black, suggesting reduction of the molecular RhI complex to Rh0 metal. Actually, this turned out to be an excellent approach to generate Rh NPs directly inside the core of CCMs (Figure 11), triggering further investigations of biphasic hydrogenation with Rh NPs [127].

TEM image of Rh NPs generated in the core of CCM-C. Reprinted with permission of The Royal Society of Chemistry from [127]. Copyright 2021.
The RhNP@CCM-C system was initially tested for aqueous biphasic hydrogenation of acetophenone, but only low conversions were obtained, unless high temperature (90 °C) was used. The slow reaction rate was attributed to a poor mass transport of the acetophenone into the CCM-C core, resulting from a possible electrostatic interaction between its carbonyl group and the cationic pyridinium functions present in the shell [127]. Conversely, the catalytic performances were excellent when the catalyst system was applied to the hydrogenation of 1-octene and styrene, with full conversion and full selectivity towards the hydrogenation of the olefinic group. Notably, the catalytic activity was superior to that of molecular RhI systems. The RhNP@CCM-C system proved also recyclable for styrene hydrogenation up to four runs without loss of activity and with the Rh NPs remaining well-dispersed when toluene was used for product recovery/catalyst recycling. However, when using diethyl ether as extraction solvent a dramatic loss of activity was observed. This phenomenon was attributed to facilitated extraction of the Rh NPs from the CCM-C core by the oxygen-based solvent (Figure 12).
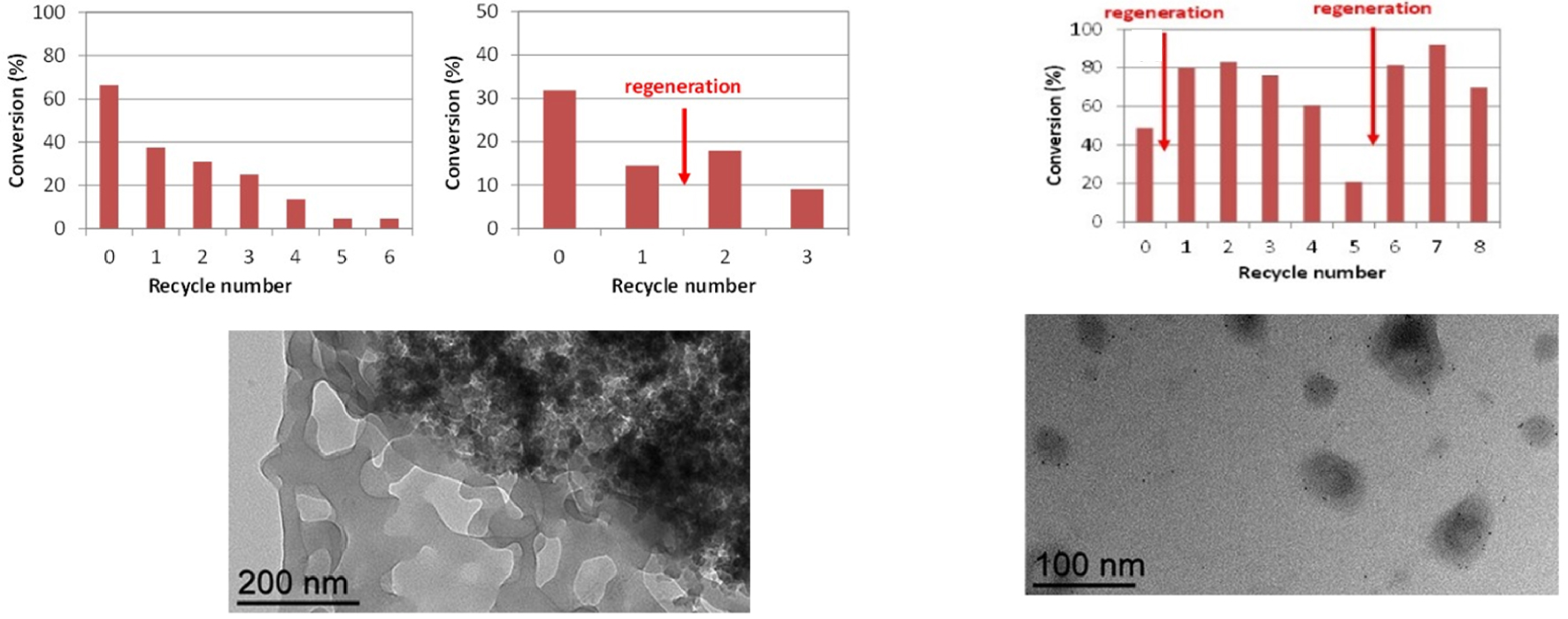
Recycling tests in the hydrogenation of styrene with RhNP@CCM-C using diethyl ether (left) and toluene (right) as extraction solvent (25 °C, 0.5 h, styrene/Rh = 2000/1) (top). TEM images of the recovered catalyst latexes after the final run (bottom). Reprinted with permission of The Royal Society of Chemistry from [127]. Copyright 2021.
