



 CC-BY 4.0
CC-BY 4.0
1. Introduction
In the past decade, advances in protein engineering, genomic database mining, and computational methods associated with high-throughput screening tools and DNA library synthesis have enabled a step change in biocatalysis. As a consequence, the nature of biocatalytic transformations can be nearly infinitely tunable and optimizable with billions of combinations of alterations to tailor specific reactivity and selectivity [1]. Due to modern directed evolution techniques [2, 3], advanced artificial intelligence (AI) [4], machine learning (ML) techniques [5, 6], the introduction of non-natural amino acids and new reactivity [1], the potential of biocatalysis to transform modern drug manufacturing has been expanded. The field has moved beyond traditional like-for-like enzyme replacements to wholly new routes designed around system biocatalysis [7, 8]. Sanofi, among others, has identified biotransformations as one of the most transformative technologies to date in the synthesis of today’s complex new drug candidates. In this article, we describe how Sanofi is accelerating the implementation of biocatalysis in its pharmaceutical research and development portfolio. For details, original publications and patents are referenced.
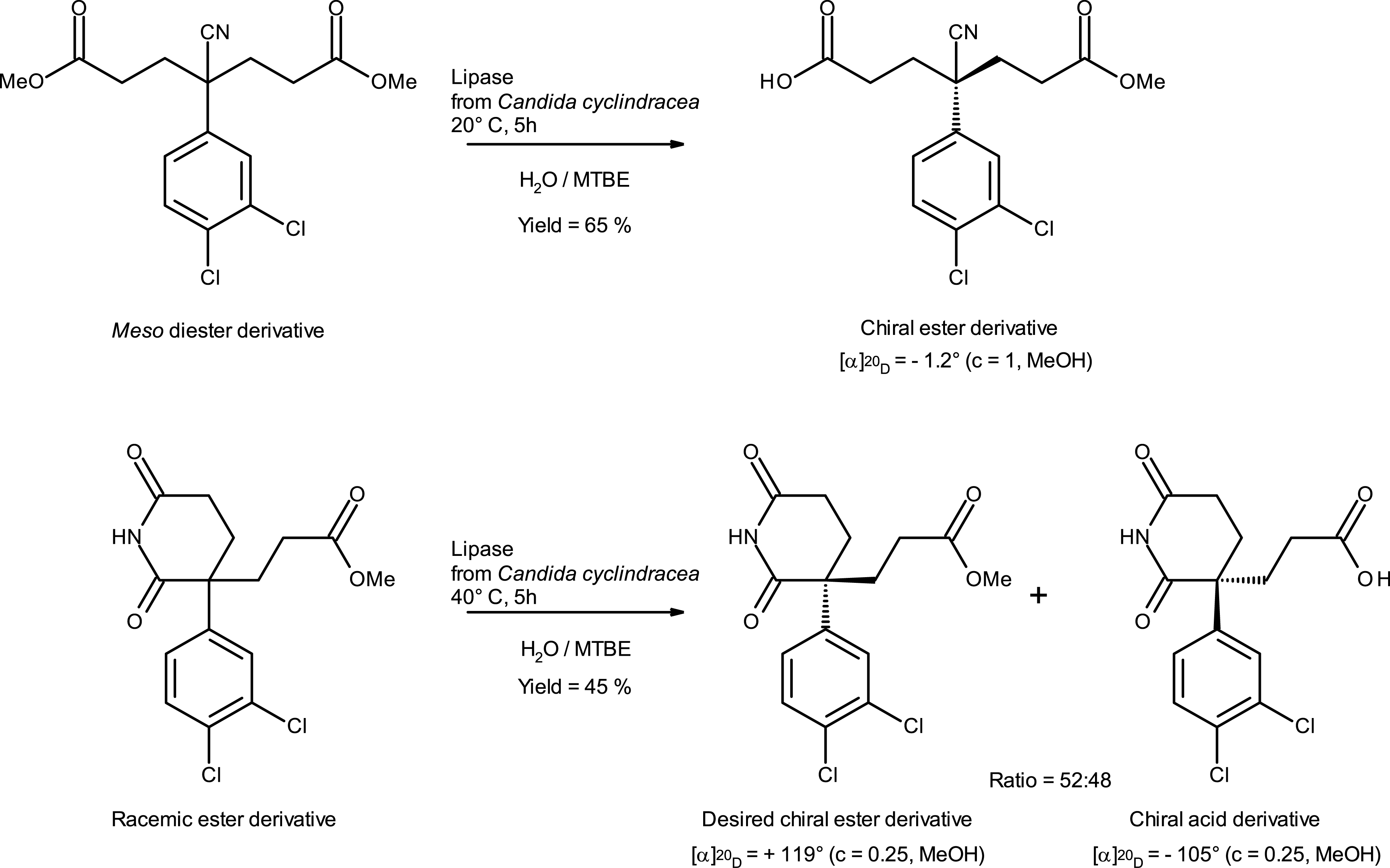
Examples of biocatalytic transformation at Sanofi [9].
2. History of Sanofi use and production of enzymes
Sanofi has extensive experience in the synthesis of molecules of interest, including small molecules and recombinant enzymes, through fermentation processes.
Sanofi also used enzyme technologies to produce small molecules at various stages of their development. Discovery capabilities were developed to produce drug metabolites for quantification and profiling of in-vivo generated compounds. The CYP450 enzymes of microorganisms are well described for their metabolizing ability, as “the microbial model for drug metabolism”. This type of technology was developed internally, and the use of such enzymes has been proven to be an efficient way to produce either Phase I or Phase II metabolites [10] in their labeled [11, 12] or non-labeled form. Another class of enzyme used for the synthesis of Carbon-14-radiolabeled metabolite is nitrilase. We showed that radiolabeled nitrile derivatives can be hydrolyzed to get the corresponding radiolabeled acid, using mild enzymatic conditions [13, 14]. In addition to the use of biocatalysis for small-scale pharmaceutical metabolite synthesis, implementation of enzyme technologies for the scalable synthesis of pharmaceutical advanced intermediates was also developed. Castro et al. [9] identified commercial lipase for the enantioselective hydrolysis of racemic carboxylic ester and for the desymmetrization of a symmetrical diester to achieve a chiral monoacid ester product (Scheme 1).

Sanofi charter on Planet Care environmental sustainability program.
By taking advantage of internal history in using enzymes for synthesizing optically molecules, and in fermentation processes, Sanofi decided in early 2020, to build a fully integrated end-to-end biocatalysis platform within the Process Chemistry department belonging to the R&D organization. This platform aims to provide chemists with efficient tools for improving the environmental impact of API synthesis and addressing the challenges associated with the growing complexity of molecules entering development.
3. Biosynthesis technology to address new challenges from the R&D portfolio
Sanofi has a strong ambition to build a sustainable environment with a global approach to minimizing impacts. Three main axes have been defined and are described in Figure 1. A key pillar toward achieving these environmental commitments revolves around reducing the environmental footprint of manufacturing processes through an ecodesign approach. This includes reduced water and solvent use, eliminating toxic metals, reagents and solvents, and lowering energy requirements. To meet our long-term environmental sustainability commitment, technological innovations such as biocatalysis, are required.
