



 CC-BY 4.0
CC-BY 4.0
1. Introduction
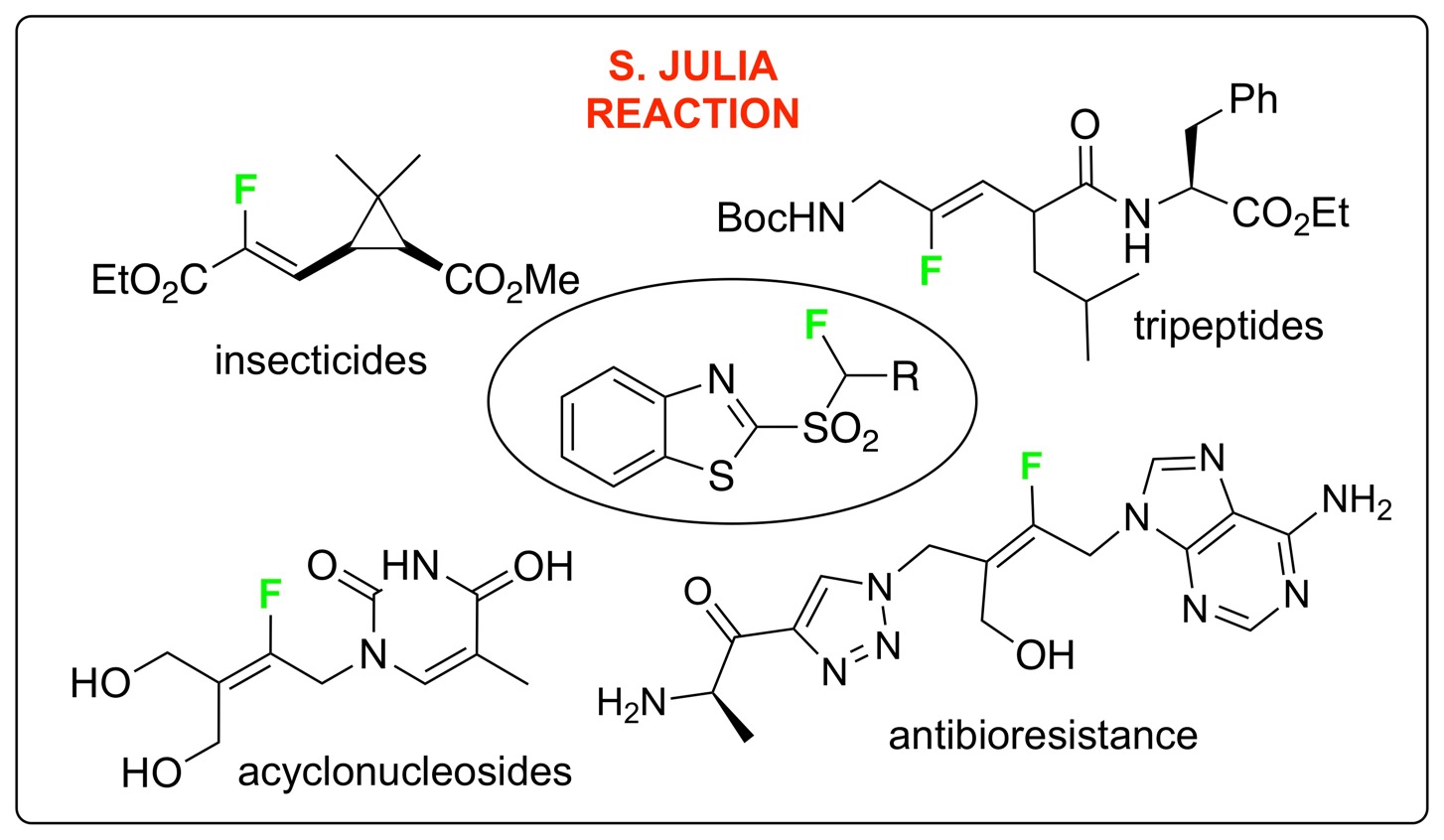
The introduction of a fluorine atom into a biologically active molecule significantly modifies its physicochemical properties. This modification has been widely used for the preparation of pharmaceuticals, agrochemicals and materials [1, 2, 3, 4].
In this field, the synthesis of fluorinated carbon–carbon double bonds has been the source of a considerable and increasing interest in recent years (Figure 1) [5, 6]. It has been found that fluoroalkenes have similar steric and electronic properties compared to amides and can therefore act as peptidomimetics [7]. For example, fluoro-olefin 2 was found to be an excellent DPP IV inhibitor with a constrained conformation.
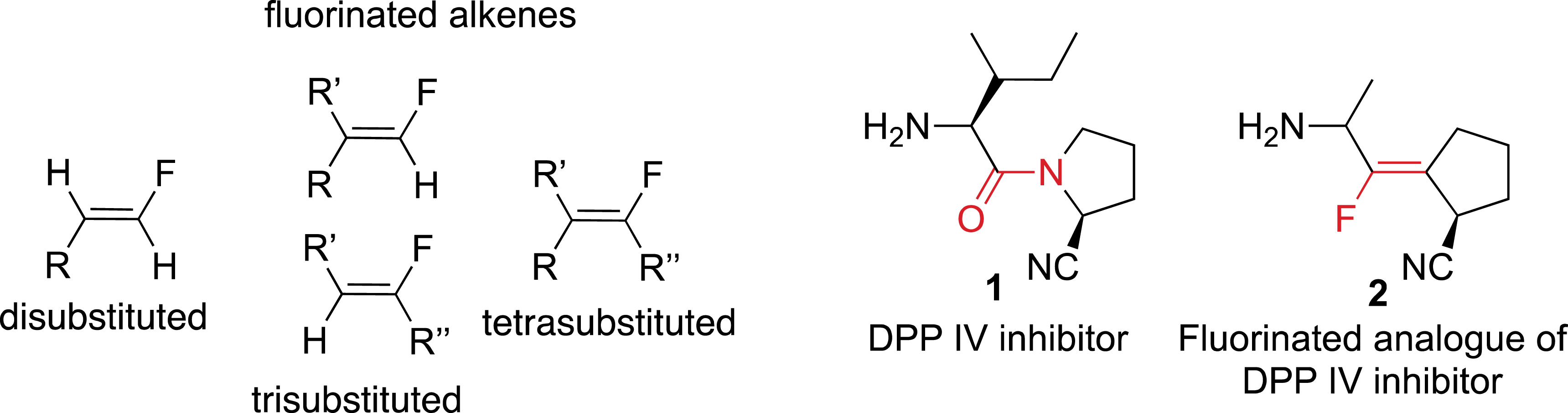
Fluoroalkenes as amide isosteres.
The most common method for the synthesis of alkenes containing an [(Alk)CF=] moiety is the reaction of an aldehyde or ketone with fluorinated Horner–Wadsworth–Emmons (HWE) reagents such as Ph3P=CHF or (EtO)2(O)PCHFCOOEt [8, 9, 10, 11, 12]. While, this approach is very efficient for the preparation of fluoroalkenes bearing electron withdrawing groups, it is not well suited for the synthesis of fluoroalkylidenes. Numerous efforts have been made in order to develop alternative methods and to overcome these drawbacks [5, 6]. In this context, the modified Julia reaction (or Julia–Kocienski reaction) [13], has emerged as one of the most effective methods since our pioneering work [14]. This reaction developed by S. Julia was extended by our group and others to fluorinated series in order to easily access fluoroalkenes from aldehydes or ketones in one step (Scheme 1) [15, 16, 17, 18]. This story began with the preparation of the potent insecticide precursor 3 from the pyrethrin family [14]. This first example has stimulated the preparation of new fluoroalkenes such as vinyl fluorides, which were later reported from the corresponding monofluoromethylsulfone [19, 20].
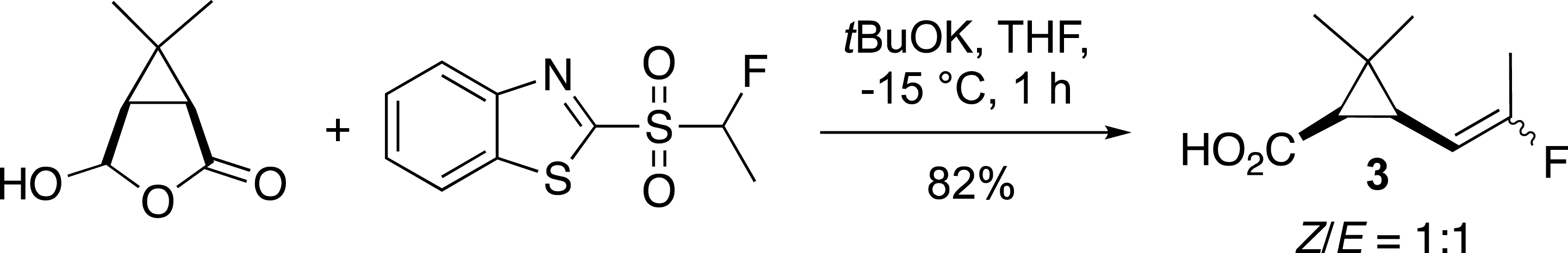
Preparation of an insecticide precursor.