To address the substrate scope limitation of the CCM-C nanoreactors, a third generation of phosphine-functionalized CCM nanoreactors was developed with a polyanionic poly(styrene sulfonate)-based shell (CCM-A) [12, 118]. The synthesis route was identical to those used for the syntheses of CCM-N and CCM-C described above, except for using the water-soluble sodium styrene sulfonate monomer in the first step (Scheme 10, Figure 13).
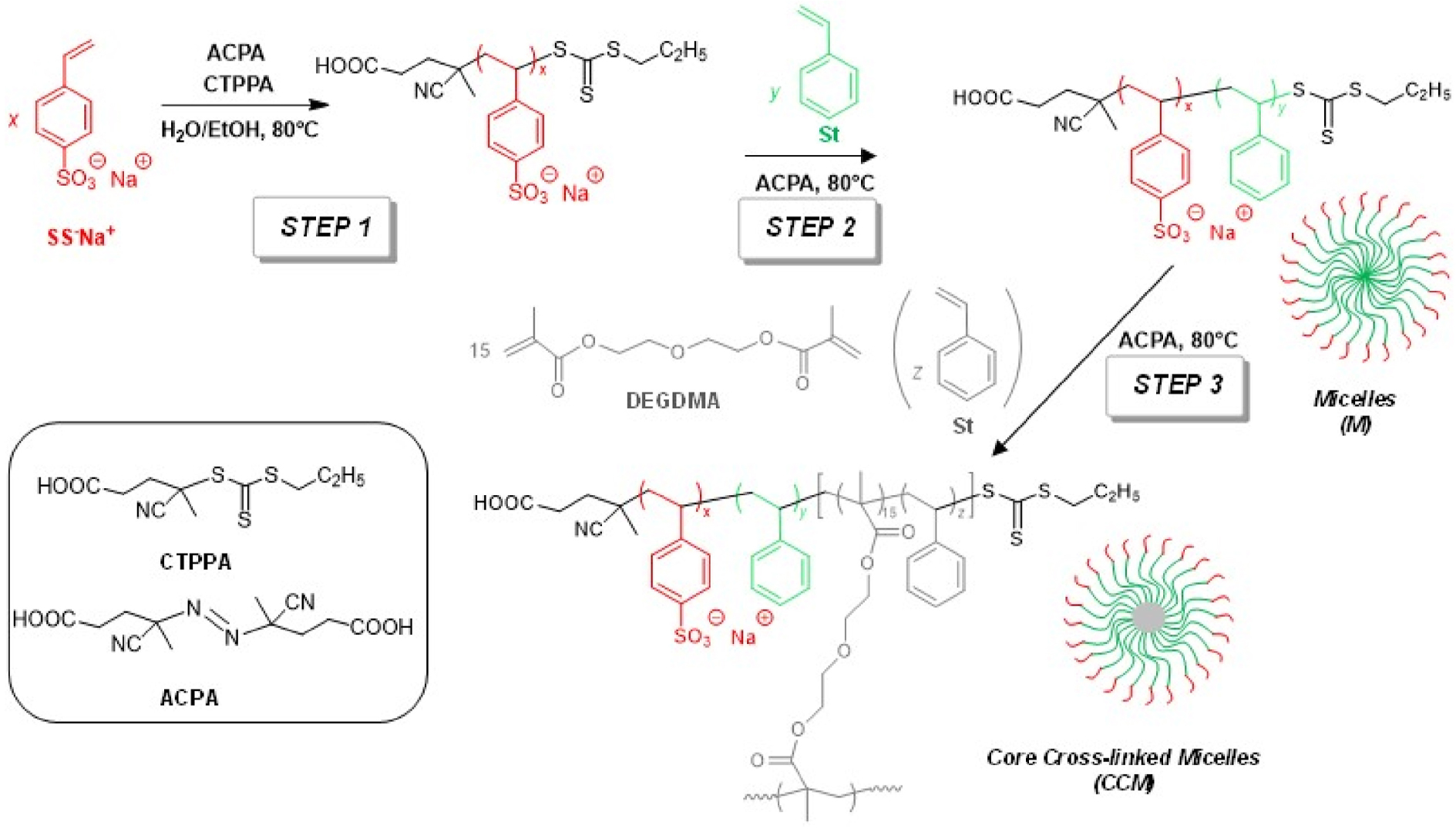
Synthesis route to CCM-A. Reprinted with permission of Elsevier Ltd [12]. Copyright 2022.