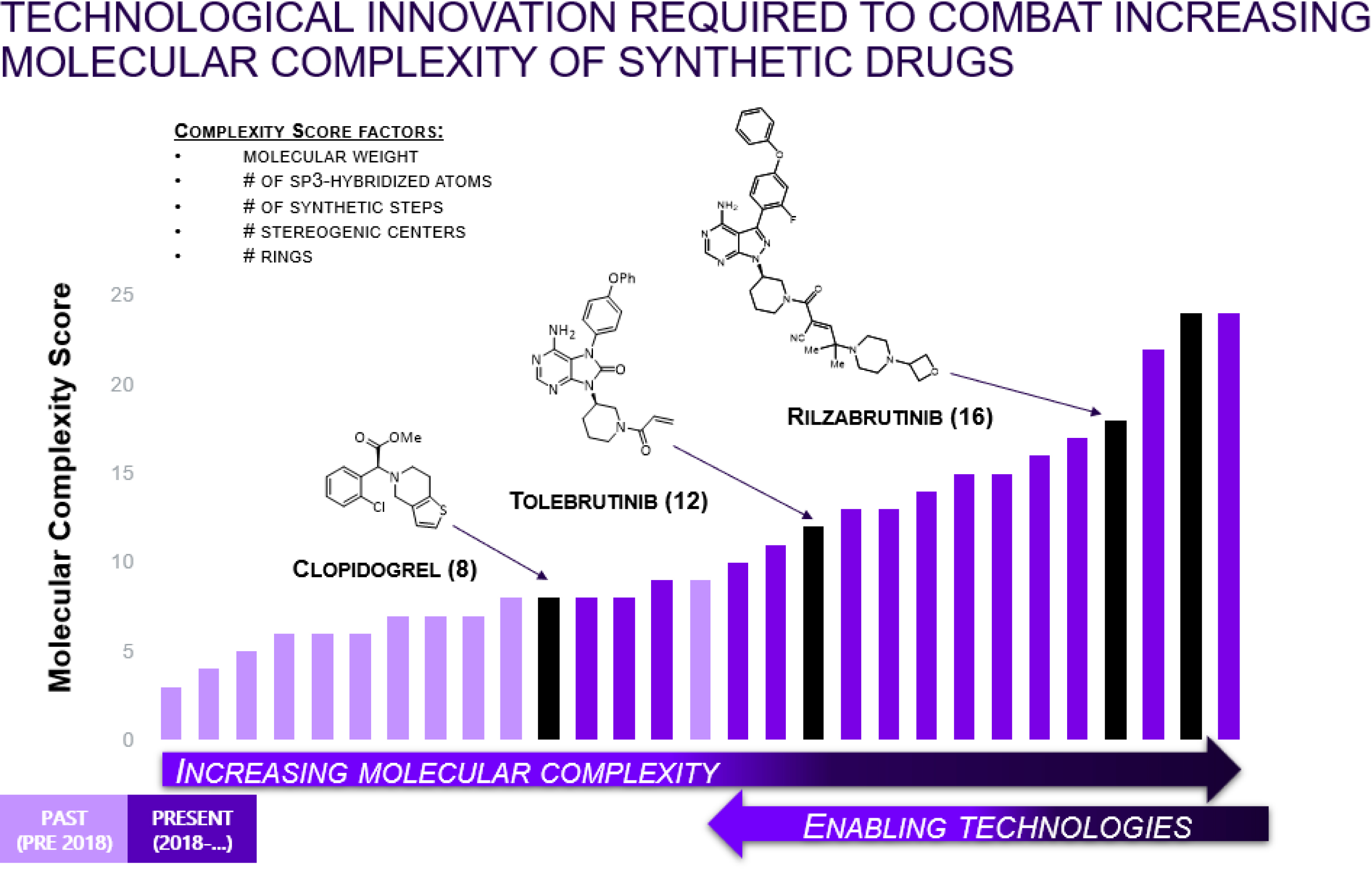
Evolution of the structural complexity in Sanofi’s small molecule portfolio.
3.1. Molecular complexity of next generation small molecule APIs
A general trend at Sanofi, and elsewhere, is that new molecular entities entering development are increasing in complexity (Figure 2) [15]. The trend is moving away from traditional activators/inhibitors toward a next-generation approach of stabilizing or disrupting complex protein–protein interactions (next-generation small molecules) to obtain the desired effects. Additionally, the introduction of bispecific small molecule degraders has also increased the overall complexity and size of the synthetic targets. With increased complexity comes chemistry manufacturing and control (CMC) risks to supply, resources, time to clinic and/or cost manufacture. The utilization of biotransformation is a key lever to help us manage the increase in synthetic complexity.
4. How we proceed to accelerate the implementation of biocatalysis in the Sanofi Pharmaceutical R&D Portfolio
Sanofi’s strategy is based on three pillars.
4.1. Development of a route-scouting mindset for an innovative route design approach
We consider that organic chemists and biochemists need to work together to design routes integrating a biocatalytic retrosynthesis approach, supported by a strong network of external experts and partners (Figure 3). The biosynthetic technologies team is integrated into the global Process Chemistry organization which favors scientific exchange through brainstorming sessions on innovative chemical route design. Furthermore, enzymatic transformations can be considered for the design of clinical and commercial routes thanks to new enzyme engineering techniques that can quickly deliver an effective biocatalyst in a short time.

Route design workflow.
4.2. Contribution all along value chain (Figure 4)
At the research stage, the main input from biocatalysis is either to produce metabolites or to generate diversity through late-stage functionalization (LSF) for accelerating compound discovery [16]. Currently, we are using biotransformation early in discovery as a diversity engine to aid candidate generation.
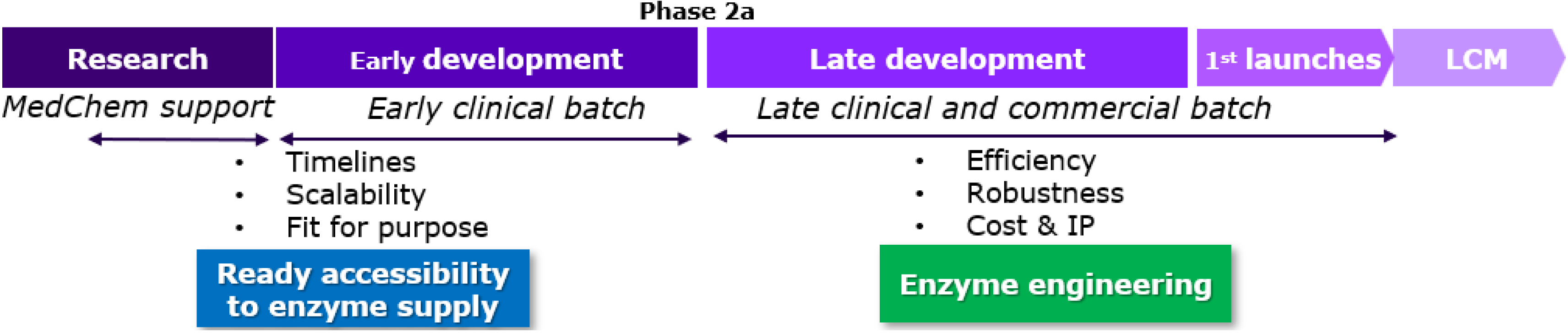
Implementation of biocatalysis all along the value chain.
Starting in preclinical development, a first generation of synthetic routes is used for delivering preclinical and clinical batches with a focus on speed to the clinic. A retrosynthetic strategy integrating biotransformation from the beginning has proven to be of great interest. Accelerating pressure on clinical timelines is squeezing development timelines and shifting our strategy toward fewer cycles of route selection and development. In some cases, this can mean keeping the same disconnection strategy throughout the product’s life cycle, but optimizing, concatenating and replacing less sustainable technologies with highly selective and efficient technologies. This could involve using an off-the-shelf enzyme, readily accessible at scale, that may not be optimal for the system but can sustain projects within a short timeframe (see example in Section 5). Another option is to utilize a non-enzymatic strategy that could be replaced by a more sustainable biocatalyzed sequence later in the development phase. An illustrative example from our R&D portfolio of such chemical transformation is the asymmetric hydrogenation catalyzed by a very expensive chiral ruthenium catalyst (Scheme 2) [17].
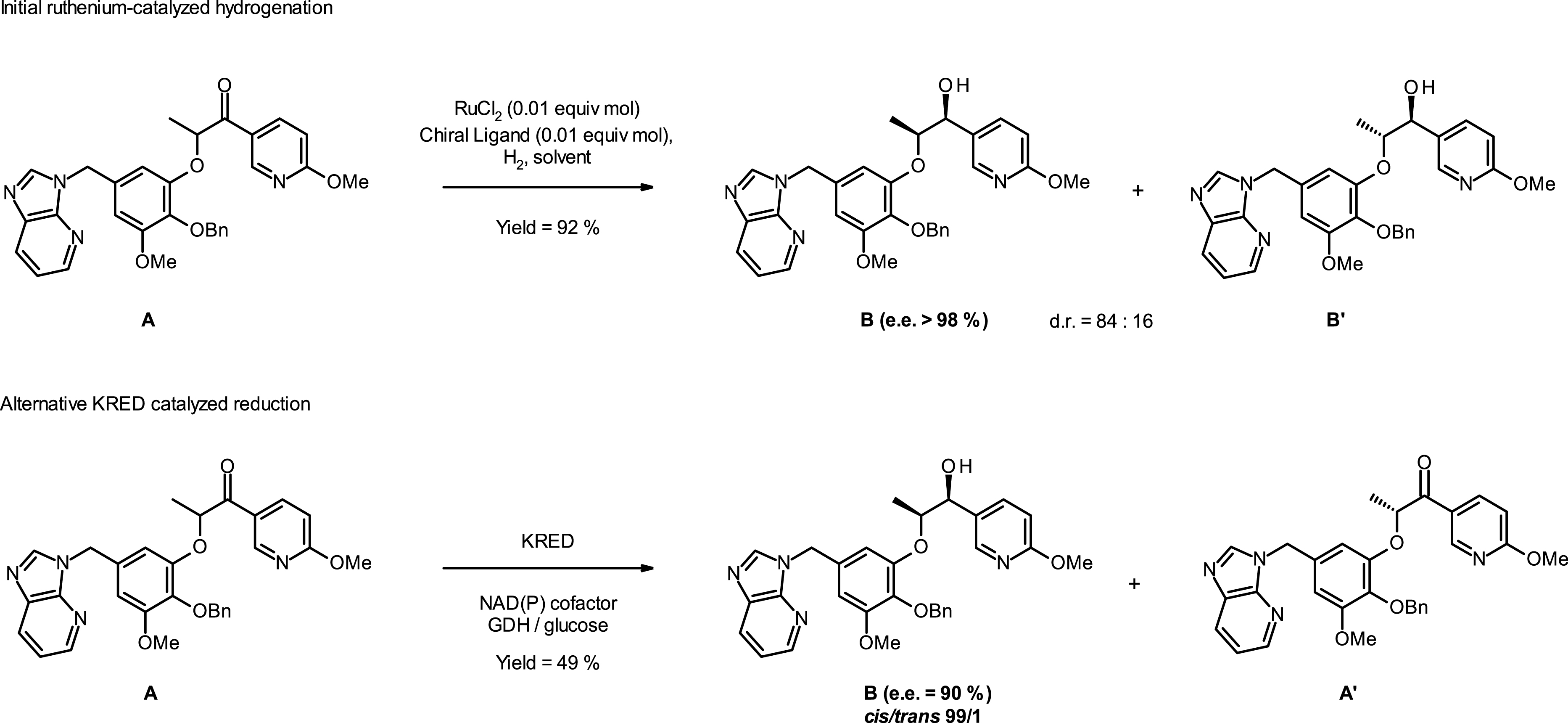
Initial ruthenium-catalyzed hydrogenation [17] and KRED-mediated biocatalytic reaction [18] affording chiral hydroxy intermediate B.