In this account, we will focus on the preparation of fluoroalkenes via the modified Julia reaction that was reported by our group. The aim is to report the main methods for the preparation of di-, tri- and tetrasubstituted alkenes, with a particular emphasis on highly functionalized fluoroalkylidenes acting as peptidomimetics and nucleoside surrogates.
2. Synthesis of fluoroacrylate derivatives
The effect of the fluorine was important for the preparation of acrylate derivatives 5 (Scheme 2). Indeed, the destabilization of the intermediate carbanion by the fluorine atom increased the reaction rate. The reaction with fluorosulfone 4 is 30 times faster than with the corresponding non-fluorinated sulfone [21, 22]. It was completed in only 30 minutes with the fluorosulfone at 20 °C, whereas the same reaction took at least 16 h with the non-fluorinated sulfone.
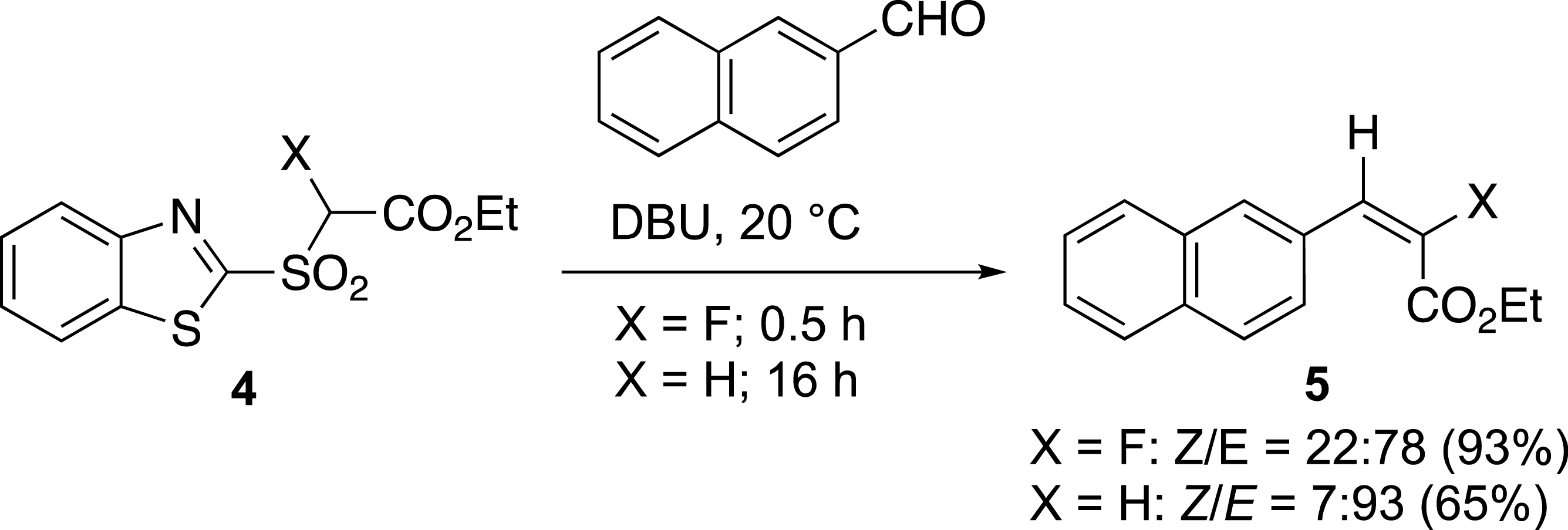
Yields and ratios for the olefination of fluorinated and non-fluorinated aldehydes.
In order to control the geometry of the newly formed carbon–carbon double bond, we found that the reaction had to be carried out in the presence or absence of metalated bases (Scheme 3). Indeed, in the presence of DBU, the (E)-alkene was the main product, whereas the (Z)-alkene was mainly obtained in the presence of NaHMDS [23]. The selectivity is strongly dependent on the nature of the aromatic sulfone, and in particular on the π-deficient character of the heterocycle. In this way it is possible to control the geometry of the carbon–carbon double bond by replacing the benzothiazolyl (BT) [21, 23] 6 with a pyrimidine (Pym) 8 [24] or a bis-trifluoromethylphenyl (BTFP) [25] ring 7.
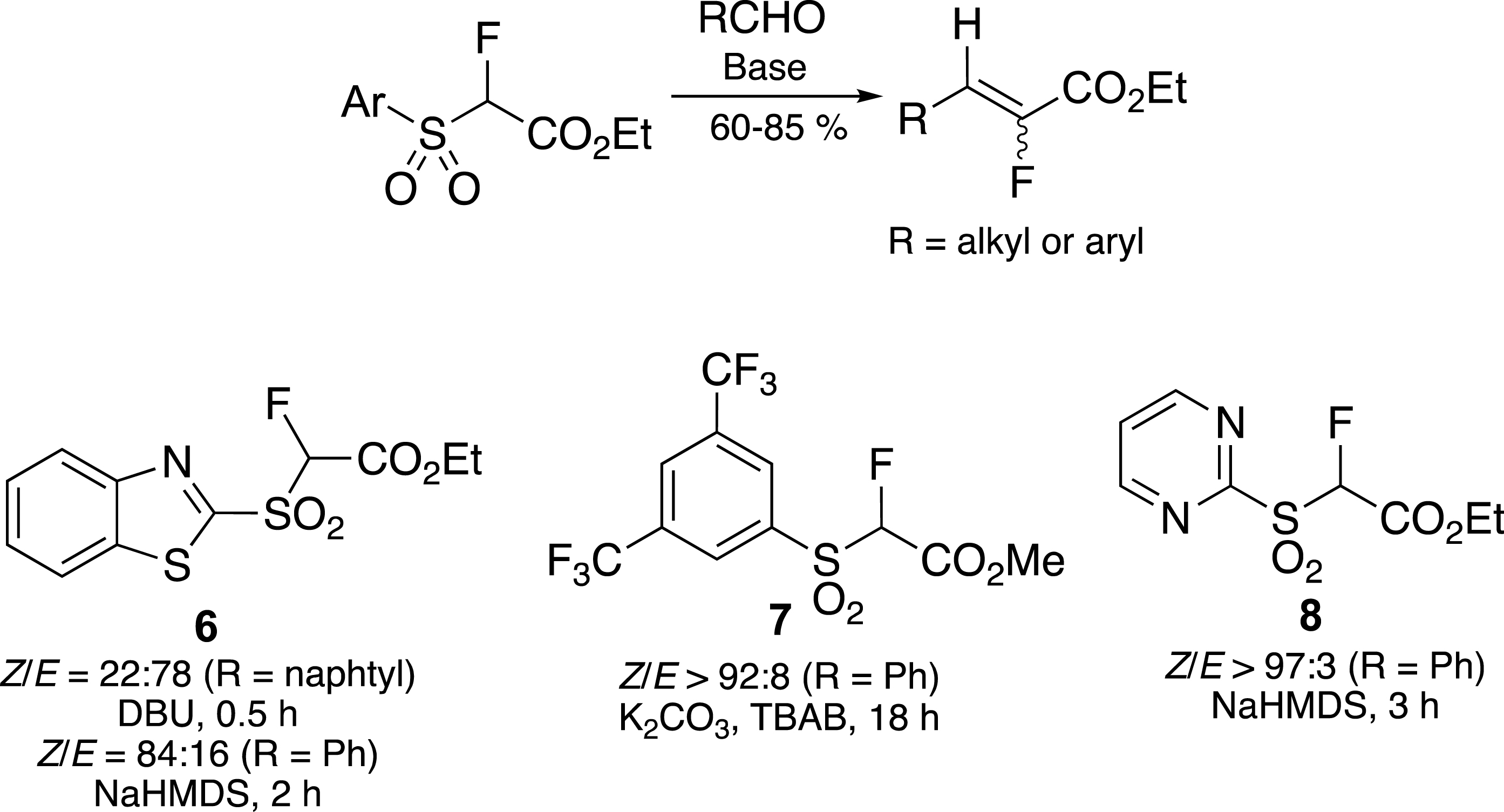
Control of the carbon–carbon double bond geometry.
Other electron-deficient fluoroalkenes were prepared by the modified Julia reaction using BT [26, 27] or BTFP [25] fluorosulfones. Depending on the experimental conditions Z isomers were preferentially obtained. For example, fluorinated Weinreb amides 10 were formed from bis-trifluoromethylphenyl-sulfones 9 and aldehydes with an excellent Z-selectivity in contrast to α,β-unsaturated fluoroacrylates reported in the same work (Scheme 4) [25].
