



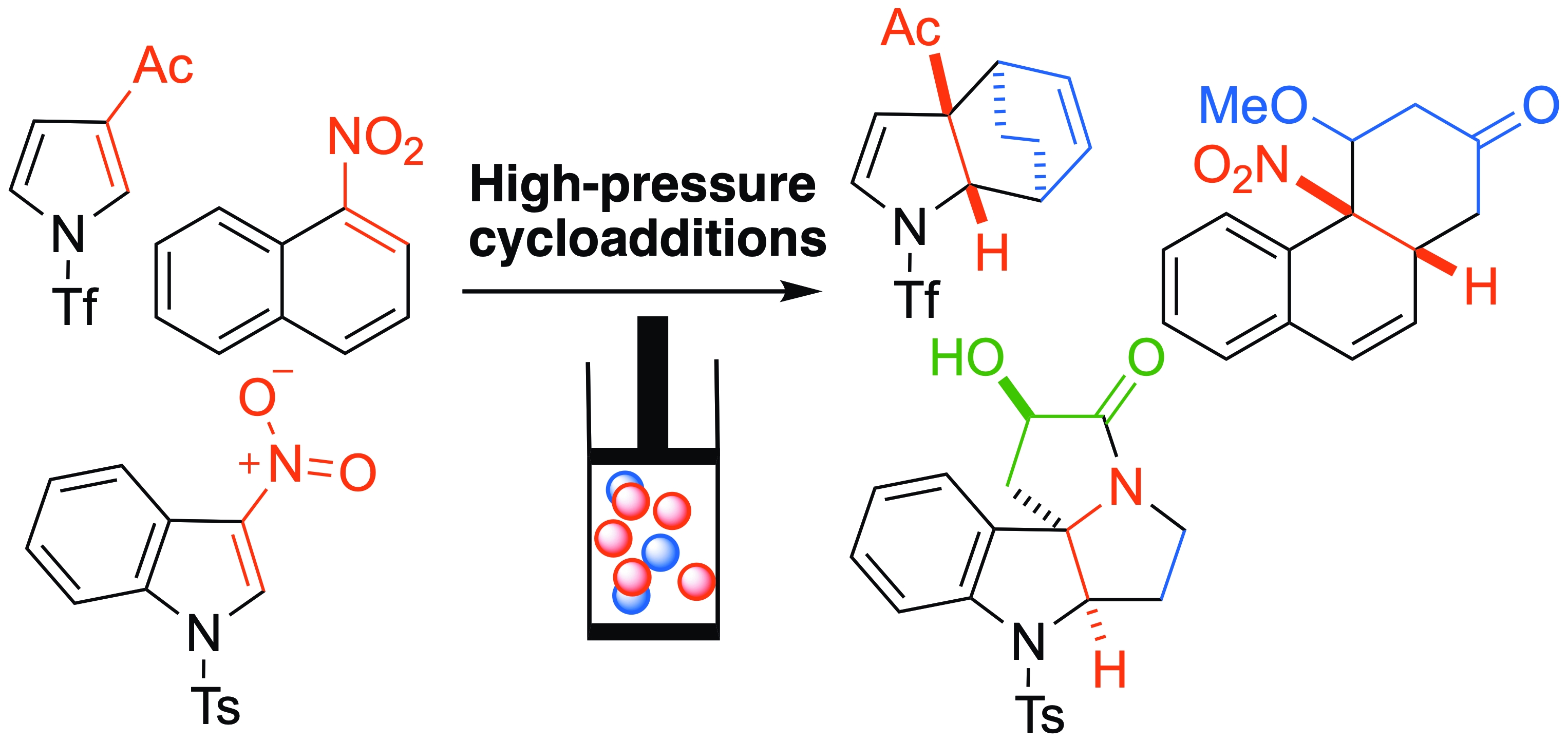
 CC-BY 4.0
CC-BY 4.0
1. Introduction
Pressure is a parameter frequently overlooked by the community of organic synthetic chemists, except in cases involving gases, such as hydrogenation or carbonylation, where it typically reaches values between 1 and 50 bars. In organic chemistry, reaction media are most often liquids, and compressing a liquid is generally considered challenging, if not impossible, especially when aiming to attain pressures above 1 kbar, such as those needed to significantly affect the reaction outcomes. For clarity, the term “high pressure” (HP) used in the following refers specifically to the 1–20 kbar range (i.e., 2000–20 000 atm or 0.2–2.0 GPa).
On the basis of the Evans–Polanyi transition state theory, it has been established that the influence of pressure on the activation energy of chemical reactions (ΔG‡), and thus on their kinetic constants, is a function of the sign and absolute value of the activation volume (ΔV‡), which corresponds to the volume of the transition state (VTS) minus the volume of the substrate(s) (VSM) (ΔV‡ = VTS − VSM). A chemical reaction characterized by a negative ΔV‡ is accelerated when pressure increases, while in contrast it is decelerated when ΔV‡ is positive [1, 2, 3].
Cycloadditions are among the most useful reactions in synthesis, giving rapid access to functionalized (poly)cyclic compounds from simple starting materials such as, for example, alkenes, dienes, or dipoles. As the cycloadducts incorporate the entire starting materials, the process generates no waste and exhibits high atom economy. However, the application of these reactions is often hampered by the unfavorable steric or electronic characteristics of the reagents. The typical activation methods used for promoting cycloadditions are thermal, microwave, or chemically catalyzed ones (acid, base, metal, or organo-catalyst). Because cycloadditions are characterized by highly compact transition states, they are generally associated to negative activation volumes, comprised between −10 and −50 cm3⋅mol−1, and are thus significantly accelerated when the pressure increases [4].
Under HP, deactivated and/or sterically hindered substrates can become good partners in cycloadditions, making HP a valuable alternative to conventional heating or chemical catalysis [5]. An increase in selectivity comes as an extra bonus since this activation mode disfavors uncontrolled side reactions, especially when they are characterized by a positive activation volume. For instance, HP tends to prevent retrocycloadditions or eliminations that are difficult to control under more classical conditions. Energy is required to compress the reaction medium, but HP remains constant once established, and the overall energy input remains low. Consequently, HP represents a sustainable and effective strategy to enhance the yields of sluggish or inefficient cycloadditions [6].
Electron-deficient aromatic compounds are easily accessible, through SEAr reactions (electrophilic aromatic substitution) for example. Their involvement in dearomatizations is particularly attractive, facilitating a straightforward increase in the complexity of the molecular structure. In general, when electron-deficient aromatics interact with nucleophilic species, they undergo nucleophilic aromatic substitutions (SNAr), which involve an addition/elimination sequence restoring the aromaticity of the original ring [7]. In some cases, the dearomatized Meisenheimer intermediate resulting from the nucleophilic addition step can be trapped intramolecularly, in a formal cycloaddition process, affording a dearomatized cycloadduct [8]. Complementarily, concerted cycloadditions between electron-poor aromatics and electron-rich dienes or dipoles can also deliver the dearomatized cycloadducts. Because the latter processes involve transition states that are more compact than the starting materials, they are efficiently accelerated under HP. At the same time, HP helps to prevent potentially competitive retroadditions or rearomatizations, which are typically associated with positive activation volumes.
Over the past two decades, our laboratory has explored dearomatizations of electron-deficient (hetero)arenes in different cycloaddition processes. While the involvement of the aromatics as electron-poor dipolarophiles in the presence of electron-rich 1,3-dipoles is typically performed at atmospheric pressure [9, 10, 11, 12, 13, 14], HP activation has proven very useful to promote (4+2) dearomative cycloadditions. Two different types of reactions have been developed: the first one involves one double bond of the aromatic compound acting as an electron-poor dienophile, while the second exploits the C=C–N=O system of nitroarenes acting as an electron-poor heterodiene. Both approaches are described below.
2. Electron-deficient arenes as dienophiles in HP-promoted Diels–Alder cycloadditions
We have developed (4+2) dearomative cycloadditions in which the double bond of the aromatic bearing an electron-withdrawing substituent reacts as dienophile, through its lowest unoccupied molecular orbital (LUMO), with an electron-rich 1,3-diene involved through its highest occupied molecular orbital (HOMO). This typically leads to a polycyclic adduct bearing a tetrasubstituted carbon atom at the ring junction, with a relative cis stereochemistry of the junction due to the pericyclic character of the reaction (Scheme 1).