In addition to the costly precious ruthenium catalyst, the diastereoisomeric ratio obtained after the chemo selective reduction step was moderate (84:16), negatively impacting the process efficiency, manufacturing costs, and waste generation. To anticipate potential constraints regarding large-scale drug supply, we initiated a study on biocatalytic transformation using ketoreductase (KRED) enzymes [18].
One enzyme was found to be highly selective toward the desired cis alcohol product B (cis/trans = 99:1 and enantiomeric excess ∼90%). Our results demonstrate that the KRED enzyme is an alternative to the ruthenium catalyst for the reduction of the very bulky ketone substrate A to the chiral alcohol product B.
Once the project has moved into late clinical development and, drug supply has been taken out of the critical path, the time and resources needed to find the most ideal way to utilize biotransformation was investigated. At that stage, the chemical route for launch is selected and we can introduce the fully engineered enzyme as if we can replace one catalyst by another one. The objective at this point is to meet the prerequisites of an industrial process, including productivity, yield, turnover number, robustness, cost. Finally, the incorporation of the biocatalytic strategy early in development, when possible, allows us to focus our efforts on enzyme engineering and optimization instead of compressing enzyme development on top of additional route selection activities.
4.3. Building efficient capacity
A biosynthetic technologies team was created, designed for end-to-end biocatalyst development and deployment, able to take in charge the different studies related to biocatalysis involved at early and late development phases.
A global workflow has been established and related capacities and expertise have been developed internally with the support of key partners to rapidly implement biocatalytic process:
- Molecular biology platform including expertise in enzyme discovery and in the design of new enzyme through protein engineering approaches such as directed evolution or more rational engineering. This is supported by internal protein physical modeling. Regarding the large computational power needed for the enzyme searches and multimodal modifications we have implemented strategic partnerships in order to enhance our in-house capabilities.
- Reaction engineering capacities including medium- and high-throughput equipment and homothetic vessels for the development and the optimization studies of biocatalytic reaction.
- Enzyme production platform including various host fermentation and formulation techniques for biocatalysts production.
- Pilot capacities for the upscaling of biocatalytic processes and the production of enzymes at medium scale by fermentation.
- Analytical platform capable of developing analytical methods for reaction monitoring, conducting structural analyses for product and impurity identification, and characterizing biocatalytic enzyme.
5. Recent examples of enzyme use in the manufacture of APIs across early- and late-stage development
One of the first projects, using biocatalysts in manufacturing and studied in the newly created biosynthesis technology platform, was the new route toward a potent and selective LRRK2 inhibitor via chiral intermediate 1 [18, 19, 20, 21, 22]. One key intermediate of the route, for the early preclinical batches, was the chiral 3-hydroxy 4-methyl tetrahydrofuran 4, which could be prepared by an enantio- and diasteroselective reduction of ketone 5 (Scheme 3) [19, 23].
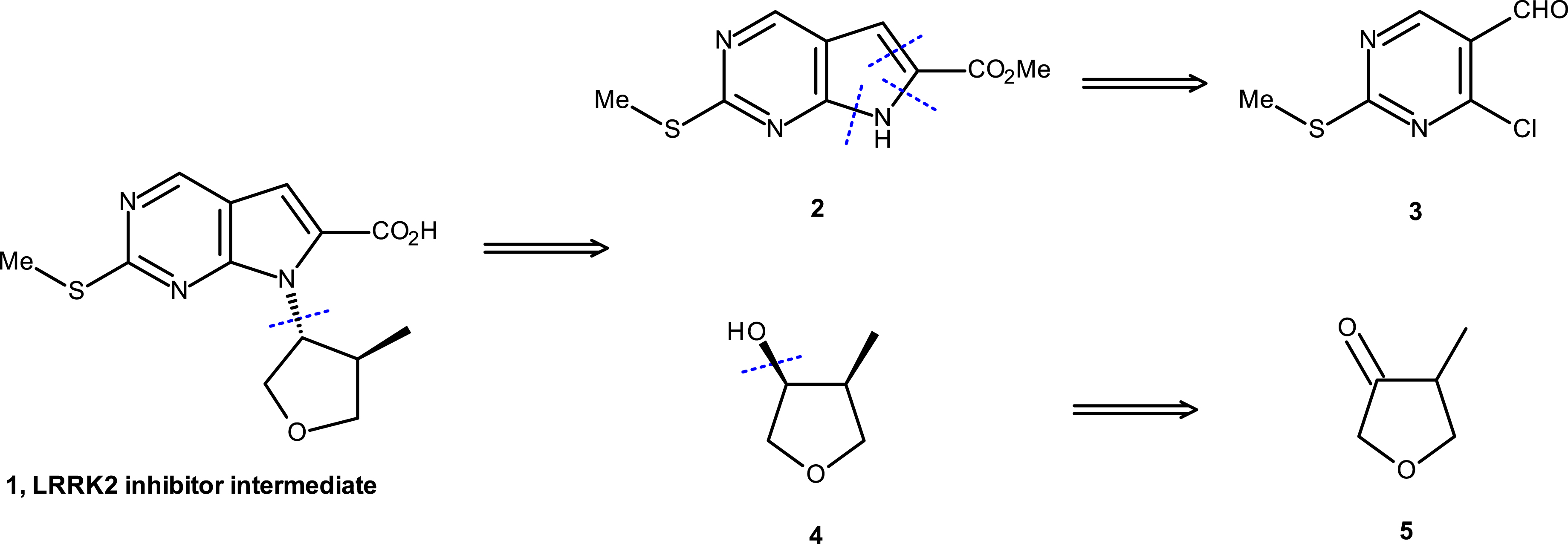
Disconnection of 1 toward simpler fragments.
Access to the challenging fragment 4 for early clinical deliveries was first achieved through the enabling route presented in Scheme 4.
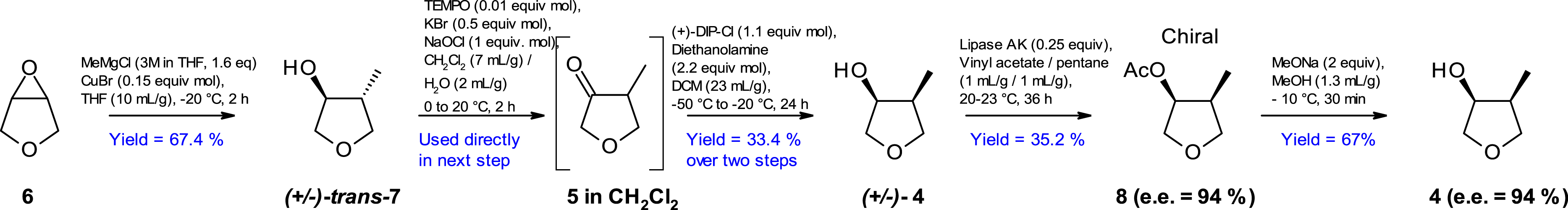
Enabling route to chiral tetrahydrofuran derivative 4.
In this first-generation route, it should be noted that to increase the enantiomeric excess of 4, a lipase-catalyzed kinetic resolution (esterification) was performed, using Lipase Amano AK [24] from Pseudomonas fluorescens (commercially available from Sigma-Aldrich) in the presence of vinyl acetate. This resulted in an isolated enantio-enriched (ee = 94%) O-acetylated intermediate 8 which was then deprotected to generate enantio-enriched secondary alcohol 4 (ee = 94%). However, this suboptimal enantiomeric excess necessitates performing a chiral SFC later in the synthesis, as requested by the project.