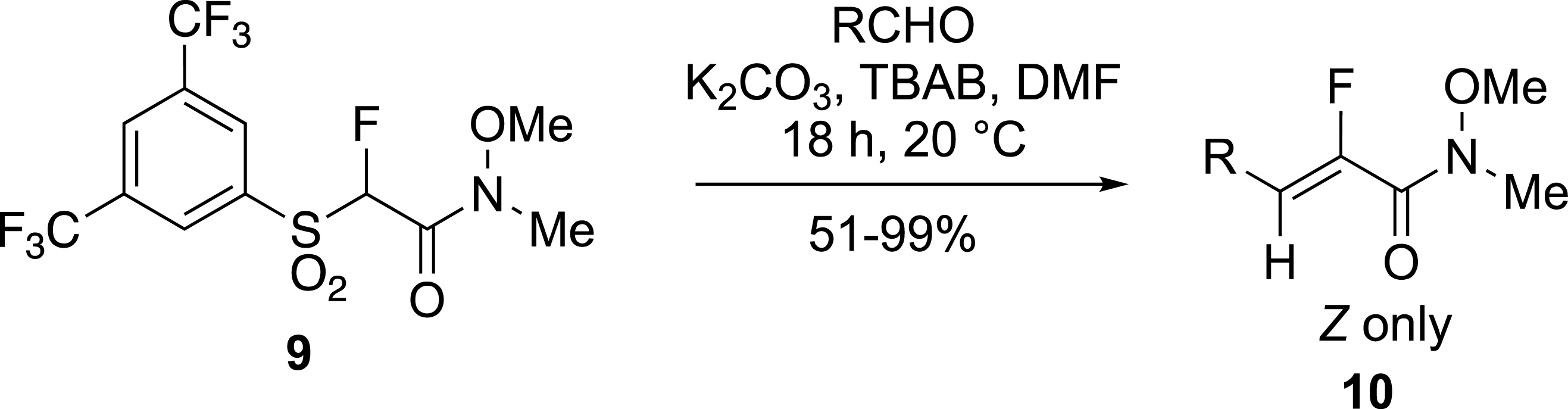
Selective formation of unsaturated Weinreb amides.
3. Fluoroalkylidenes substituted by an aromatic ring
The most important advance in the field of the preparation of fluoroalkenes has been the use of the modified Julia reaction to prepare fluoroalkylidenes that cannot be obtained directly from carbonyl compounds (Scheme 5). Indeed, our initial work on the modified Julia reaction opened up an easy access to tri- and tetra-substituted fluoroalkenes bearing an aryl ring, alkyl chain or an alkylamine. The main difficulties encountered were in the preparation of the fluorosulfones involved in this reaction. As mentioned above, the Z/E selectivity is strongly dependent on the nature of the carbonyl compound, the sulfone and the experimental conditions. In general, bulky substituents on both reagents favoured the formation of the (Z)-alkenes.
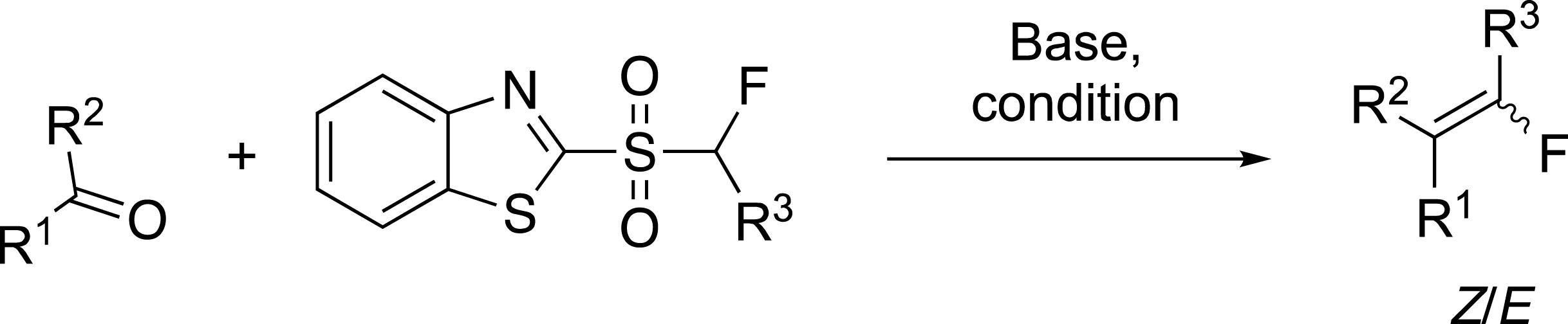
One step synthesis of fluoroalkylidenes.
The preparation of monofluoroalkenes substituted by an aryl was reported by the group of Zajc from monofluorosulfones 11 (Scheme 6) [28, 29]. (E)-alkenes 12 were the main product of the reaction, and the use of polar solvents (DMF:DMPU) could drive the reaction in favour of the Z isomers. This reaction can also be carried out from ketones.
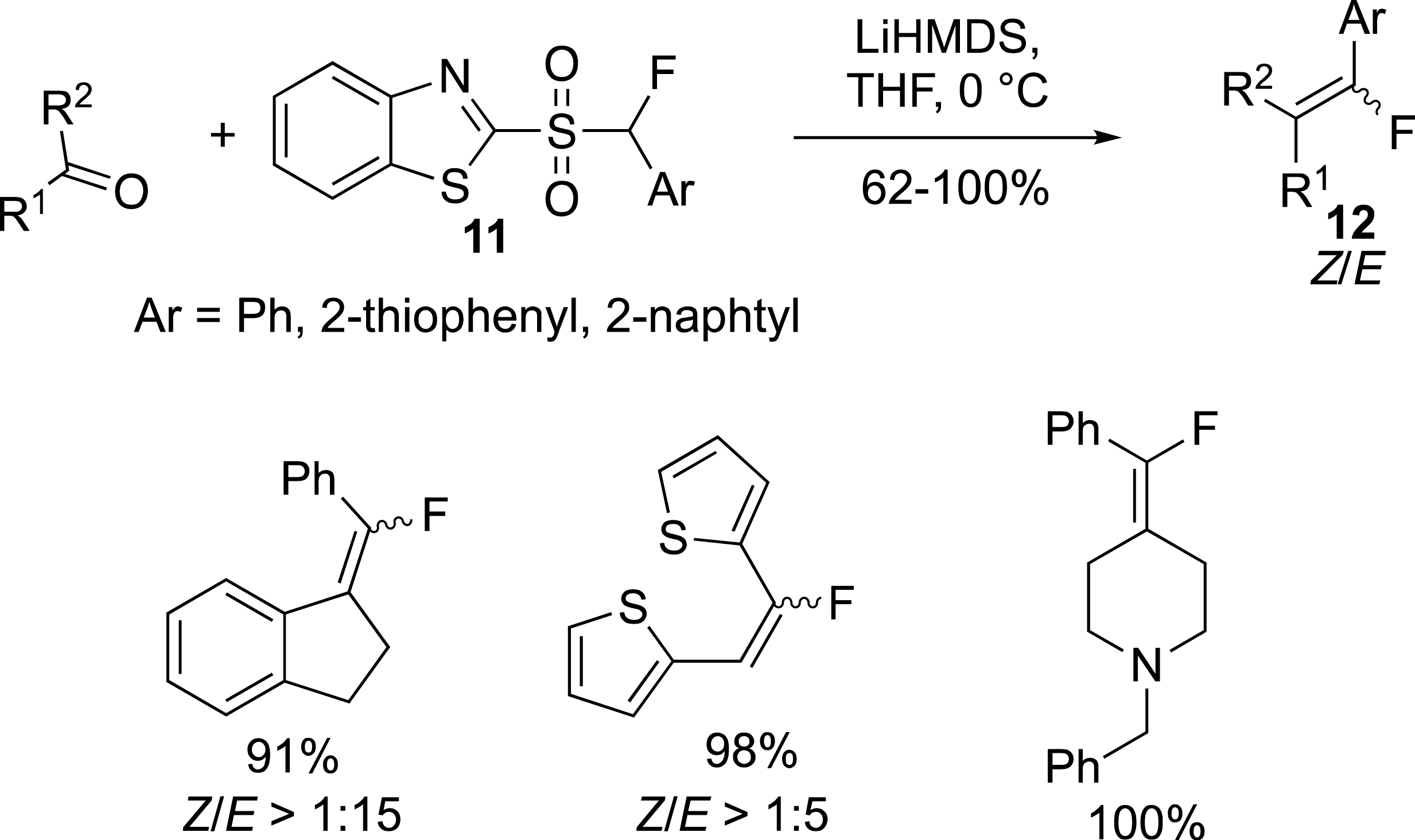
Fluoroalkenes substituted by an aromatic ring.
4. Fluoroalkenes substituted by an alkyl chain or a saturated ring
The synthesis of fluoroalkylidenes is the most challenging topic. As mentioned above, the fluoroolefination reactions of carbonyl compounds were limited and the modified Julia reaction opened a new way to access a wide range of fluoroalkenes.
Our first example on the pyrethrin analogue was the cornerstone of the fluoroolefination of carbonyl compounds based on the modified Julia reaction [14]. For example, the expected alkenes 14 and 15 were obtained in high yield but with a moderate Z/E selectivity (Scheme 7). This olefination reaction was applied to the synthesis of exo-glycals which were obtained in moderate to good yields and with moderate selectivity. Importantly, the reaction carried out with lactones required a stepwise process. In this case, the alcoholate intermediate had to be captured before the Smiles rearrangement could be carried out at room temperature [30, 31]. The modest selectivity observed in these examples was related to the small size of the methyl group even for the preparation of 16. This reaction has been applied to the preparation of glucosidase inhibitors.