Electron-deficient arenes as dienophiles in Diels–Alder cycloadditions.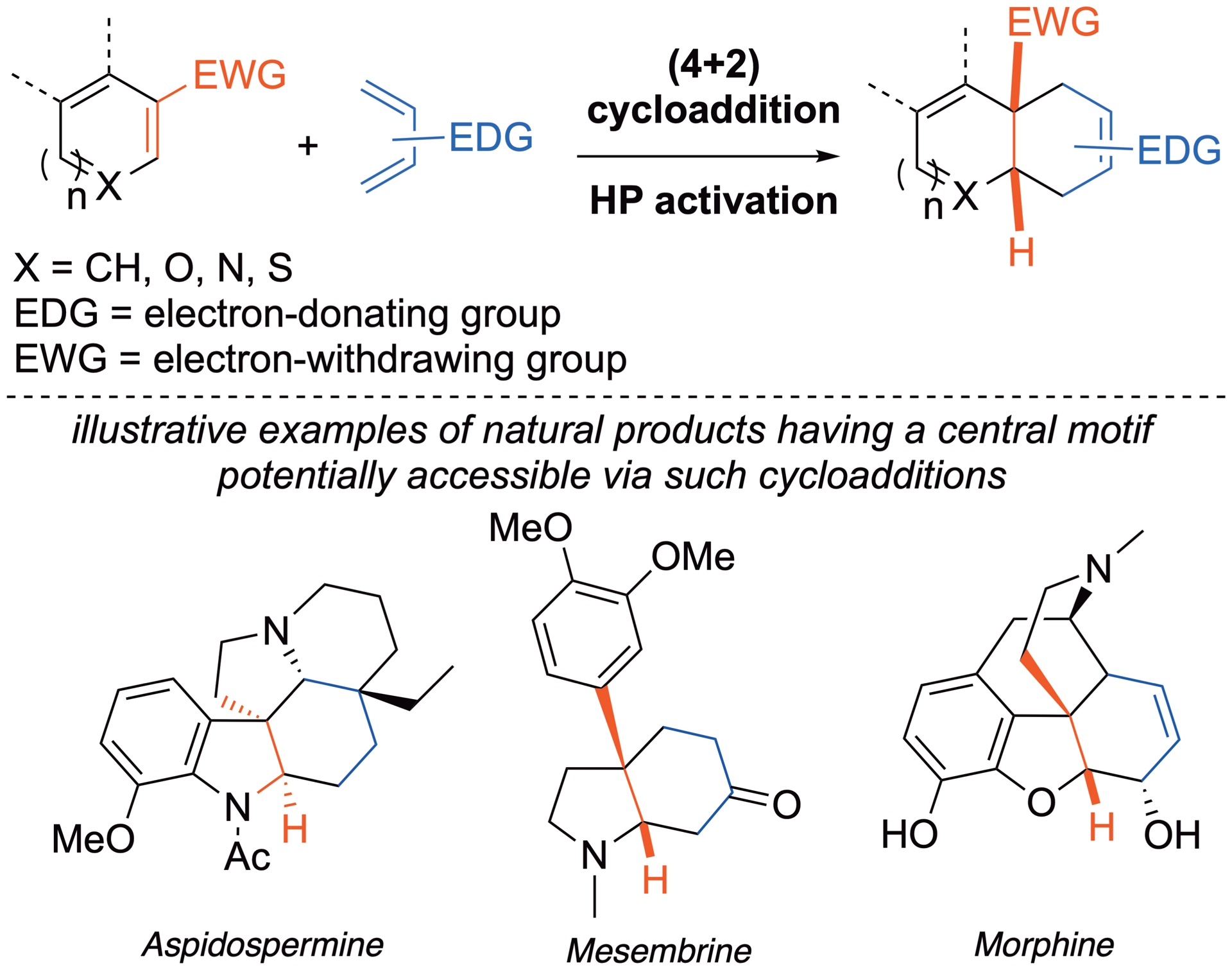
Different electron-poor (hetero)aromatics have been involved in these reactions with electron-rich dienes, and the cycloaddition conditions depend on the nature of the aromatic (indole, pyrrole, naphthalene, quinoline, or benzene), the electron-withdrawing group(s) (nitro, cyano, (alkoxy)carbonyl, or ketoamide) and the electron-rich diene. The following classification is based on the nature of the aromatic.
2.1. Electron-depleted indoles as dienophiles in Diels–Alder cycloadditions
Indoles are crucial structural frameworks present in a wide range of natural products, many of which display notable biological activities. From a reactivity perspective, indoles are widely recognized as moderate nucleophiles [15], and their reactivity with various electrophiles has been extensively studied in organic synthesis. Recently, in a complementary approach, the electrophilic reactivity of electron-deficient indoles has been largely investigated [16, 17]. Intramolecular reactions have been developed some time ago, to generate interesting scaffolds [18, 19]. In the case of the intermolecular version of the cycloadditions, it has been demonstrated that introducing a nitro group at the C3 position of this heterocycle decreases its aromaticity, thereby facilitating the addition of electron-rich species [20]. In this context, different research groups have contributed to exploit the “umpolung” reactivity of electron-depleted indoles and demonstrated that 3-nitroindoles can react as C2=C3 electron-poor dienophiles with electron-rich dienes in Diels–Alder cycloadditions (Scheme 2) [21]. Note that thermally activated processes generally lead to partial or complete rearomatization of the cycloadducts by nitrous acid elimination, a thermodynamically favored process [21, 22, 23, 24, 25, 26, 27].
3-Nitroindoles as dienophiles in Diels–Alder cycloadditions.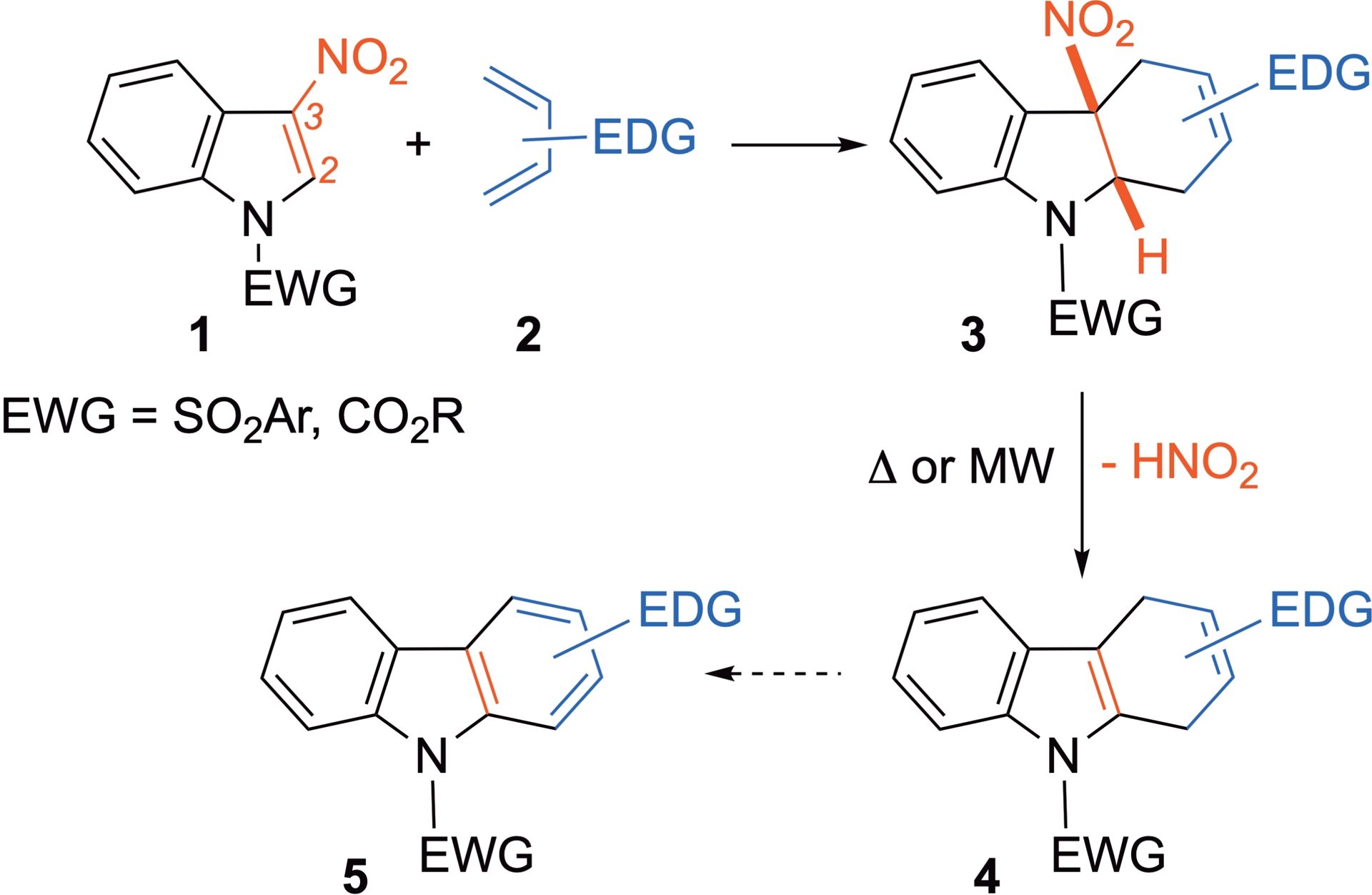
Thanks to the relatively high electrophilic character of 3-nitroindoles bearing an electron-withdrawing group at the nitrogen position [20], the presence of the Danishefsky’s diene 2a, doubly activated by two oxy groups at positions N1 and C3, renders the cycloaddition possible at room temperature (r.t.) in the absence of any promoter. Under these smooth conditions, rearomatization is prevented and the indoline cycloadduct isolated in good yields [20]. Diastereoselectivities are low, probably because of the conflicting influences of the stabilizing secondary orbital interactions favoring the endo approach, and of the steric hindrance favoring the exo one. The reaction can also occur with thiourea- or squaramide-based organocatalysts, including chiral ones. However, the stereoselectivities observed with these catalysts are generally low, likely due to the competing non-catalyzed process [28].