This synthetic route faced CMC risks related to the supply of 50 kg of intermediate 4, resources, time to clinic and cost of manufacture. Therefore, alternate chemistry was quickly required to advance the program.
Having solved the challenge associated with the preparation of 5 through a safe process [19], we then turned our attention to the challenging task of improving both the yield and the enantiomeric excess of compound 4, while diminishing the number of steps.
Starting from methyl ketone 5, we explored the synthesis of 4 through asymmetric reduction with KRED enzymes (Scheme 5) which are known to catalyze the reduction of sterically hindered ketones into the corresponding chiral alcohol [25, 26]. Due to short project timelines, we went for commercial or readily available enzymes for first deliveries while postponing the expensive and time-consuming enzyme evolution studies until later. Following an internal screening of KRED enzymes from commercially available kits, a very selective and active enzyme was found and selected for further process development.
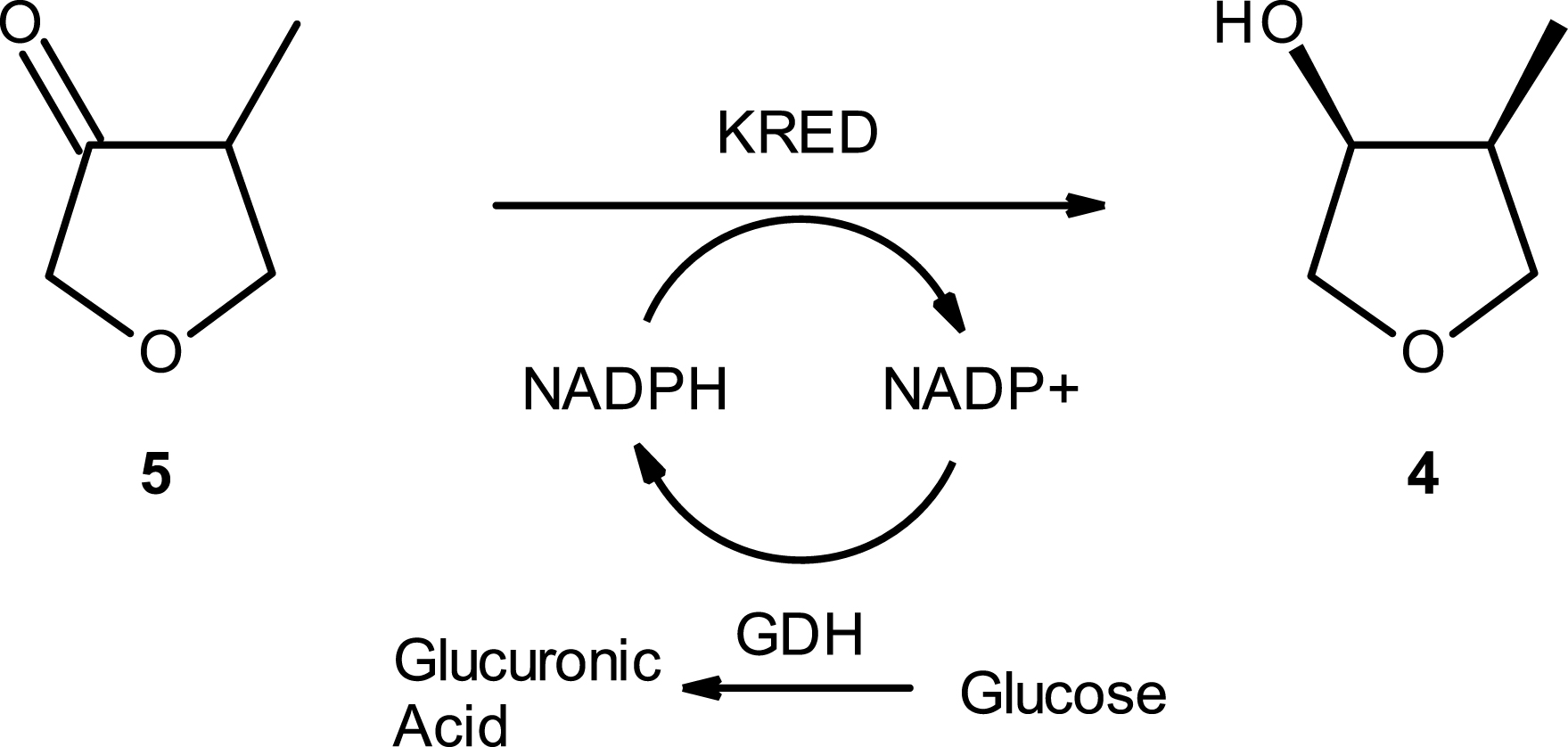
Synthesis of 4 via KRED reaction: global reaction scheme principle.
Interestingly, the complete conversion of the two enantiomers of the ketone into one main diastereoisomer of the hydroxy methyl tetrahydofuran, 4 demonstrated that a dynamic kinetic resolution is occurring under the reaction conditions (Scheme 6).
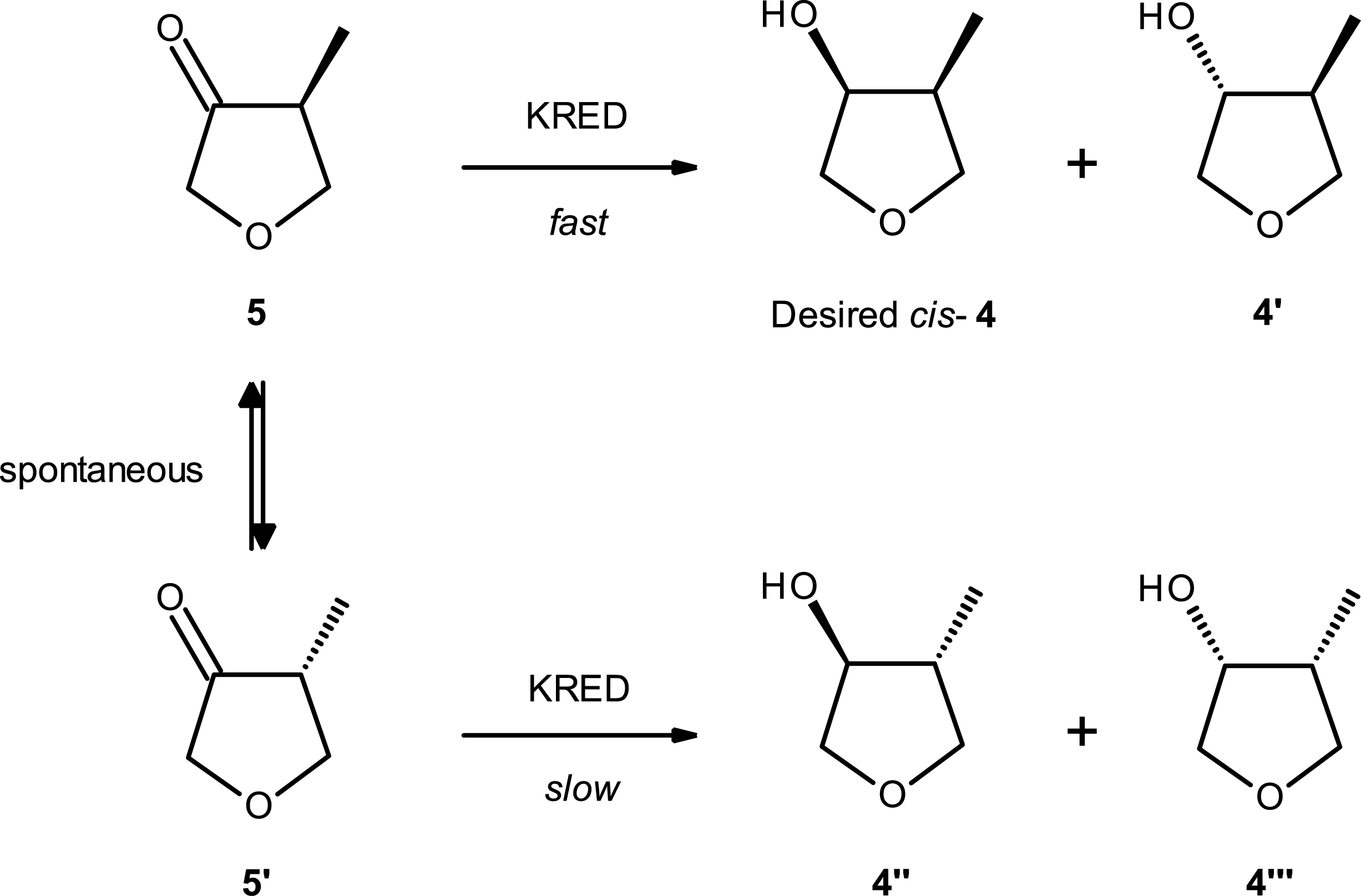
Kinetic resolution (KR) and dynamic reductive kinetic resolution (DRKD) based on ketoreductase-catalyzed reduction.