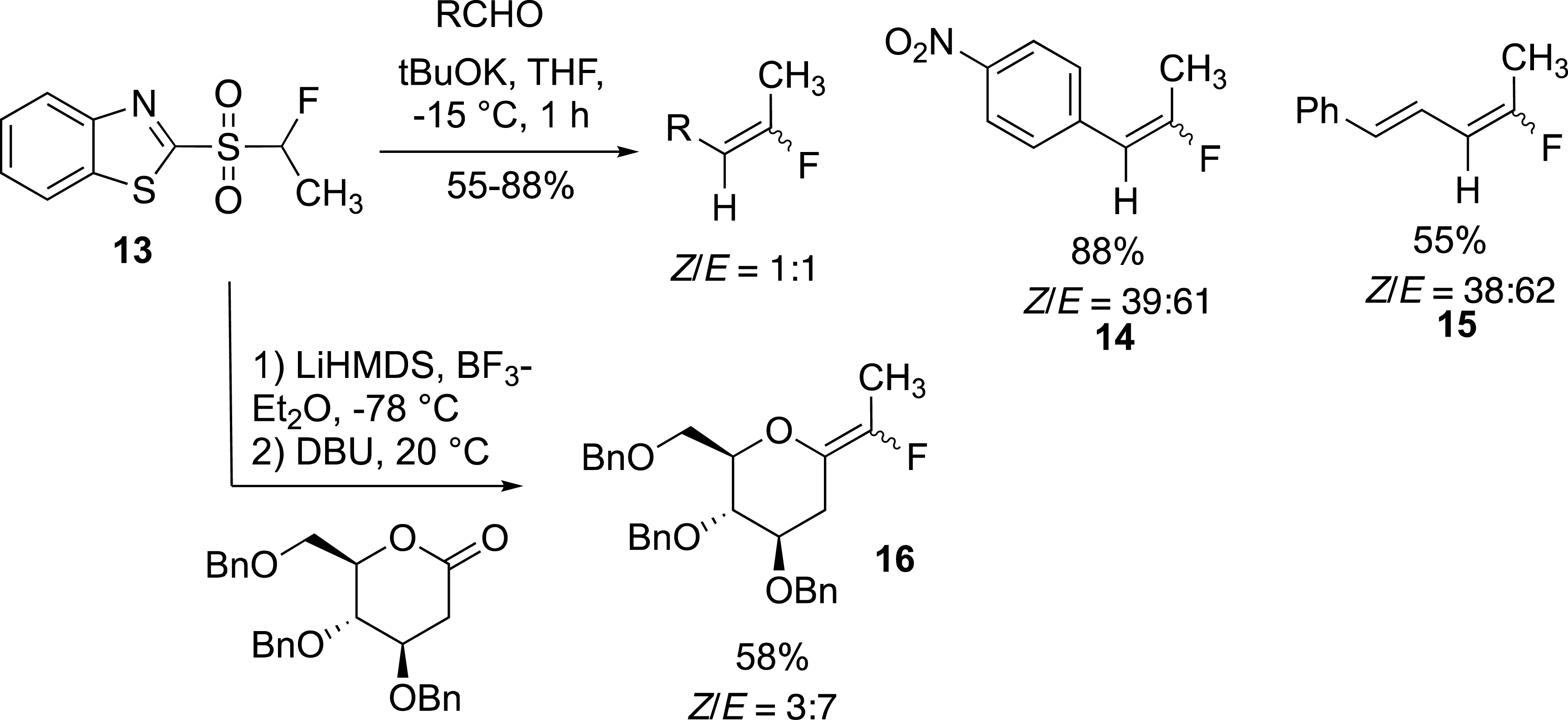
Preparation of fluoroethylidene derivatives.
To partially address the stereoselectivity issue, phenyltetrazolyl sulfones 17 were tested instead of benzothiazolyl sulfones (Scheme 8) [32]. A rational analysis of the observed results was not easy to perform as the Z/E ratio was strongly dependent on the carbonyl compounds and the nature of the alkyl chain.
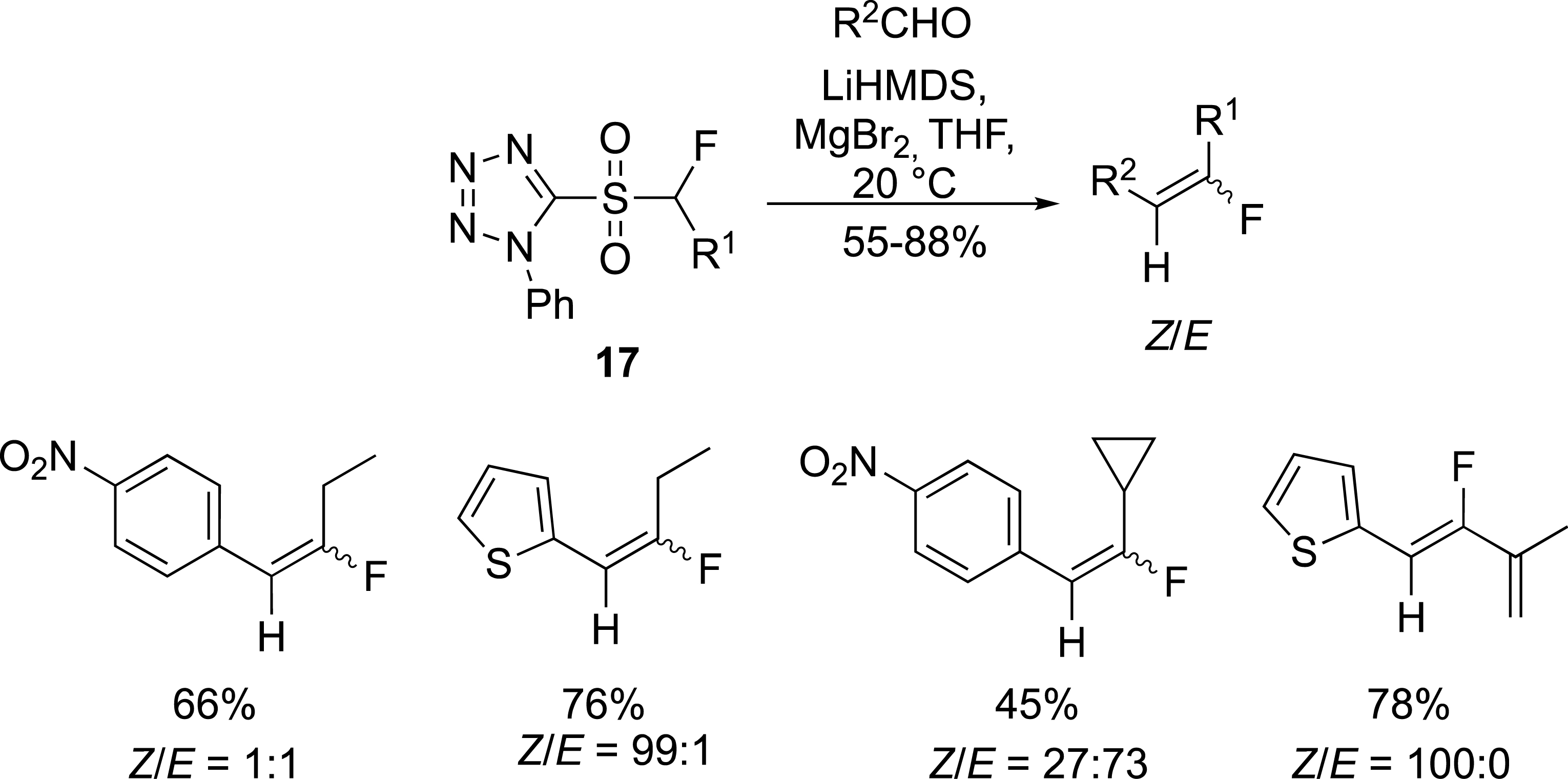
Preparation of fluoroalkylidenes with tetrazolyl sulfones.
We have also reported the preparation of enantiomerically pure allylic α-branched fluoroalkenes from chiral fluorosulfones in the benzothiazolyl series (Scheme 9) [33]. The chiral sulfones 18 were prepared by a Mitsunobu reaction followed by a Krapcho decarbethoxylation reaction [34]. These sulfones were involved in the modified Julia reaction which, despite a moderate control of the carbon–carbon double bond geometry, provided an easy route to chiral fluoroalkenes 19.
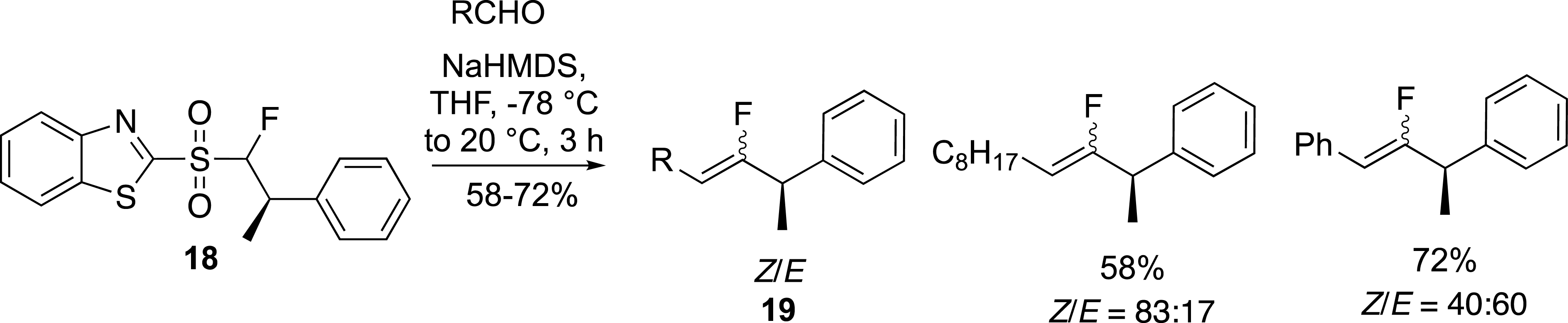
Preparation of enantiopure allylic α-branched fluoroalkenes.