With less reactive dienes such as 2-trimethylsilyloxycyclohexadiene 2b, the reaction is not possible at atmospheric pressure. In such cases, HP activation proves efficient, and the reaction proceeds under 16 kbar at r.t. [29]. These smooth thermal and chemical conditions lead to the preservation of the three-dimensional nature of the dearomatized cycloadduct, which is isolated in good yields (Scheme 3). Notably, the diastereoselectivity of the cycloaddition is complete with this diene, leading to the exquisite formation of the exo cycloadduct. The corresponding approach is characterized by lower distortion of the substrates in a late transition state and by more favorable orbital interactions, presumably between the nitro group and the dienic part, explaining the stereoselectivity [29]. Under these conditions, 7-aza-3-nitroindoles also react well, leading to azaindolines in a similar manner.
HP-promoted Diels–Alder reaction of 3-nitro(aza)indoles with diene 2b.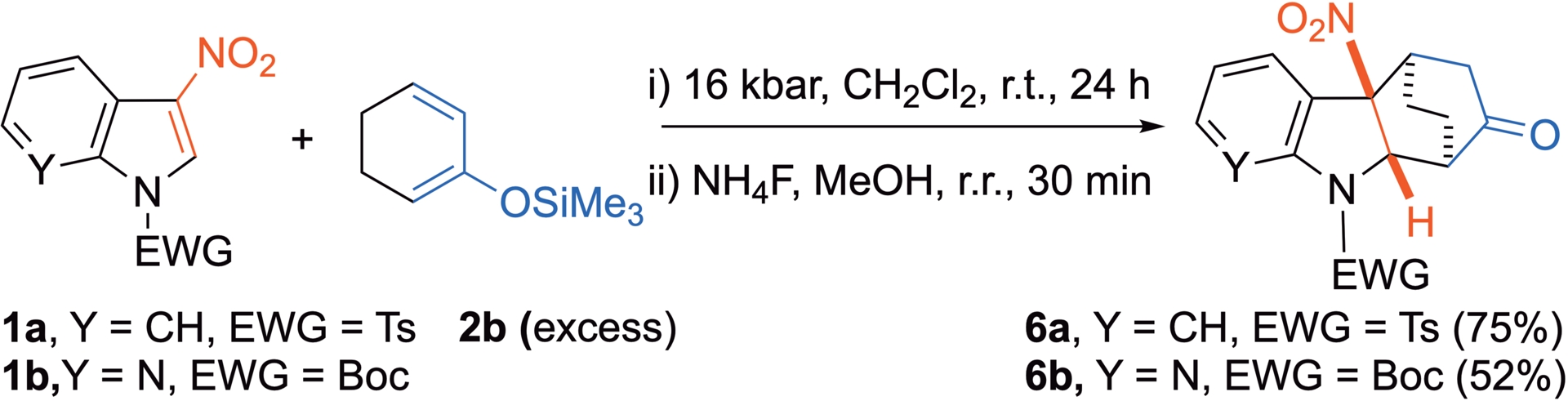
Other possible indolic dienophiles include 3-carbonylated substrates. In this case, the Diels–Alder (DA) reaction involving the C2=C3 double bond of the heteroaromatic competes with the hetero-Diels–Alder (HAD) reaction, where the carbonyl C=O bond acts as a competing dienophile. The reaction outcome depends on the carbonyl substituent, with tertiary ketoamide favoring the DA process, while formyl, ketoester, and secondary ketoamide favor the HDA one, all using Danishefsky’s diene 2a (Scheme 4) [30].
HP-promoted Diels–Alder reaction of 3-carbonylated indoles with 2a.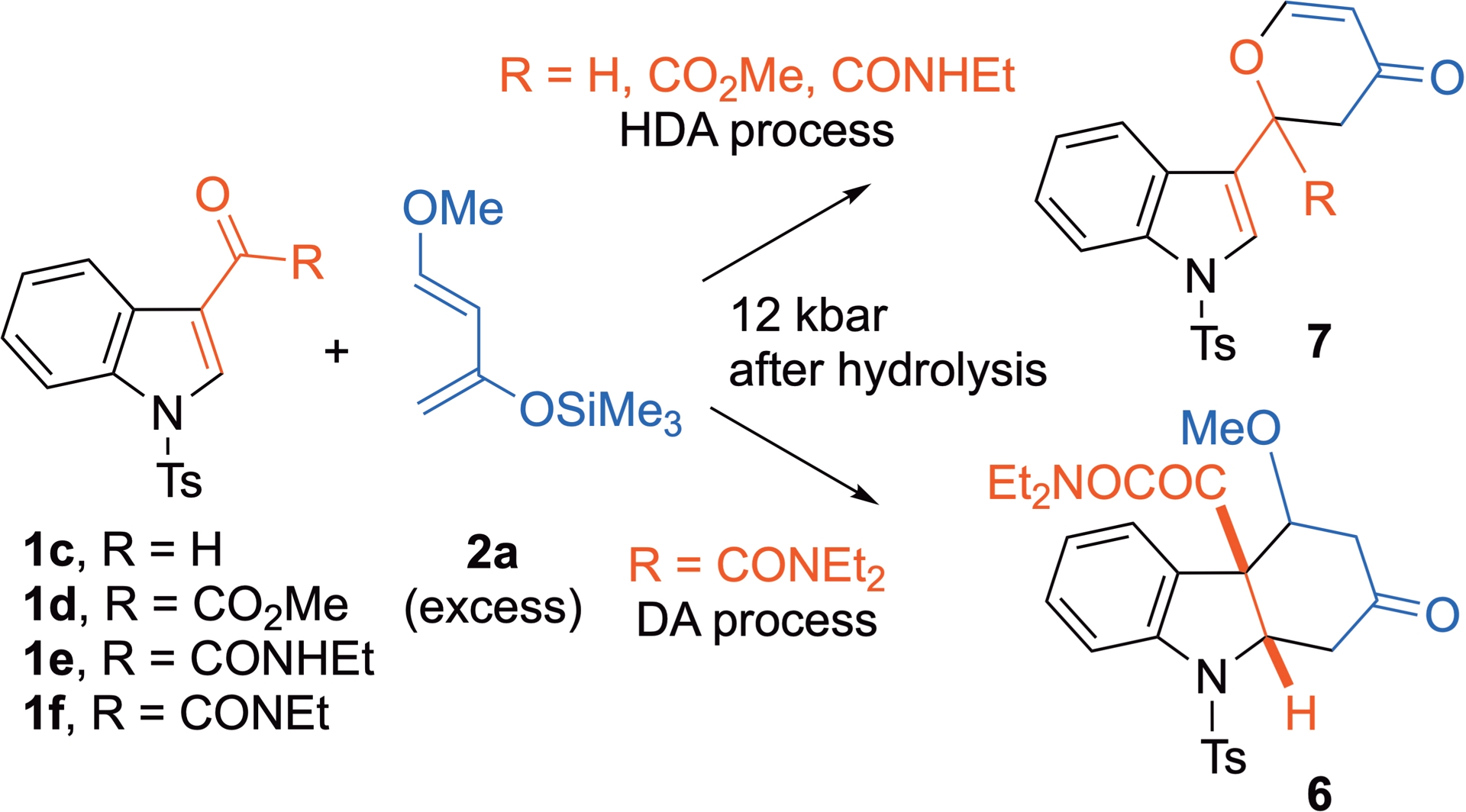
When less polarized, all-carbon dienes 2c and 2d are involved in the process with 3-formylindole 1c, chemoselectivity favors the DA cycloadduct. Owing to the lower reactivity of these dienes, Lewis acid activation, such as zinc chloride catalysis, can be beneficial. However, this acidic catalyst also promotes competitive polymerization of the diene used in excess under HP, and this can sometimes complicate the isolation of the desired cycloadduct. Under thermal conditions (195 °C), cycloadduct 6c is obtained in a good yield. However, when diene 2c is used, HP activation triggers a subsequent reaction, leading to the formation of bis-cycloadduct 8 (Scheme 5) [30, 31].