The optimized laboratory process was further refined to fit the scale and was successfully scaled in the pilot plant to 40 kg of ketone 5 per batch (Table 1). Yield and enantiomeric excess like those observed at laboratory scale were obtained, demonstrating the robustness of the biocatalytic process.
Scale up results for preparation of alcohol 4
| Batch type | Starting material | Product | ee (%)a | cis/trans Ratioa | Yield (%)∗ |
|---|---|---|---|---|---|
| Demo batch | 1.155 kg | 0.86 kg | >99.9 | 16.7 | 73.2 |
| Pilot plant #1 | 39.95 kg | 31.7 kg | >99.9 | 13.4 | 78.0 |
| Pilot Plant #2 | 33.75 kg | 25.6 kg | >99.9 | 14.9 | 75.0 |
∗Assay yield. aCalculation: Ratio cis/trans: R = ∑[cis(S,S) + cis(R,R)]/∑[trans(R,S) + trans(S,R)]. Enantiomeric excess: ee (%) = 100 × [cis(S,S) − cis(R,R)]/∑[cis(S,S) + cis(R,R)].
A qualitative comparison between the enabling route (3 steps from ketone 5) and the biocatalytic alternative (1 step from ketone 5) is shown in Table 2 [19].
Qualitative comparison between enabling and biochemical processes from ketone 5 to alcohol 4
| Route | Number of steps | Work up Chromato. | Operating temp | Solvent with constraints | Isolated yield (%) | cis/trans ratio | ee (%) | PMI (kg/kg) |
|---|---|---|---|---|---|---|---|---|
| Enablinga | 3 | 1 | −65 °C | CH2Cl2 | 8 | >30 | 93.0 | 1142 |
| Enzymaticb | 1 | 0 | 30 °C | None | 75 | 14.9 | >99.9 | 151 |
aKilolab campaign, bPilot plant campaign #2.
The one step enzymatic process outperforms the three steps chemical reactions in terms of yield, waste generated, scalability, and timelines. Ultimately, the enzymatic route was the only viable alternative for large scale clinical batch manufacturing to meet timelines and quality.
After unlocking the production of preclinical and clinical batches with the implementation of the ketoreductase process to produce the chiral hydroxy methyl tetrahydrofuran derivative, we looked for an optimal synthesis route in anticipation of the commercial launch (Scheme 7)1 .
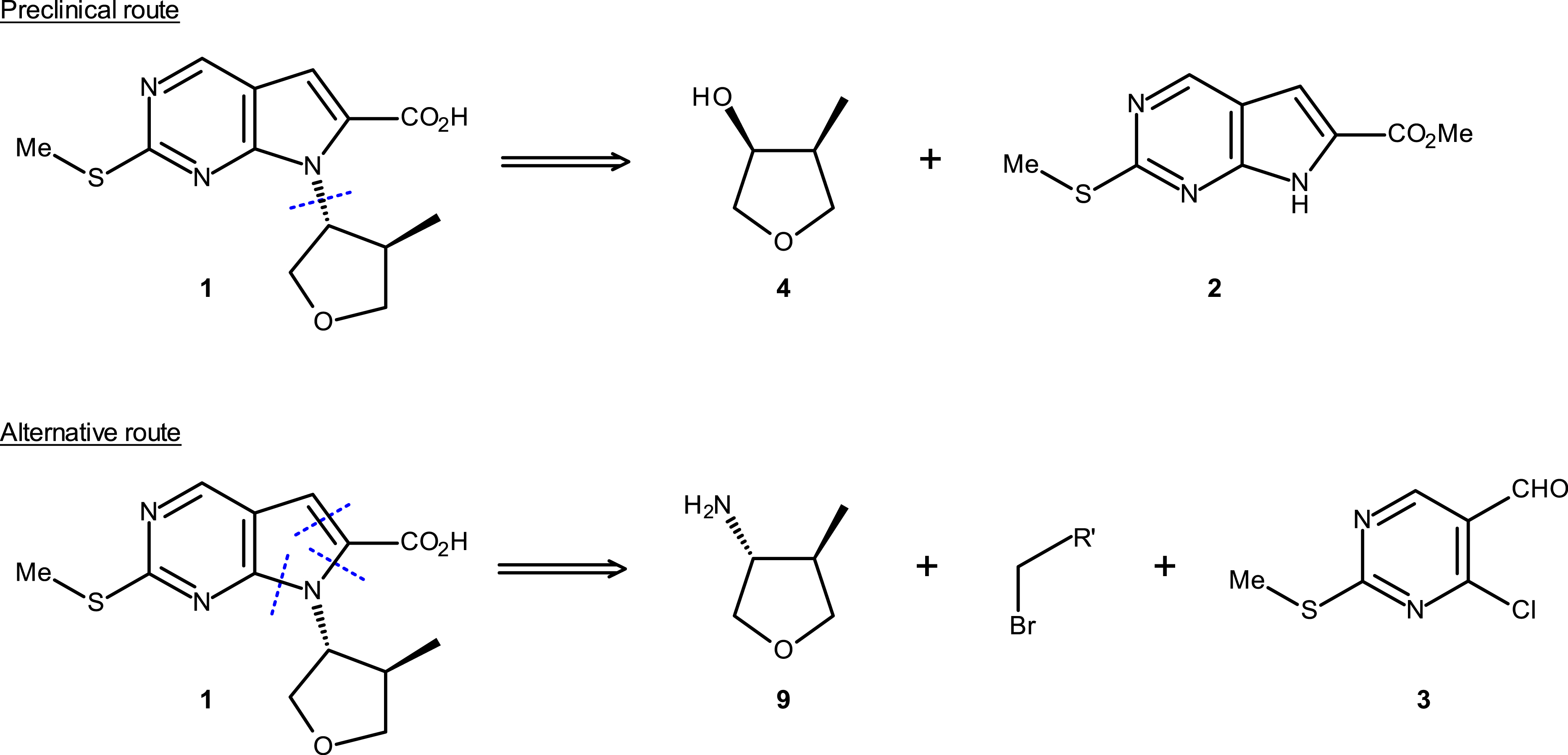
Disconnections integrating hydroxy methyl tetrahydrofuran 4 and amino methyl tetrahydrofuran 9.
The preclinical route based on the chiral hydroxy methyl tetrahydrofuran derivative intermediate 4 was first assessed [19]. The enantiomerically pure compound 4 obtained from KRED-mediated reduction was introduced at the next step of synthesis under Mitsunobu conditions. It quickly became apparent that this transformation was not efficient enough due to the formation of by-products such as triphenylphosphine oxide as well as reduced form of DIAD used in the Mitsunobu reaction. Additionally, an excess of compound 4 (3 equiv) was necessary, likely due to decomposition under the reaction conditions [27].
To install the chiral fragment into the molecule, the trans-(3R,4R)-amino methyl tetrahydrofuran derivative 9 was identified as a potential intermediate. It was demonstrated that it could be introduced into the global synthesis through an efficient nucleophilic substitution reaction without degrading the diastereoselectivity. Having solved the challenge associated with the introduction of the chiral fragment onto the molecule, we then turned our attention to the challenging task of preparing the chiral amino methyl tetrahydrofuran derivative 9.
Two alternative routes have been evaluated in parallel, namely alternative 1 (chemical pathway) and alternative 2 (enzymatic pathway) starting from the same methyl ketone intermediate 5 (Scheme 8). The chemical route initially studied produced the first grams of the chiral (3R,4R)-amino methyl tetrahydrofuran derivative 9, which allowed validation of the end of the synthesis. It should be noted that diastereo- and enantioselectivity were improved through (S)-mandelate salt formation but remained moderate (trans/cis = 88:12 and ee ∼ 93%). The overall yield of this four-step synthesis was low (∼15%) and generated high quantities of waste. This chemical route was not suitable for commercial batch manufacturing, and it was decided to investigate enzymatic approaches.