Notably, the group of J. Hu reported a rapid synthesis of Z- and E-fluoroalkenes by phase separation with pyridine sulfone 20 (Scheme 10) [35]. In this case, the anti-periplanar elimination is slow and depends on the relative configuration of the sulfinate intermediates. It has been suggested that the anti-lithium sulfinyl arylether, which gives gives the (Z)-alkene 21, decomposes more rapidly than the syn-lithium sulfinyl arylether 22, which gives the (E)-alkene 23. Because of the aqueous solubility of the intermediate sulfinates, the kinetic control of the reaction allowed the (Z)-alkene to be extracted first with an organic solvent. The aqueous layer containing the syn-lithium sulfinyl arylether was then treated with an acid (TsOH) to give after extraction the corresponding (E)-alkene.
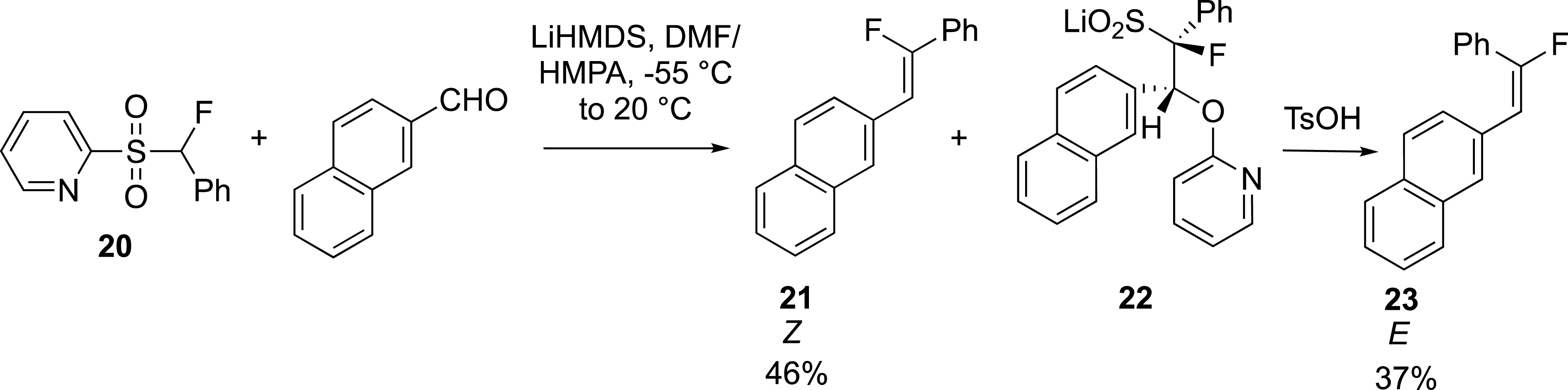
Kinetic resolution for the synthesis of fluoroalkenes.
While the modified Julia reaction was efficient for the synthesis of fluoroalkenes, the preparation of functionalized fluoroalkenes was limited by the access to starting alkylfluorosulfones. In this area, we were focused on the challenging synthesis of nitrogen-containing fluoroalkenes. The preparation of aminosulfones was achieved from a fluorovinyl sulfone, and involved the olefination reaction. The aza-Michael addition of amines and amino acids to vinylsulfones 24 afforded the corresponding sulfones 25 as new fluoro-olefination reagents (Scheme 11) [36], whose reactivity in the modified Julia reaction was studied with carbonyl compounds and allowed us to prepare dipeptide isosteres 26 [37, 38], and functionalized exo-glycal derivatives. Regarding the access to 26, the N-terminal amino group was introduced by a conjugate addition of phtalimide onto fluorinated vinylsulfones containing an α-amino-acid side chain. The C-terminal motif was attached to the fluorovinylic peptide bond mimic via the Julia–Kocienski reaction between fluorosulfones and substituted aldehydes with α-amino-acid side chains (Scheme 11). In all cases, the diastereoselectivity was in favour of the Z isomers due to the steric demand of the bulky substituents.
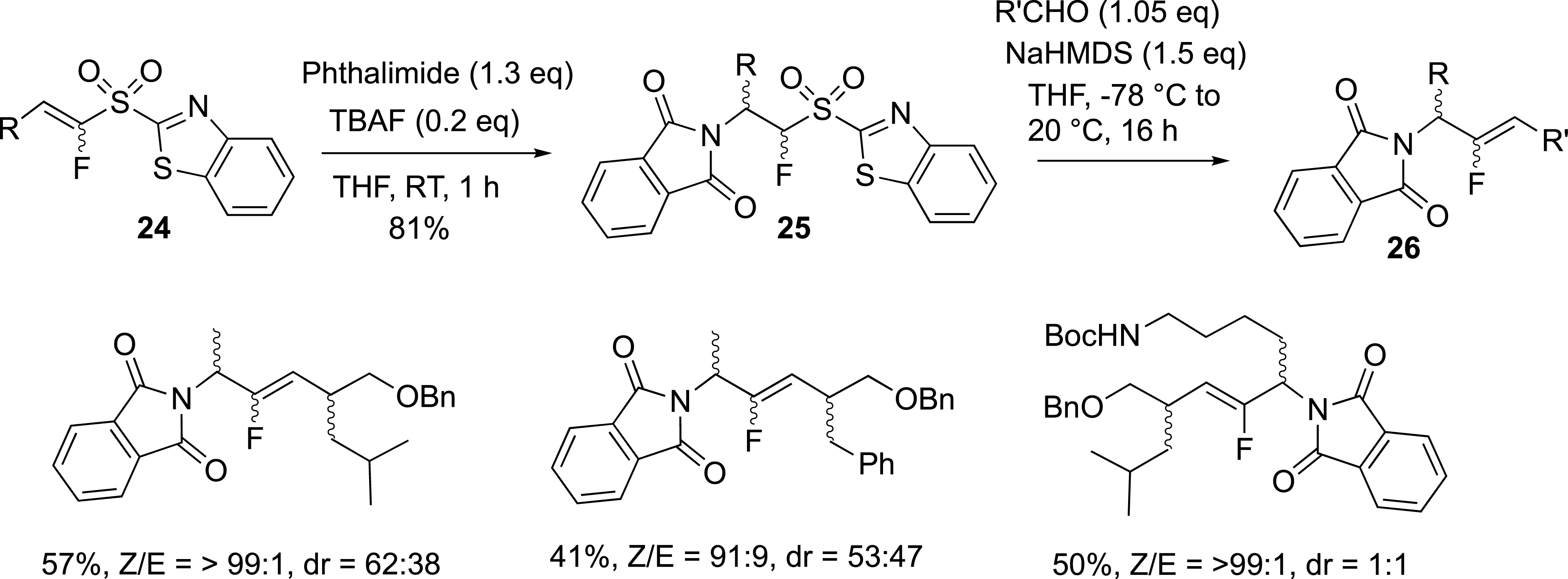
Preparation of dipeptide isosteres.
Finally, the incorporation of monofluoroalkene-based dipeptide isosteres into peptide synthesis was tested (Scheme 12). A coupling reaction in solution between racemic Boc-Gly-ψ[CF=CH]-Leu 27 and (L)-phenylalanine ethyl ester was performed, yielding the corresponding tripeptide isostere Boc-Gly-ψ[CF=CH]-Leu-Phe-OEt 28.
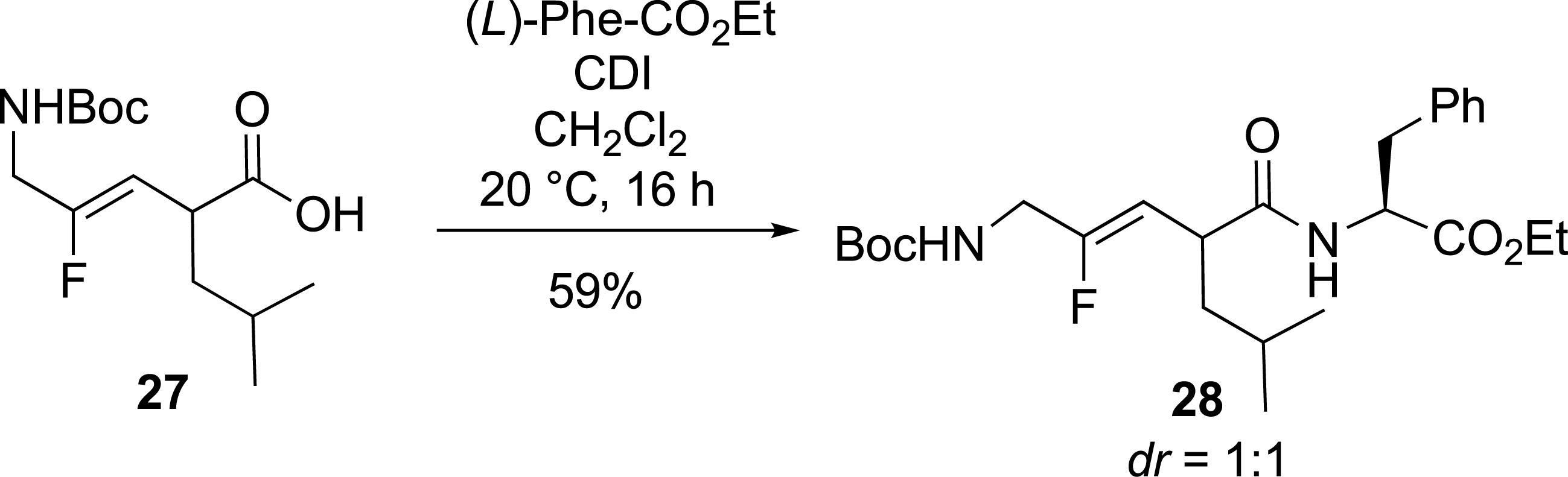
Fluorinated tripeptide analogue.