Thermal versus HP-promoted Diels–Alder reaction of 3-formylindole with 2c.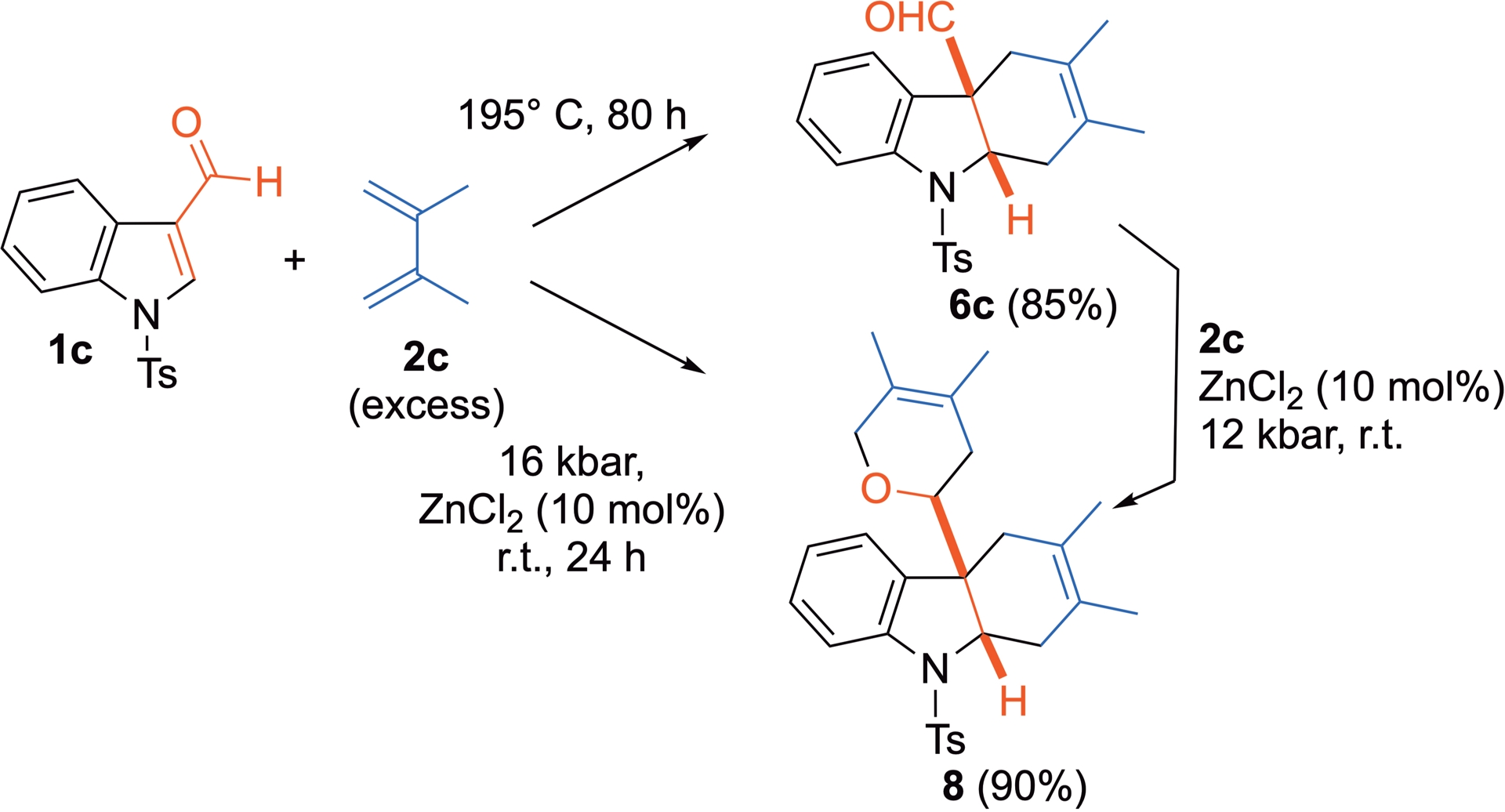
In contrast, when cyclohexa-1,3-diene 2d is involved, the reaction leads to cycloadduct 6d regardless of the reaction conditions (Scheme 6) [31]. The conversion, yield and diastereoselectivity of the reaction under HP conditions are greater than under thermal activation.
Thermal versus HP-promoted Diels–Alder reaction of 3-formylindole with 2d.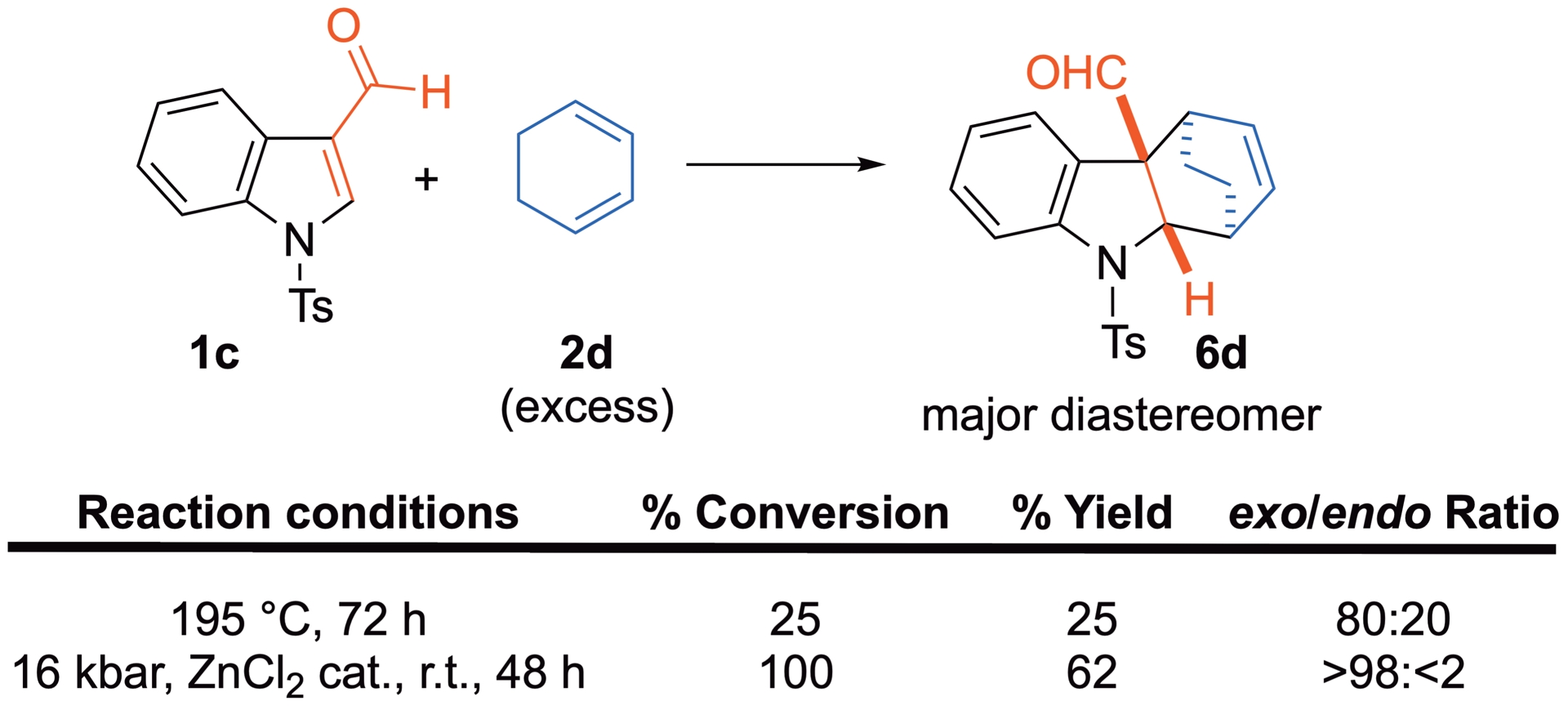
The type of electron-withdrawing substituent on the nitrogen atom of the indolic dienophile plays a crucial role in the cycloaddition. Although sulfonyl groups are known to be stronger electron-withdrawing groups than (alkoxy)carbonyl groups, the cycloaddition of 3-ketoamidic indoles with diene 2a occurs more readily when the nitrogen atom is activated as an amide or carbamate rather than as a sulfonamide (Scheme 7) [32]. This fact is ascribed to a greater ability of the carbon-centered groups to achieve delocalization of the nitrogen’s lone pair, resulting in stronger global withdrawing effects. The sulfur-centered functional group is less efficacious in decreasing electron density on the nitrogen atom, even when inductive effects are considered. This results in a lower aromaticity of the (alkoxy)carbonyl indoles and a higher reactivity at C2=C3 electron-poor dienophile [32].