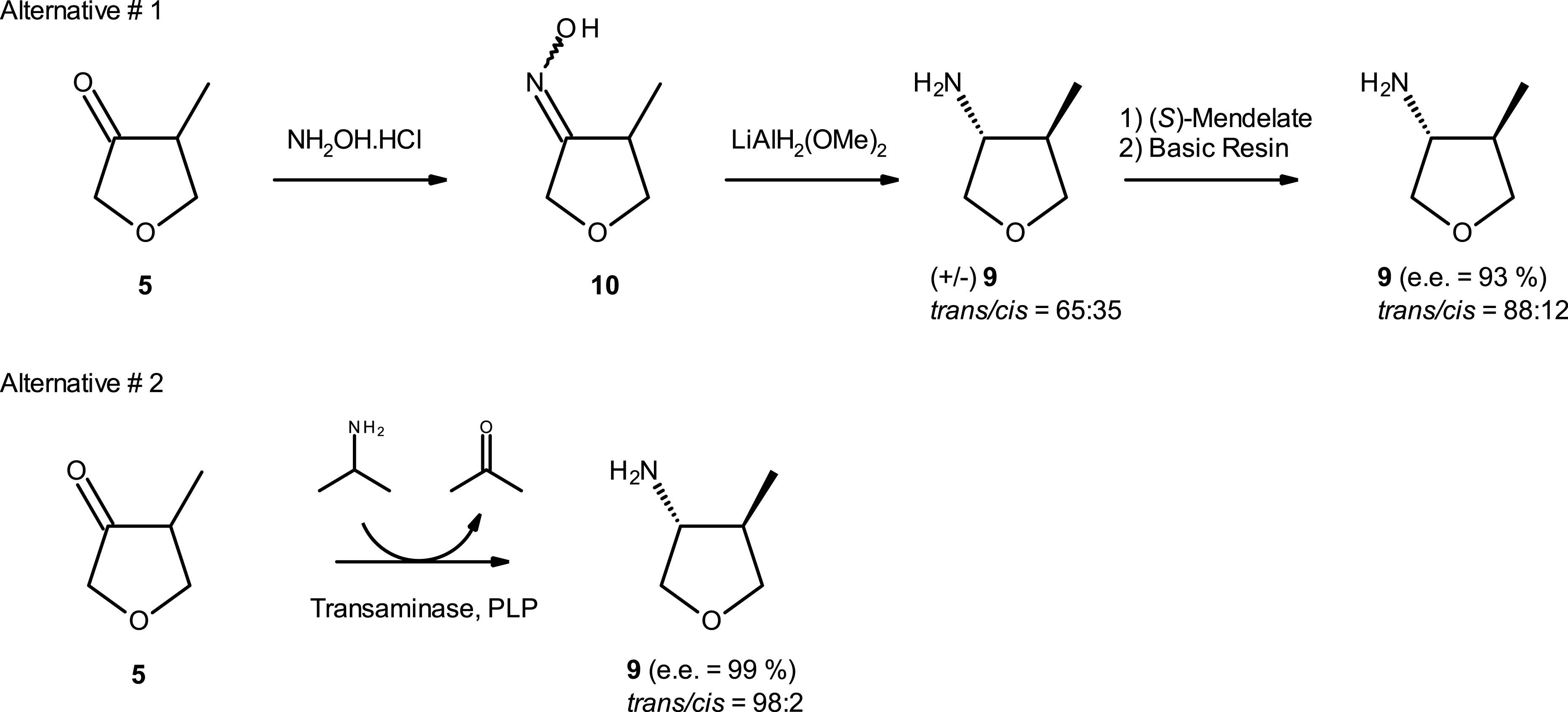
Chemical pathway and enzymatic transamination pathway toward the synthesis of trans amino methyl tetrahydrofuran 9.
Starting from methyl ketone 5, we explored the synthesis of the amino methyl tetrahydrofuran derivative 9 through asymmetric trans amination with transaminase enzymes (see footnote 1) [28, 29].
At first, commercial enzymes were used for the proof-of-concept. Following an internal screening of 83 transaminase enzymes from commercially available kits, 19 enzymes demonstrated good biocatalytic activity (assay yield > 50%). We identified one enzyme (transaminase ATA-047) that was able to deliver the amino methyl tetrahydrofuran derivative 9 with high selectivity toward the desired trans (3R,4R)-diastereoisomer (assay yield ∼ 99% after 24 h reaction, ee > 99%, trans/cis > 30) under screening conditions. Interestingly, the reaction occurred under dynamic kinetic resolution and only the trans (3R,4R)-diastereoisomer 9 was produced starting from the racemic ketone 5. To demonstrate the potential of the transaminase approach, a scale trial in 100 mL thermostated glass reactor with overhead stirring containing was performed. Applying a low enzyme/substrate ratio (1:10), a high conversion rate (assay yield 9 = 92.7%) was observed after 48 h reaction with excellent stereoselectivity (Figure 5).
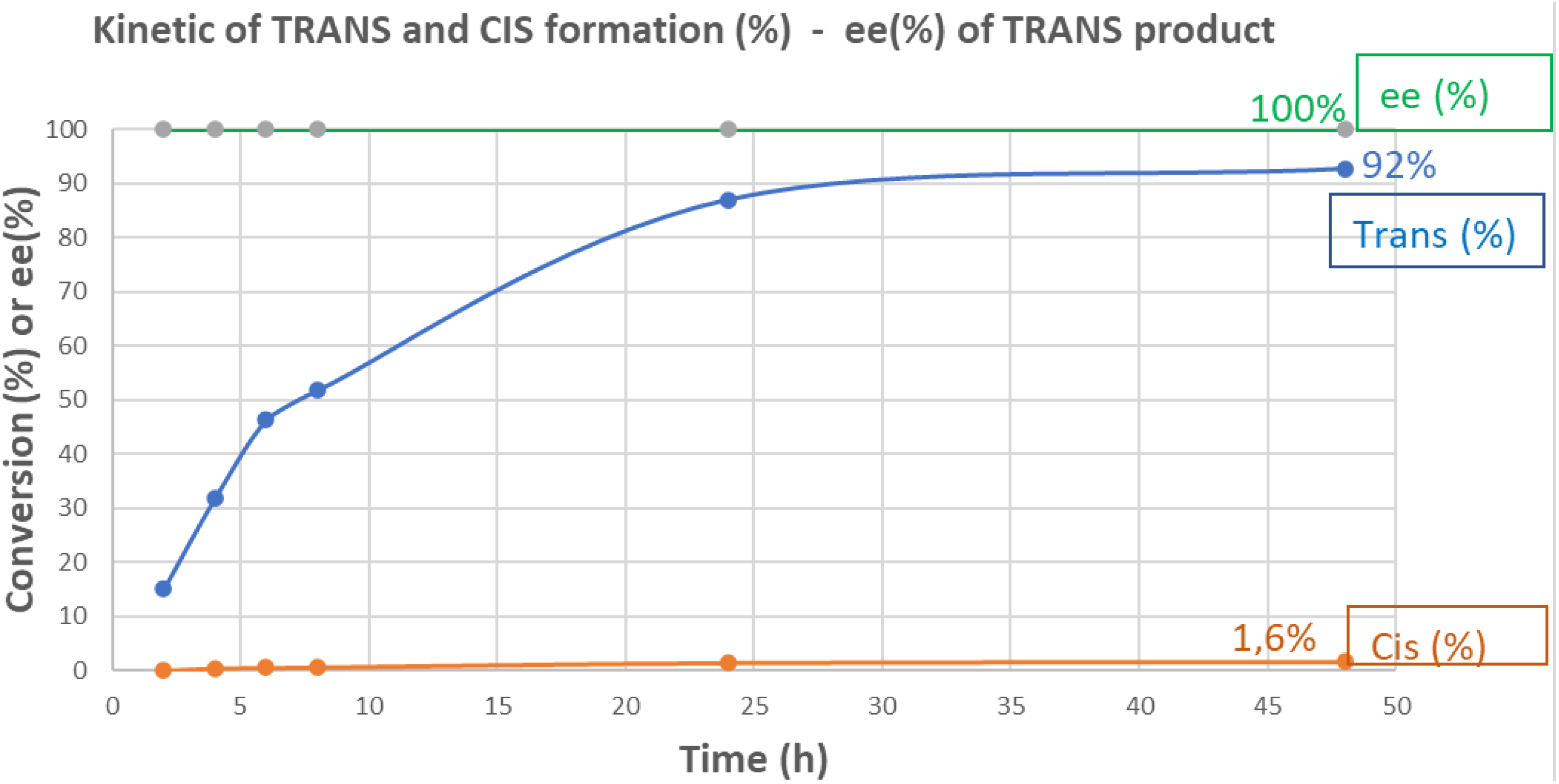
Time course of transamination reaction of 5 with ATA-047 at 100 mL scale.
Conditions: transaminase ATA47 1 mg/mL; ketone 5 = 100 mM; isopropylamine = 0.9 M; PLP = 10 mM; sodium phosphate buffer 100 mM, pH 9; temperature = 30 °C; volume = 100 mL. See footnote for calculation2 .
Although the commercial enzyme performed well, industrial objectives required a more efficient biocatalyst with better stability and higher substrate loading compatibility. Thus, in parallel with the testing of commercial kits, we carried out a genome mining approach through literature search and database in-silico screening. Fifty wild-type transaminases were selected for cloning and screening assays. Two enzymes were found to be very active in the formation of the amino methyl tetrahydrofuran derivative 9 (screening conditions), e.g., transaminase from Chromobacterium violaceum (assay yield amino methyl tetrahydrofuran ∼ 90%), providing mainly the trans-(3R,4R)-diastereoisomer and Arthrobacter sp (assay yield amino methyl tetrahydrofuran ∼ 50%) providing mainly the cis-diastereoisomers. Enantioselectivity could be reversed during enzyme evolution. Thus, with such results in hand, we considered these two enzymes to be good hits to start an enzyme evolution campaign to achieve the industrial goal of an efficient biocatalyst.