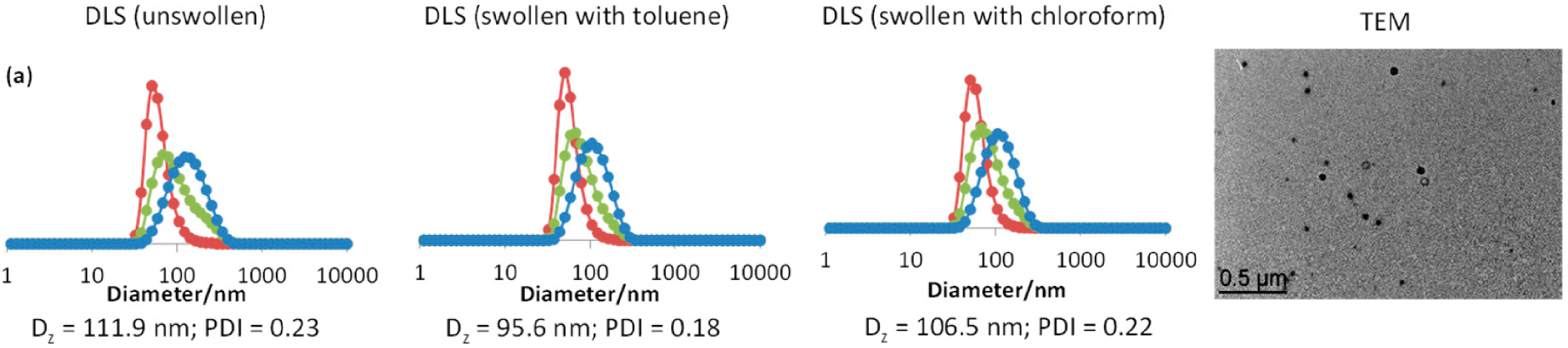
DLS (before and after swelling with toluene or chloroform) and TEM image of the CCM-A. Color coding for the DLS size distributions: number (red), volume (green) and intensity (blue). Reprinted with permission of [118]. MDPI (https://creativecommons.org/licenses/by/4.0/).
For application in aqueous biphasic styrene hydrogenation, the CCM-A nanoreactors were loaded with [RhCl(COD)]2, as previously done for the CCM-N and CCM-C counterparts (vide supra). However, interaction between the Rh dimer complex and the outer shell of the CCM-A limited the transfer of the metal complex to the core, presumably via chelating RhI-sulfonate complex formation. This bottleneck was circumvented by “dilution” of the shell with a neutral monomer (PEOMA), forming a mixed polyanionic-neutral CCM, which allowed RhI complexation in the core. Nevertheless, the mixed polyanionic-neutral CCM nanoreactors had inferior performance compared to equivalent nanoreactors with neutral and polycationic shells, which seemed to stem from catalyst alterations induced by migration of the Rh centers towards the shell sulfonate groups [12, 118]. Thus, despite significant progress in the development of CCMs, further improvements are still needed to obtain efficient and durable nanoreactor systems for aqueous biphasic hydrogenation catalysis.
4. Conclusion
Nanostructured materials with high specific surface area offer many advantages, attributed to their unique properties, paving the way for advances in various fields such as nanotechnology, materials science, electronics, energy storage, and catalysis. This review has highlighted major developments in the synthesis and catalytic applications of self-assembled nanoreactors made from amphiphilic macromolecules such as polymer micelles, polymersomes and unimolecular nanoreactors. The approaches using macromolecular nanoreactors as potential catalyst supports, hold many unique advantages owing to the diverse synthesis strategies for the generation and tuning of the nanoreactors, which could lead to unique industrial applications.
In particular, unimolecular nanoreactors have been the topic of extensive recent research. The perspectives of these systems are numerous, owing to their modularity and simple syntheses, notably by the PISA RAFT protocol, which provides access to ligand-functionalized CCM nanoreactors. In such nanoreactors, the chemical nature and the degree of polymerization of the core and the shell, the type and density of the core-anchored ligands, as well as the nature of the coordinated metal pre-catalyst can all be readily varied. This makes the unimolecular nanoreactors highly adaptable and applicable to aqueous biphasic catalysis, thus providing new avenues of research with several potential applications.
Declaration of interest
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work has been supported by the European Research Council, as part of the Horizon 2020 research and innovation program under the Marie Skodowska-Curie grant agreement No. 860322.