Following a similar approach, pyrrolidine fluorosulfone 29 was also prepared and used in the olefination of carbonyl compounds to give alkenes 30 in moderate to good yields with low E/Z selectivity (Scheme 13) [39].
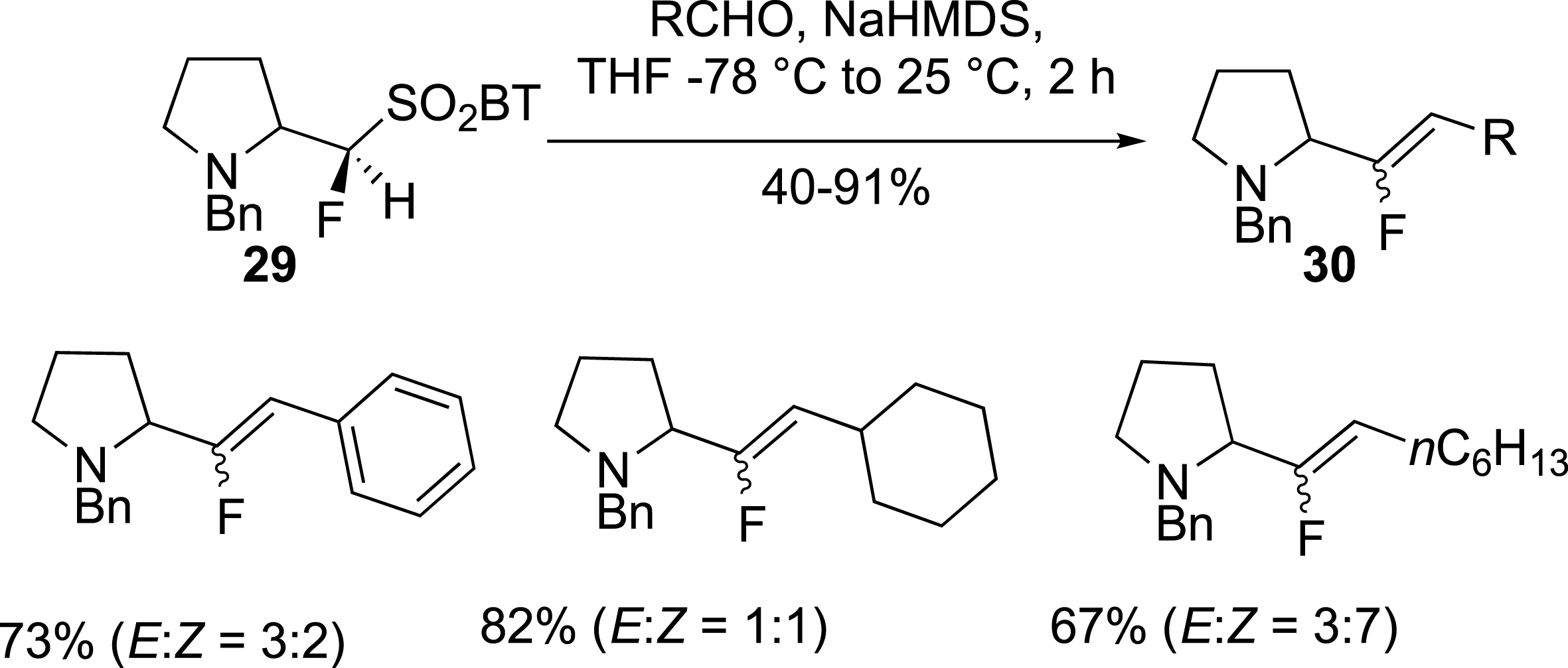
Pyrrolidine as potential proline isosteres.
Finally, this aza-Michael addition onto vinyl sulfones provided access to nucleic base-containing benzothiazolyl sulfones. For example, the thymine-derived sulfone 31 was used in the modified Julia reaction to prepare the acyclonucleoside precursor 33 (Scheme 14) [40].
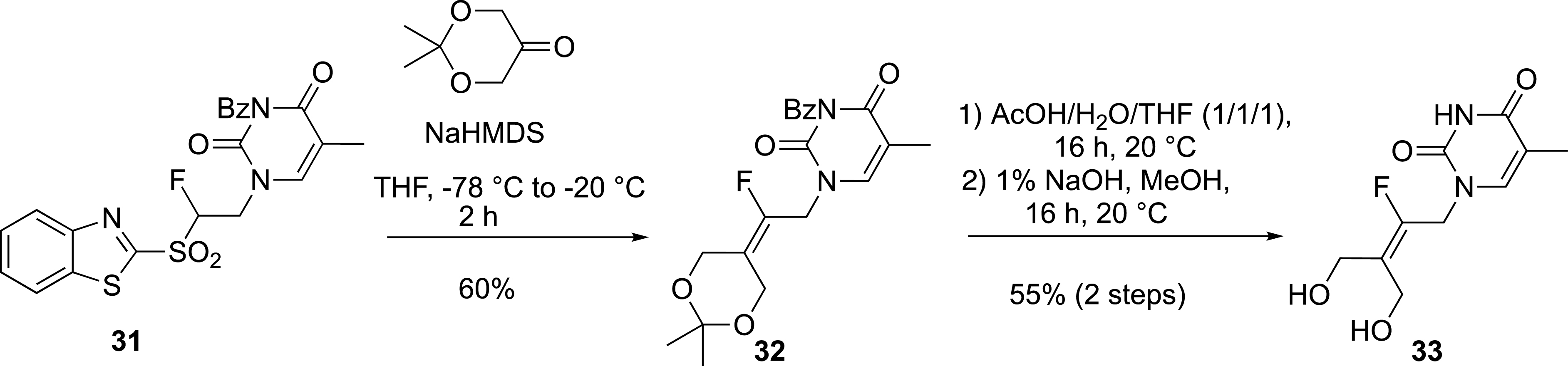
Preparation of an acyclonucleoside.
5. Access trisubstituted fluoroalkenes for the preparation of acyclonucleosides
For the synthesis of trisubstituted fluoroalkenes, we explored another alternative route using alkylidene oxetanes 35 as key intermediates (Figure 2) [41]. The selective ring–opening reaction of fluoroalkylidene oxetanes was controlled by the presence of the fluorine atom, providing a two-step access to tetrasubstituted alkenes 34 with excellent geometry control (Figure 2) [42].
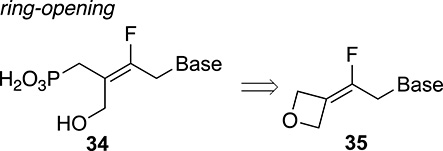
Ring–opening of oxetane derivatives.
The preparation of a series of fluoroalkylidene oxetanes was realized from 3-oxetanone through an olefination reaction with benzothiazoyl sulfones (Scheme 15) [41]. With these fluoroalkylidene oxetanes 36a–f in hand, the selectivity of the ring–opening reaction with bromide ion to access to tetrasubstituted fluoroalkenes was investigated.