The third example deals with the optimization of a sulfotransferase enzyme for the synthesis of one of our late-stage assets [30]. The biocatalytic process used 3′-phosphoadenosine-5′-phosphosulfate (PAPS) as a donor of the SO3 group and was viewed as unsustainable and a significant cost driver. As an alternative to reduce the process cost, a PAPS recycling system was envisaged. We demonstrated how it was possible to rapidly increase the activity of an enzyme for a non-natural reaction in a few optimization rounds through enzyme evolution campaign. In the literature, it is known that the enzyme Aryl sulfotransferase IV (AST-IV) from rat can produce the PAPS from a SO3 donor much cheaper, the p-nitrophenyl sulfate (p-NPS) (Scheme 9).
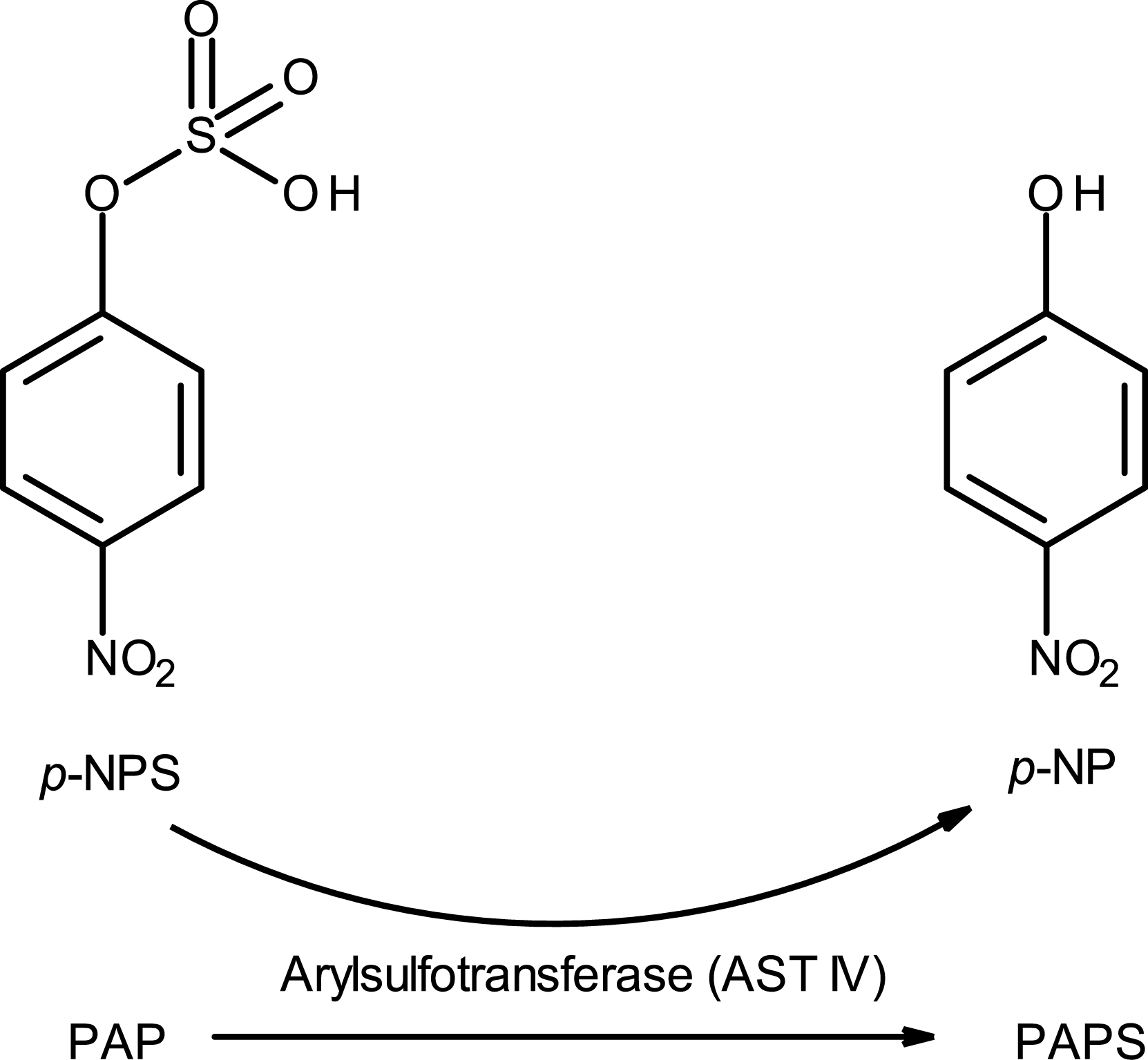
PAPS regeneration system. AST-IV catalyzes the transfer of the sulfo group from the less expensive donor, p-NPS.
However, this is the reverse reaction of this enzyme since its natural function is the sulfation of p-NP starting from PAPS. Therefore, the challenge was to force an enzyme to be efficient against its natural reaction equilibrium.
An optimization program was started with a directed evolution approach by recombination of homologues. This method consists of taking advantage of some homologues of the starting gene and recombining them randomly to produce new sequences among which we might find some improved versions of the enzyme. In this first round, 12,000 clones were screened, and we achieved a twofold activity increase in PAPS production. Even though we found more efficient variants, this method did not lead to an enzyme active enough to reach the goal, and we decided to use a more structure-guided rational design approach.
By using computational tools and molecular modeling, it is possible to predict mutations that might enhance the enzyme performance. Thanks to the 3D modelling tools, we were able to reconstitute the structure of the protein and identify the binding region for the substrates and products (PAP/p-NPS and PAPS/p-NP) in the catalytic site. The challenge was then to find how to increase PAPS formation. We decided to create variants to destabilize the binding between the enzyme and the p-NP in order to favor the binding of p-NPS as a substrate. In this case we found one mutant able to increase the activity of the enzyme sevenfold (Figure 6).
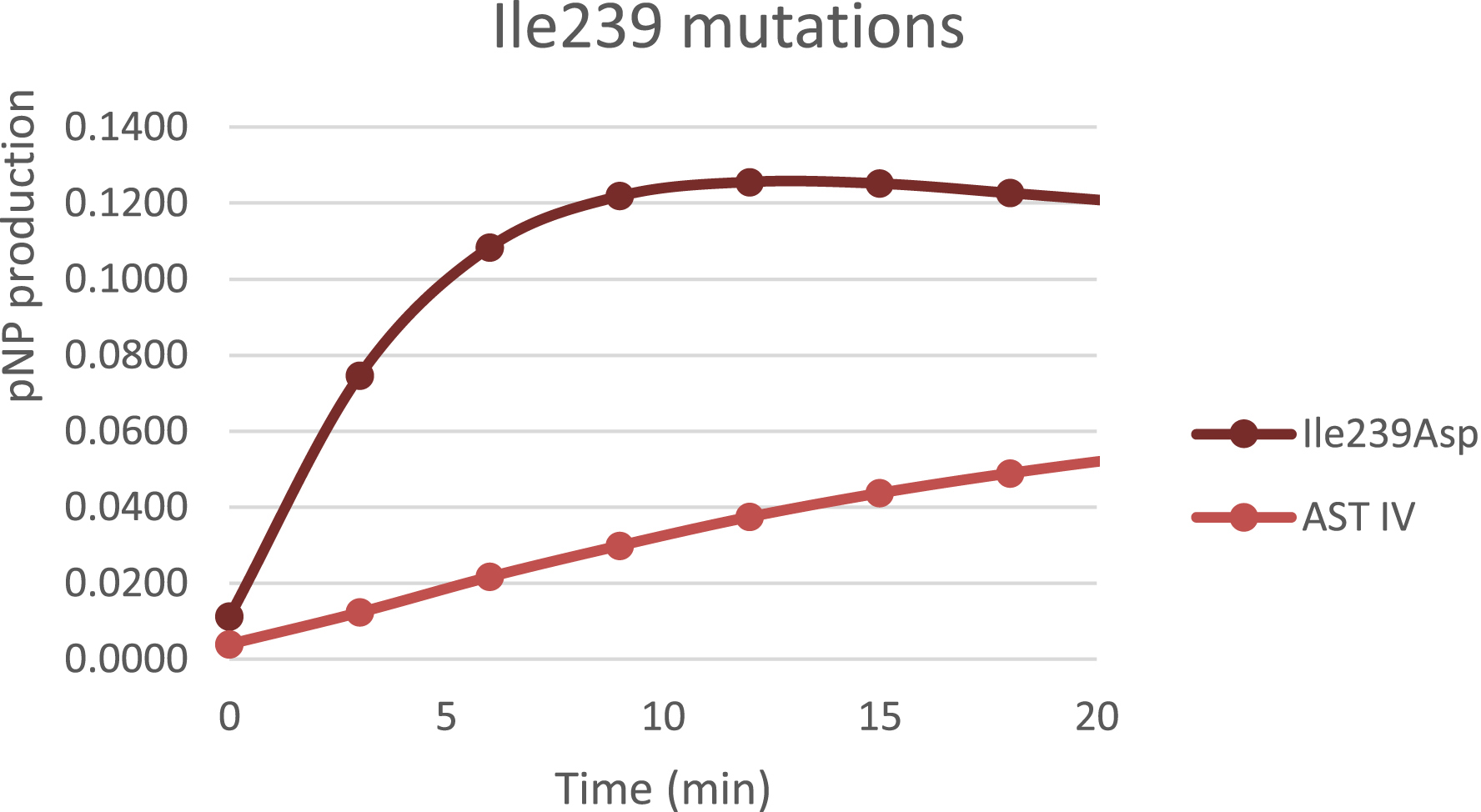
p-NP production achieved by Isoleucine 239 mutant compared to wild type enzyme.