Influence of the nitrogen atom substituent on the cycloaddition.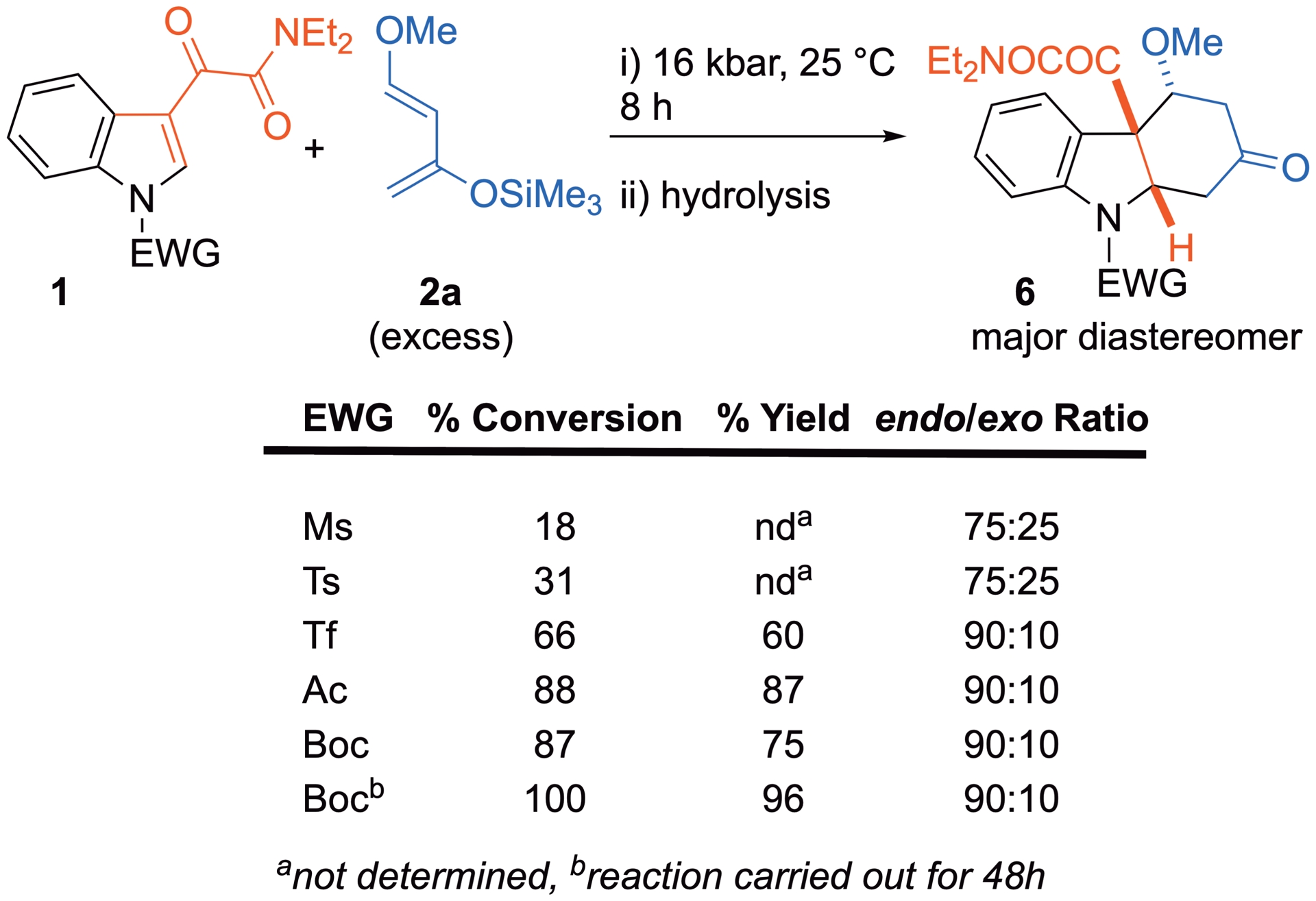
2.2. Electron-depleted benzofurans as dienophiles in Diels–Alder cycloadditions
Other possible heteroarenic dienophiles include benzofuranic compounds, bearing an electron-withdrawing substituent at position C3. Here again, while 3-nitrobenzofurans react as C2=C3 dienophile at r.t. and ambient pressure with Danishefsky’s diene 2a to yield the corresponding cycloadduct [20], reactions with the less depleted 3-carbonylated substrates require a stronger activation. With 2,3-dimethylbuta-1,3-diene 2c or cyclohexa-1,3-diene 2d, the cycloadditions can be thermally activated. However, this activation mode is less efficient when more sensitive silyloxydienes are involved and HP conditions are then to be preferred (Scheme 8) [33]. Here again, competitive HDA processes, involving the carbonyl substituent at position C3 as heterodienophile, are observed with benzofurans bearing a ketoester group in the presence of Danishefsky’s diene 2a [33].
3-Carbonylated benzofurans as C2=C3 dienophiles under HP activation.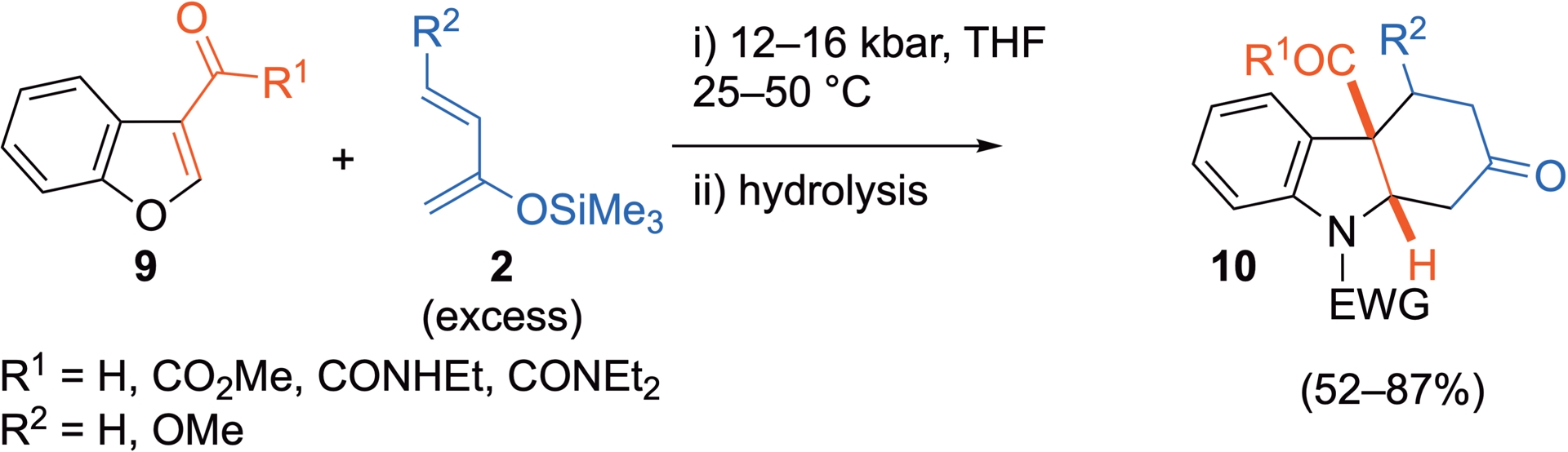
2.3. Electron-depleted pyrroles as dienophiles in Diels–Alder cycloadditions
When reacted under thermal conditions or Lewis acid activation, 3-nitro-N-tosylpyrrole 11 remains inert toward Danishesfky’s diene 2a and this sensitive diene is generally destroyed. In contrast, it reacts efficiently as an electron-poor dienophile with 2a under HP activation [34]. The cycloadduct generated is unstable after decompression and leads to the corresponding carbazole 12, resulting from a complete rearomatization (Scheme 9).