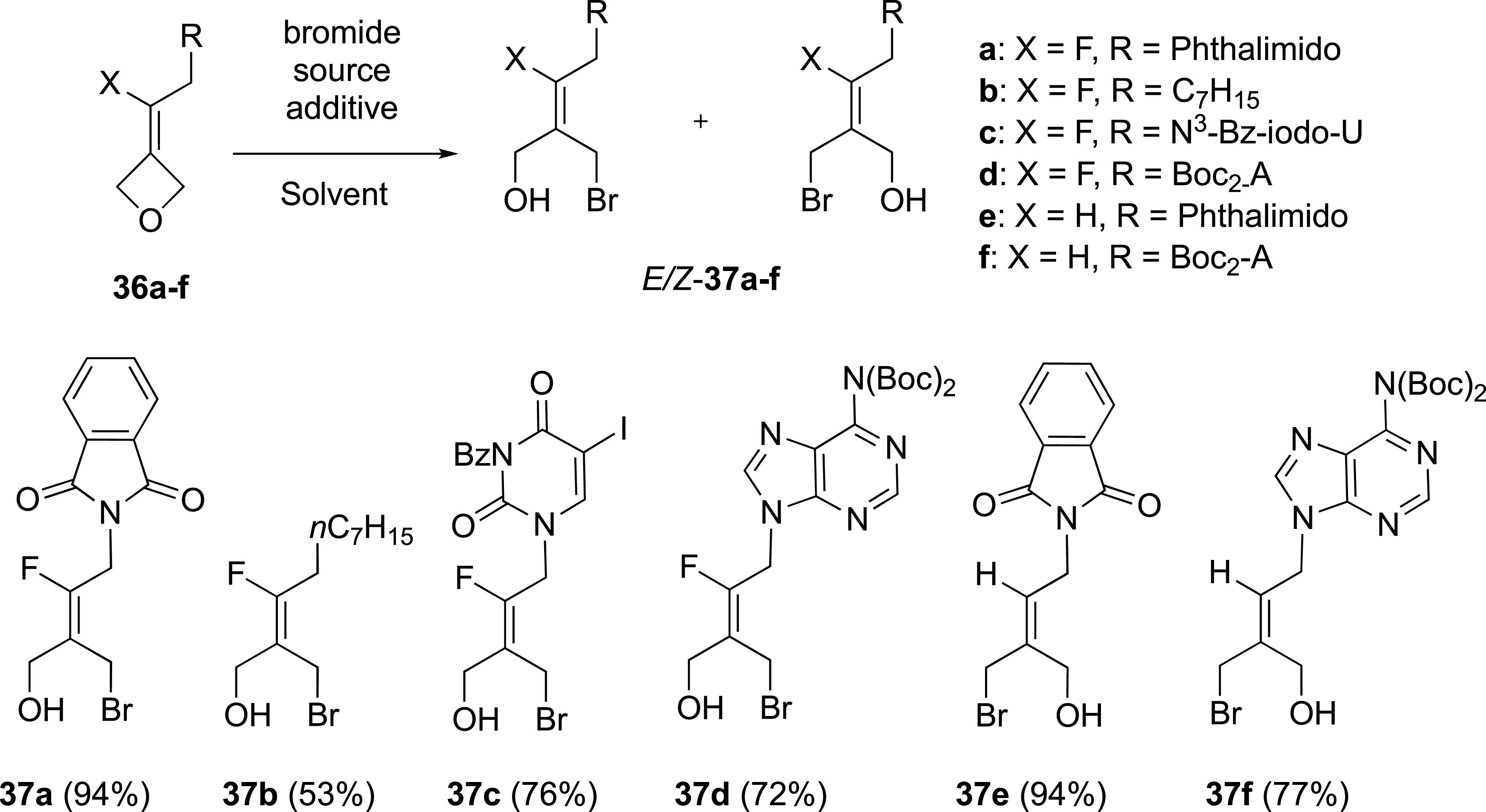
Ring-opening reaction of alkylidene oxetanes.
The ring-opening reaction of oxetane 36a, substituted by a phthalimido group, carried out with bromide ion sources (HBr 33 wt% in AcOH or Bu4NBr in the presence of BF3-Et2O) afforded the corresponding (E)-alkene 37a in an E/Z ratio of 89:11 (Scheme 15). This reaction was extended to other fluoroalkylidene oxetanes 36b–d substituted with bulky substituents (an alkyl chain or a nucleic base) and different sources of bromide ions [42, 43]. The ring-opening of these oxetanes to (E)-alkenes was also highly selective, thus confirming our initial observation. The reaction tested with oxetanes 36b–d selectively gave the corresponding E-fluoroalkenes 37b–d, in up to 9:1 E/Z ratios. On the other hand, when the reaction with oxetanes 36e,f was carried out in the absence of a fluorine atom, only (E)-alkenes 37e,f, were formed. In this case, the attack of the bromide ion occurred on the opposite side of the bulky phthalimido or protected adenine substituents.
The functionalization of alkene 37a allowed an easy access to a large series of tetrasubstituted fluoroalkenes. Indeed, the oxetane ring-opening reaction was restricted to the bromide ion (Scheme 16).
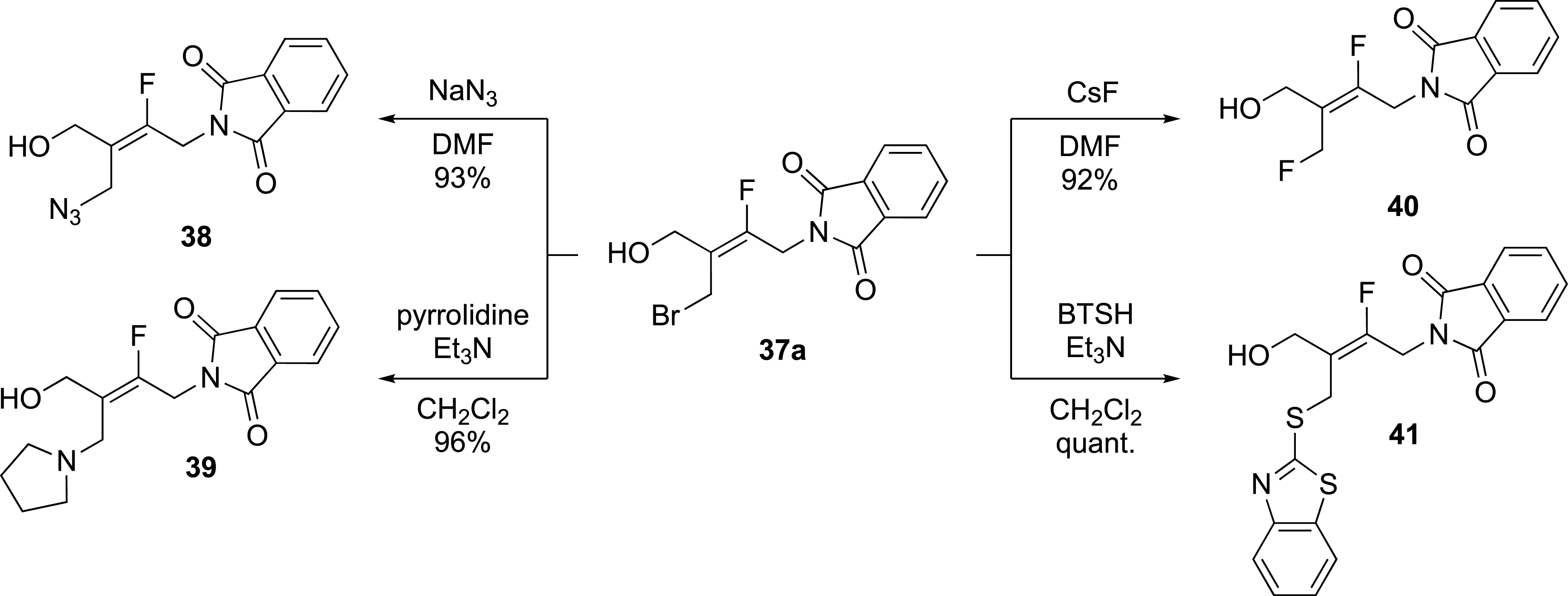
Functionalization by substitution reaction of the bromide.
Functionalization of alkene 37a by displacement of the bromine atom, with nucleophiles such as CsF and NaN3 afforded products 40 and 38 in 92% and 93% yields, respectively. Reactions with amines and thiols such as pyrrolidine and 2-mercaptobenzothiazole, afforded products 39 and 41 in high yield.
Finally, this rapid synthesis of tetrasubstituted alkenes by sequential ring-opening and nucleophilic substitution reactions was intended to test the robustness of a selective preparation of acyclonucleoside precursors. In connection with our work on the preparation of inhibitors of a protein involved in antibioresistance [44, 45, 46], we focused on the synthesis of acyclic analogues of functionalized adenosine derivatives as enzyme inhibitors. Our aim was to determine the influence of the replacement of the ribosyl ring by a fluoroalkenyl chain on the biological activity of the resulting nucleoside analogue (Scheme 17). To access the final target compound 46, the preparation of fluorinated azide 44 was realized from the tetrasubstituted (E)-alkene 42. The substitution of the bromine atom was carried out with KOAc to give the intermediate acetylated alcohol 43, which was mesylated and then treated with NaN3 to produce azide 44 in 74% overall yield. Finally, the expected fluorinated triazolyl derivative 45 was obtained in 79% yield from 44 and N-Boc 4-amino-pentynone. Compound 45 was treated by an acidic methanolic solution to afford 46 in 76% yield (Scheme 17).