After this first positive result we decided to launch a more in-depth study. We tested a list of other ∼100 variants targeting both the binding site (to directly increase the activity) and the entire protein with the idea of stabilizing the activity of the enzyme during the reaction. After this additional round, we identified 10 mutations that, in combination, were able to increase the activity of the protein more than 10-fold, maintaining a high level of activity throughout the reaction.
This final variant of the enzyme can sustain the recycling of PAPS and allow an efficient sulfonation of the target, reducing the manufacture cost of the process (Figure 7).
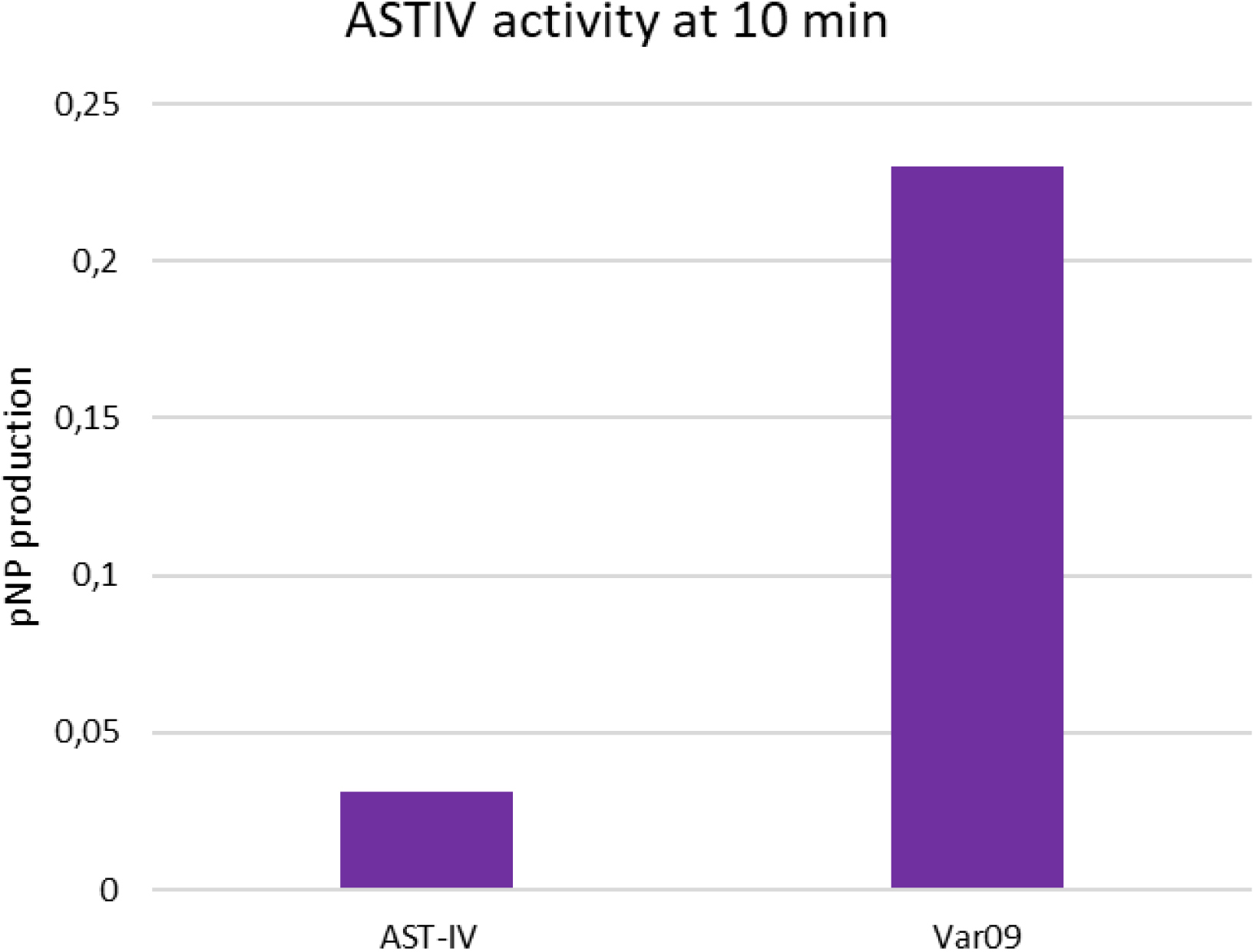
Activity of final variant (Var09) of AST-IV.
6. Outlook and opportunities
Environmental considerations drive our processes all along development and industrial phases. This is a key factor in accelerating the implementation of our internal Biosynthetic Technologies platform. A key success factor for the implementation of biocatalysis is the collaboration between organic chemists and biochemists to design new and more efficient chemical routes enabled by biocatalysis.
The examples described in this article show that the implementation of enzymatic transformations in the synthesis of complex molecules of our R&D portfolio has a positive impact on environmental footprint and can be in some cases the only viable alternative for large scale clinical batch manufacturing to meet timelines and quality.
We have built a very efficient toolbox to investigate common biocatalytic transformations using well-developed classes of enzymes (such as imine reductase (IRED), transaminase, ketoreductase (KRED), ene-reductase or nitrilase). However, the molecules entering our R&D portfolio are becoming increasingly complex. Innovative retrosynthetic pathway integrating biocatalytic step will be of great interest to overcome associated CMC risks. Diversification of our toolbox with less well-investigated classes is needed to enlarge the investigation in the repertoire of biocatalytic reaction on non-natural substrates. Currently, certain gaps in the toolbox remain. The biocatalysis community (academics and industrials) is addressing these challenges and new enzymatic platforms for industrial chemistry should arise in the near future [1].
Protein engineering is of great importance for developing enzymes for biocatalytic transformations at an industrial scale. We decided to build an in-house molecular biology platform (expertise and capabilities) that allows us to tailor enzyme performance to meet demanding industrial requirements. A proof-of-concept was achieved through the development of an efficient AST-IV enzyme. Looking to the future, the ability in expanding the field of computational methods, such as artificial intelligence, machine learning or data management will support our internal strategy toward designing highly efficient enzyme for non-natural substrates within reduced cycles time. This will be possible thanks to: (1) the better identification of suitable enzymes from protein sequence information accessible in databanks (genomic database mining); (2) smarter mutant libraries design.
Abbreviations
CMC, chemistry, manufacturing and control; DIAD, diisopropyl azodicarboxylate; DKR, dynamic kinetic resolution; DNA, deoxyribonucleic acid; IPA, isopropyl alcohol; IRED, imine reductase; KRED, ketoreductase; PMI, process mass intensity; PLP: pyridoxal 5′-phosphate; R&D, research and development; SFC, supercritical fluid chromatography.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgments
All authors would like to express their gratitude to all the Sanofi teams who contributed to the work presented in this manuscript. Their names can be found in the references.
1 A. Rabion, “Accelerating the implementation of Biocatalysis in the Sanofi Pharma portfolio: Synthetic Routes to complex chiral tetrahydrofuran derivatives”, in Oral Communication CBSO24 30th Symposium, October 7, 2024, Cap Hornu, France.
2 Calculation : ee = [trans(3R,4R) − trans(3S,4S)]/[trans(3R,4R) + trans(3S,4S)]; conversion cis (%) = [cis(3R,4S) + cis(3S,4R)]/[cis(3R,4S) + cis(3S,4R) + trans(3R,4R) + trans(3S,4S) + Ketone]; conversion trans (%) = [trans(3R,4R) + trans(3S,4S)]/[cis(3R,4S) + cis(3S,4R) + trans(3R,4R) + trans(3S,4S) + Ketone].