3-Nitropyrrole as C2=C3 dienophile under HP activation.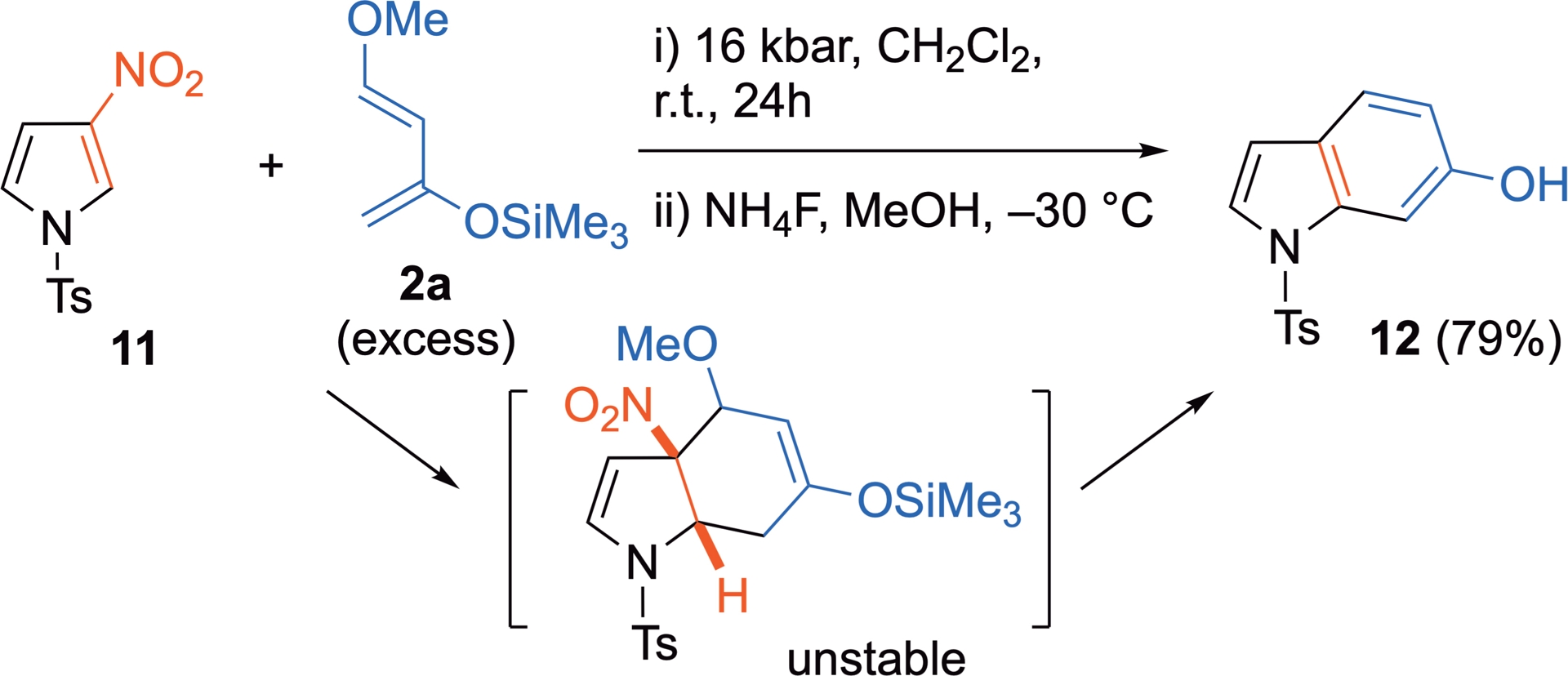
Cycloadditions with the less electron-depleted 3-acylpyrroles are also feasible under HP conditions, although Lewis acid catalysis is required alongside HP activation in this case. For example, cycloadditions between electron-poor pyrroles 13, which contain electron-withdrawing groups at least at the N1 and C3 positions and a series of electron-rich dienes, result in the formation of cycloadducts 14 (Scheme 10) [35, 36]. These products are obtained in very low yields under thermal activation in the presence of ZnCl2. Similarly, under HP and without a catalyst, this dienophile reacts poorly and enters a competitive polymerization process. However, combining HP and Lewis acid (ZnCl2) activations significantly improves the efficiency of the cycloaddition and the expected syn-tetrahydroindole 14 is recovered in good to excellent yields.
3-Acylpyrrole 13 as C2=C3 dienophile under HP activation combined with Lewis acid catalysis.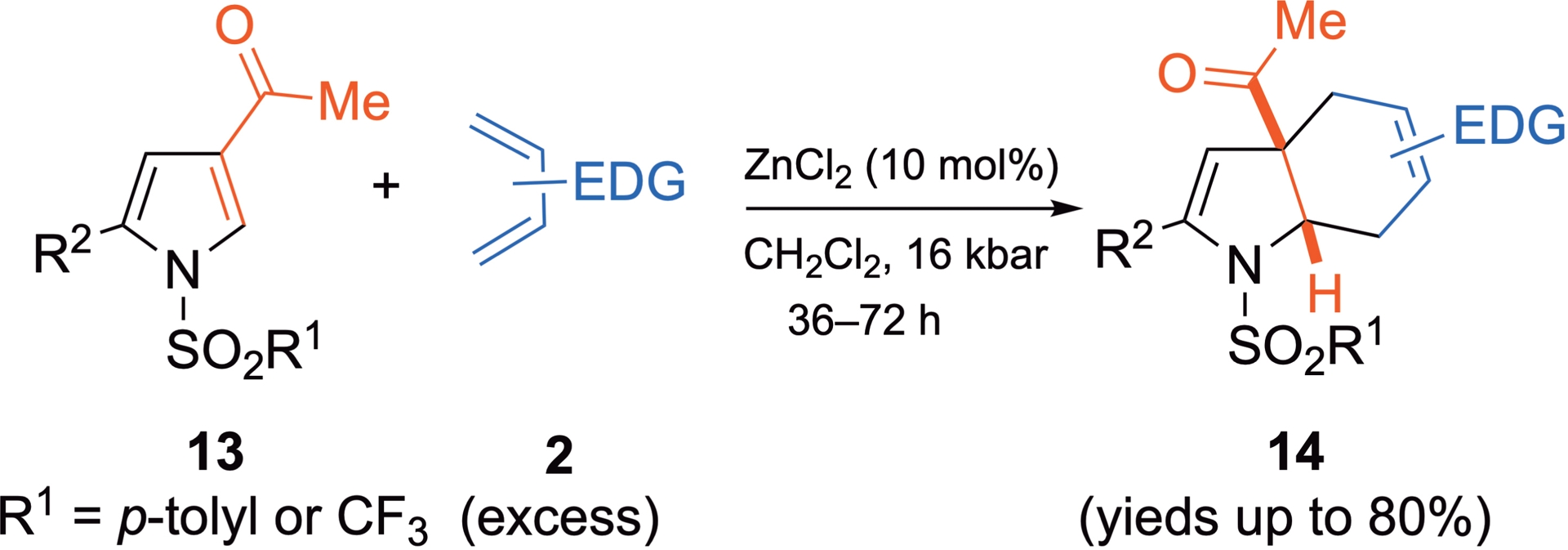
2.4. Other electron-depleted five-membered-ring heteroaromatics as dienophiles in Diels–Alder cycloadditions
Other nitrated five-membered-ring heteroaromatics can be dearomatized under HP in the presence of Danishefsky’s diene 2a, among which benzothiophene, pyrazole, or furan. The reaction does not require any additional chemical activation and smooth hydrolysis of the pressurized mixture leads to preservation of the three-dimensional structure of the dearomatized cycloadduct in these cases (Scheme 11) [34].