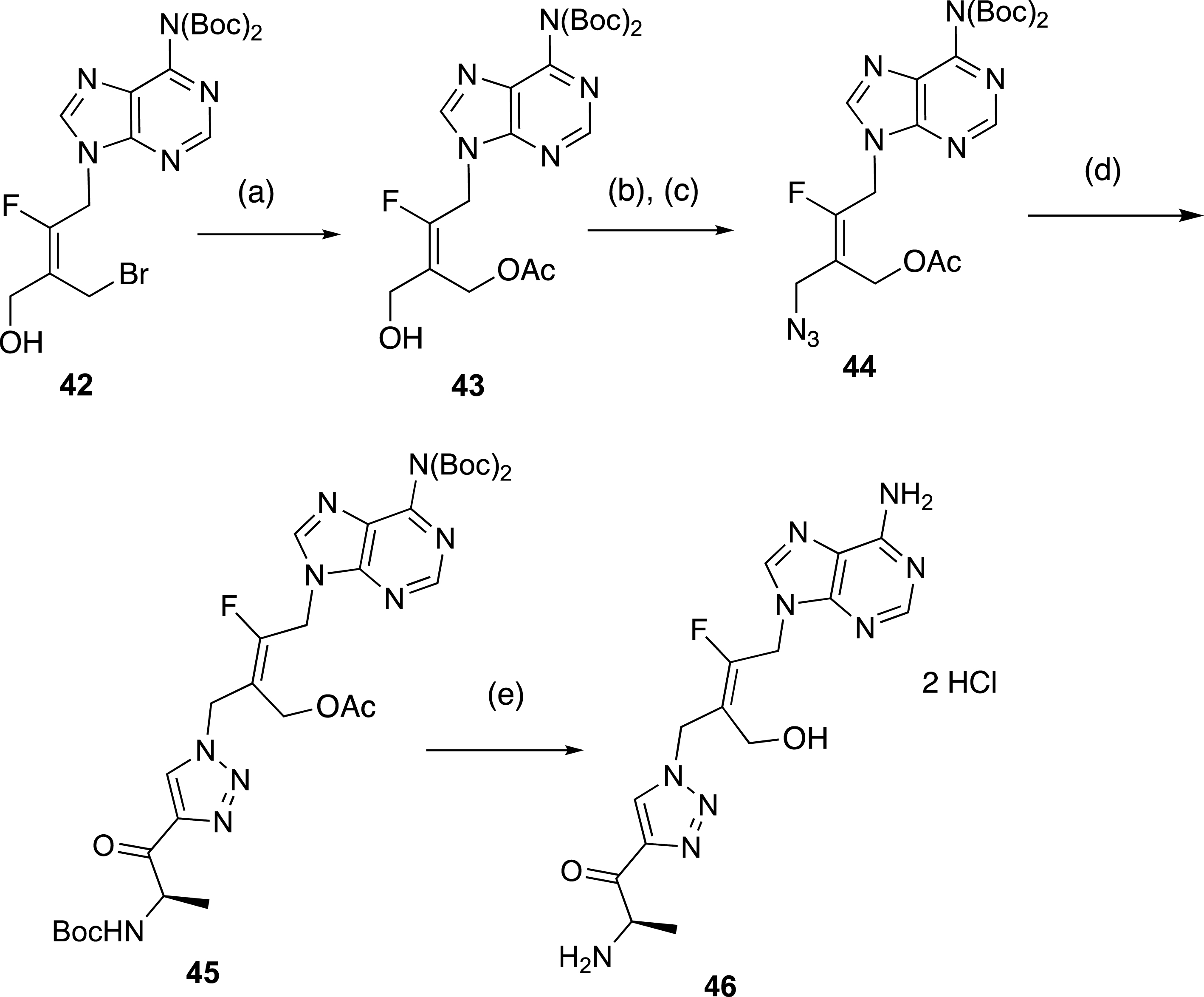
Preparation of triazolyl derivatives in F-series. (a) KOAc, DMF, 20 °C, 16 h, 86%. (b) MsCl, Et3N, CH2Cl2, 0 °C, 2 h, (c) NaN3, DMF, 20 °C, 12 h, 74% (2 steps). (d) N-Boc-amino-pentynone, CuSO4 ⋅ 5H2O, sodium ascorbate, t-BuOH, H2O, 40 °C, 24 h, 79%. (d) HClaq (3N), MeOH, 20 °C, 20 h, 76%.
The inhibitory activity of 46 against the DltA enzyme (Enterroccus faecalis) was investigated and compared with that of nucleoside 47 (IC50 = 2.5 μM) and triazolyl derivative 48 (IC50 = 3.4 μM) (Figure 3) [46]. It appeared that the introduction of a triazolyl ring did interfere with the activity, and the replacement of the ribosyl ring by a fluoro trans-butenyl moiety was also well tolerated. Compound 46 exhibited an IC50 value of 7.4 μM, slightly higher than that observed with adenosine derivative 48. This first example of the use of a fluoro trans-butenyl moiety shows that this moiety can be considered as a surrogate for the nucleoside ribosyl moiety. In the present case, the presence of a carbon–carbon double bond is important as reported for trans-butenyl substrates targeting thymidylate kinase [47, 48], In contrast, the acyclic analogue 49 containing an alkyl ether chain (2-oxa-butyl) instead of a (fluoro)alkenyl chain was the less efficient inhibitor of the DltA enzyme with an inhibition constant of 20.1 μM. The loss of activity was probably related to the high flexibility of the alkyl chain, which allows conformational changes. The (E)-fluorobutenyl moiety appeared to be the best nucleoside mimic in the butenyl series.
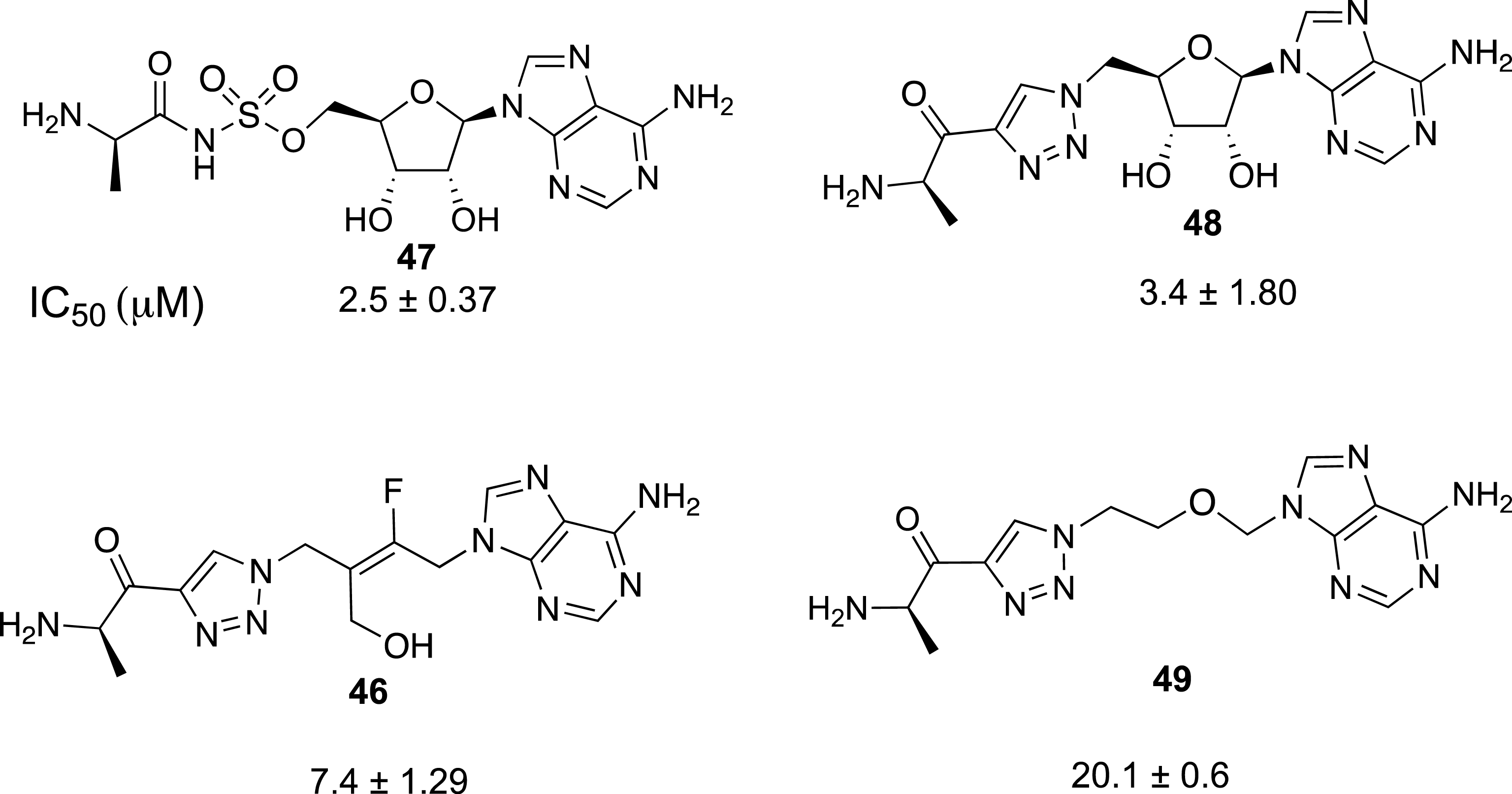
Measured IC50 values towards DltA of E. faecalis.
6. Conclusion
The modified Julia reaction has contributed to the development of new approaches for the preparation of highly functionalized fluoroalkenes including fluoroalkylidenes. The discovery of this reaction and its application to the synthesis of fluorinated biomolecules has provided access to fluoroalkene motifs as mimics of dipeptides and acyclonucleosides.
Declaration of interests
The authors do not work for advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was supported by the excellence laboratory LabEx SYNORG (ANR-11-LABX-0029), the Conseil Régional de Normandie and the European FEDER funding.
Vous devez vous connecter pour continuer.
S'authentifier