Nitrated five-membered-ring heteroaromatics as dienophiles under HP activation.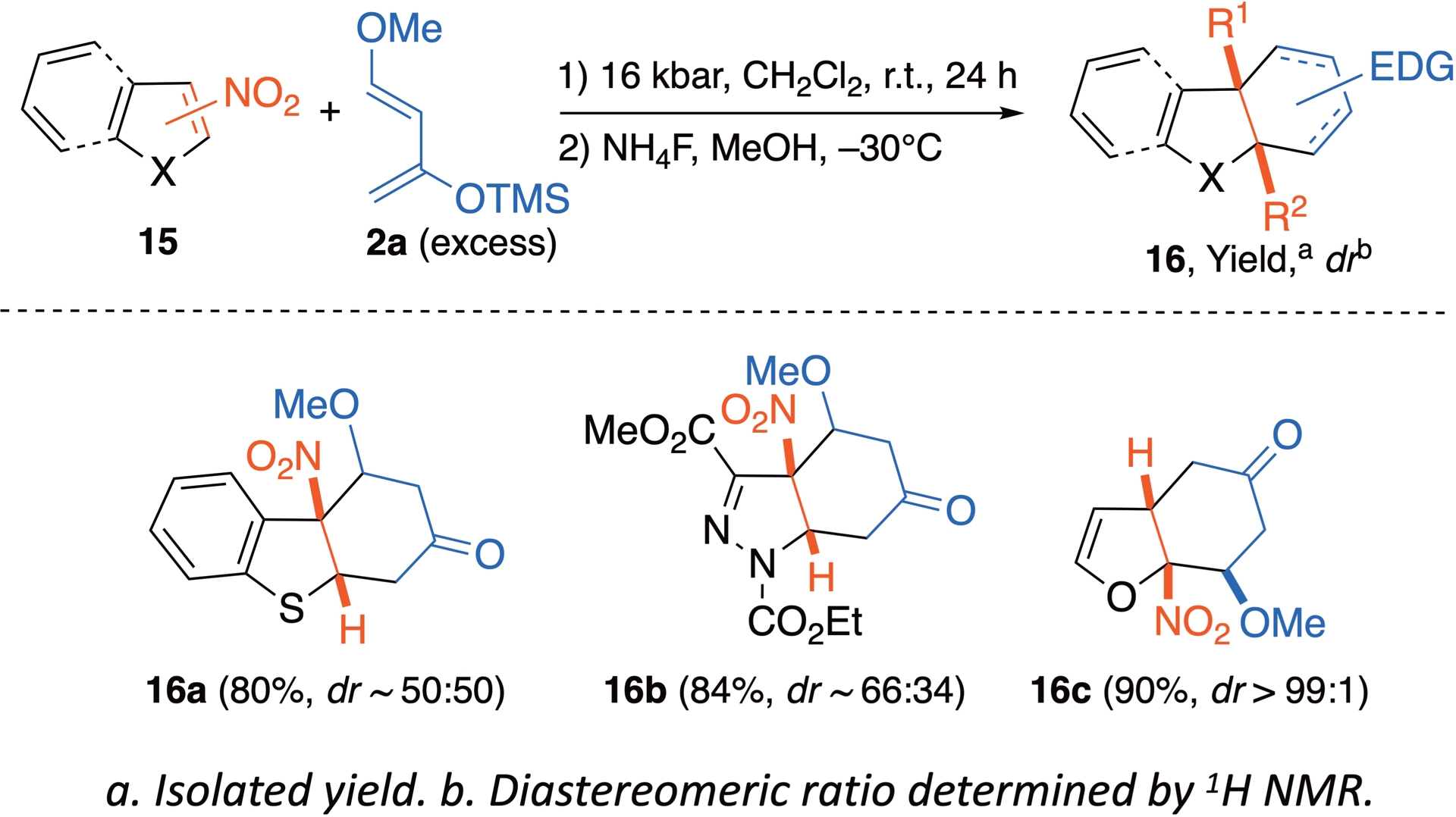
2.5. Six-membered-ring (hetero)aromatics as dienophiles in Diels–Alder cycloadditions
HP–promoted DA reactions may also involve highly aromatic six-membered rings, such as nitroquinolines or nitronaphthalenes and nitrobenzenes in the presence of Danishefsky’s diene 2a (Scheme 12) [34]. The reaction proceeds at r.t. in the absence of any chemical promoter and affords in good yields the hydrolyzed cycloadducts with their three-dimensional structures intact. Here again, the endo/exo diastereoselectivities are low with this diene, because of compensation between the stabilizing secondary orbital interactions favoring the endo approach and steric hindrance favoring the exo one. The diastereoselectivity is slightly enhanced with structurally similar but sterically more hindered dienes, especially those bearing bulky groups at position C1 or C3, but this comes at the expense of reactivity [34]. Notably, when two nitro groups are positioned on the same ring of the original aromatic substrate, two cycloadditions occur, resulting in the formation of polycyclic bis-cycloadducts 19 from 1,3-dinitronaphthalene or 1,3-dinitrobenzene.
Cycloadditions of nitrated six-membered-ring (hetero)aromatics as dienophiles with Danishefsky’s diene 2a under HP activation.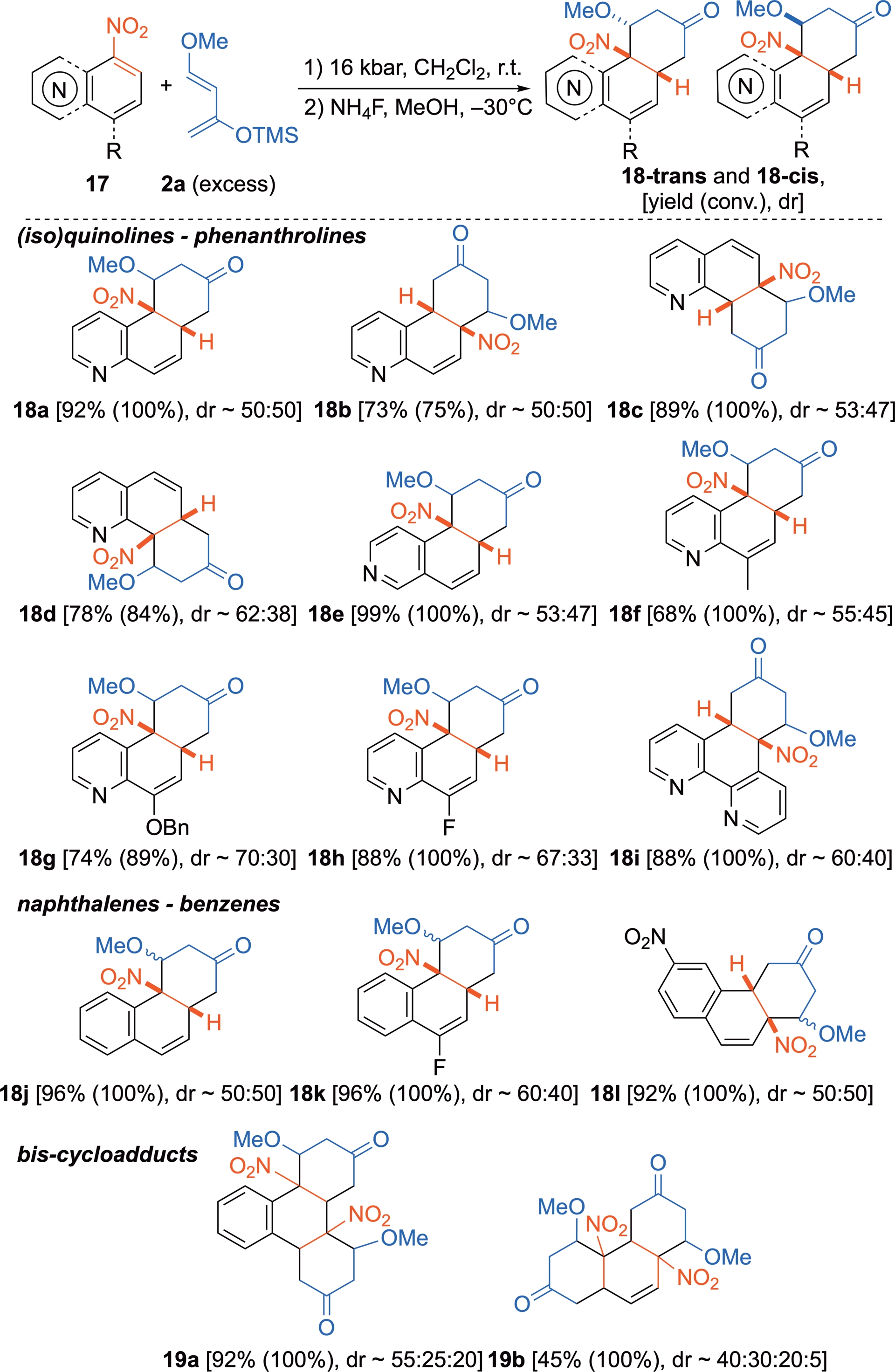
As suggested by DFT computations performed on a model reaction between 1-nitronaphthalene and Danishefsky’s diene 2a, the cycloaddition occurs in a stepwise fashion, the silylenol ether first adding at position C2 of the aromatic. The formation of an elusive zwitterionic primary adduct (PA) is quickly followed by cyclization via a Michael addition to furnish the cycloadduct (CA), prior to hydrolysis of the silyl enol ether moiety (Scheme 13). The initial dearomatization addition is associated with a significantly higher energy barrier than that of the subsequent cyclization step (∼27 versus ∼4 kcal⋅mol−1), making it the rate-determining step. Assuming that the PA undergoes no conformational change before cyclization, the diastereoselectivity of the overall cycloaddition is governed by the stereochemical outcome of the initial addition. Consistent with the experimental data, the small energy difference between the endo and exo transition states (ΔΔG‡ = 0.4 kcal⋅mol−1) accounts for the low diastereoselectivity observed in the resulting cycloadducts.
Energy profiles for the cycloadditions between 1-nitronaphthalene and diene 2a ; trans (endo) approach (red); cis (exo) approach (blue), computed at the (SMD:CH2Cl2) M062X/6-31+G(d,p) level of theory.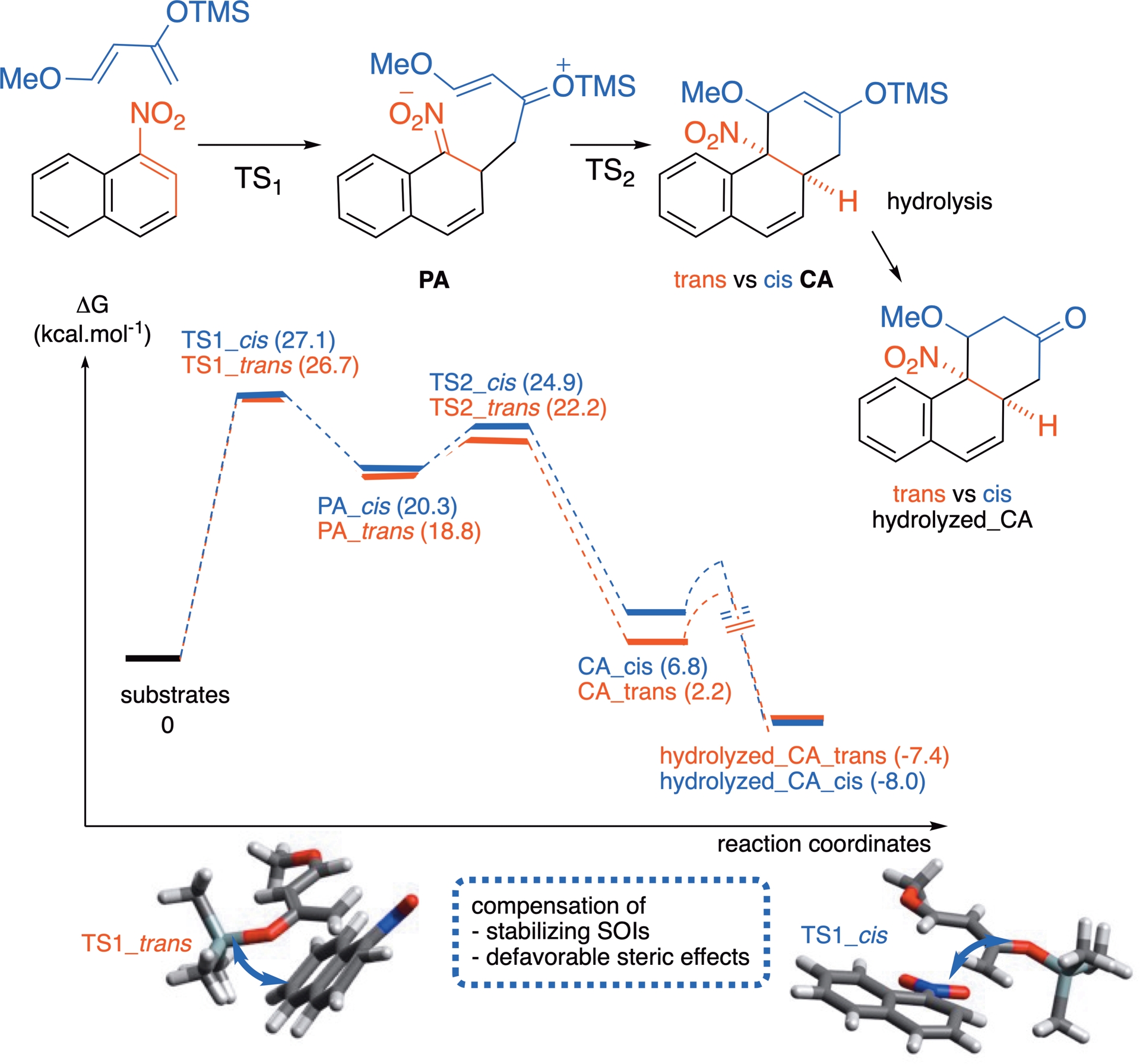
With the less reactive 2-trimethylsilyloxycyclo-hexadiene 2b, the reaction is still possible at r.t. under 16 kbar in the absence of a catalyst (Scheme 14) [29]. As observed with 3-nitroindoles, diastereoselectivity with this cyclic diene is now totally controlled and the exquisite formation of the sole exo-cycloadduct is observed. As suggested by DFT computations, the exo approach is associated with reduced substrate distortion in a late transition state, along with more favorable orbital interactions, likely between the nitro group and the diene moiety. The propensity for the cycloadducts to rearomatize by loss of nitrous acid is higher here, causing a slight lowering of the isolated yields. Here again, when two nitro groups are positioned on the same ring of the initial aromatic compound, two cycloadditions occur, resulting in the formation of the bis-cycloadduct 21 from 1,3-dinitro-naphthalene in a completely diastereoselective way.
Cycloadditions of nitrated six-membered-ring (hetero)aromatics as dienophiles with 2b under HP activation.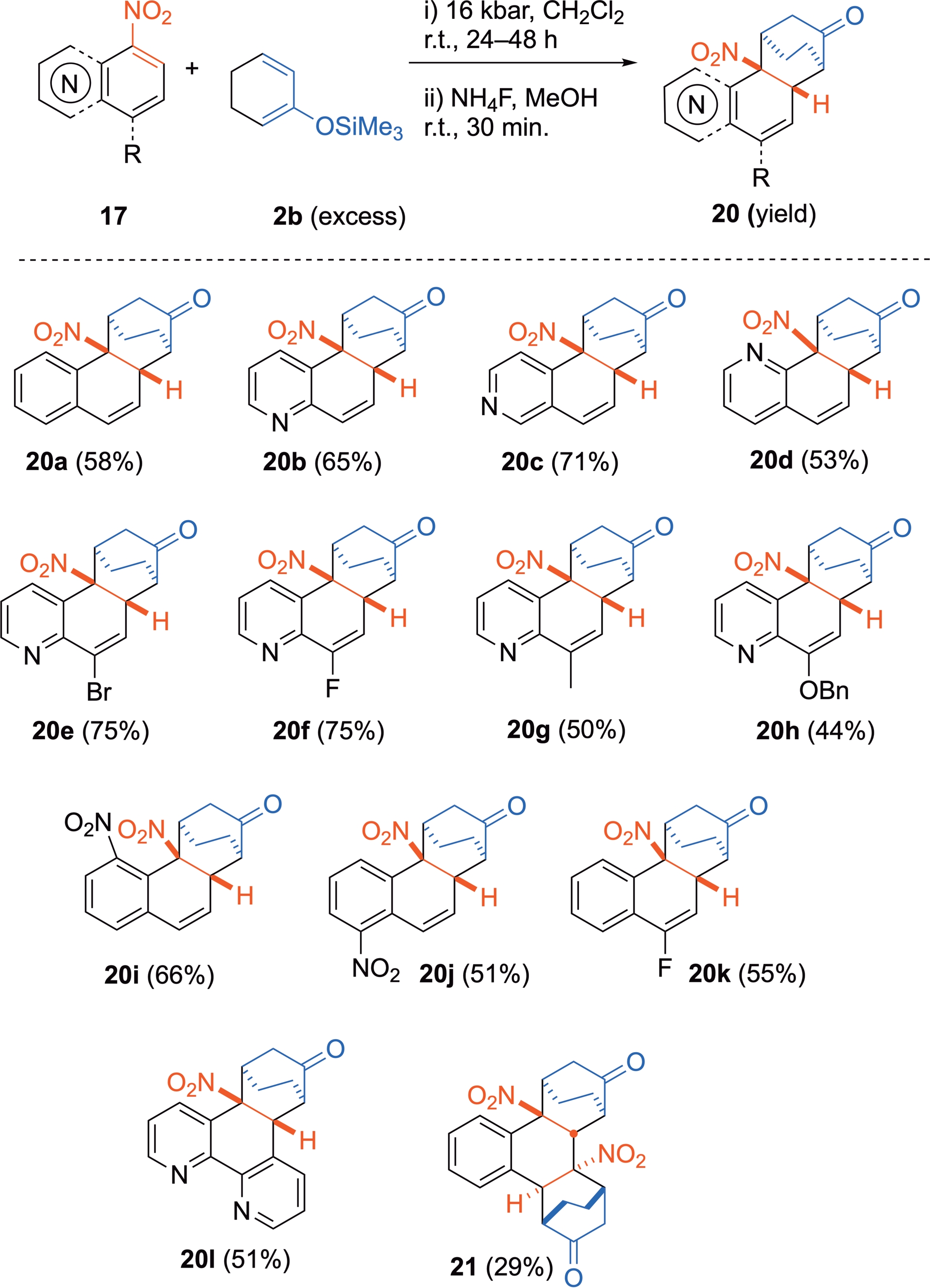
3. Nitroarenes as heterodienes in HP-promoted domino (4+2)-/(3+2)-cycloadditions
Over the years, domino processes have proven to be powerful and efficient strategies for the rapid construction of complex molecules in a minimal number of synthetic steps. Notably, Denmark et al. described a series of (4+2)-/(3+2)-cycloadditions involving nitroalkenes, and these processes have proven highly valuable for the synthesis of a wide range of nitrogen-containing compounds [37]. In these domino transformations, the nitroalkene partner 22 initially acts as a heterodiene in an inverse electron-demand (4+2)-cycloaddition, forming the intermediate nitronate 23. This species then undergoes a subsequent (3+2)-cycloaddition to afford a nitroso-ketal derivative 24 (Scheme 15). Such compounds provide useful synthetic intermediates, giving an easy access to nitrogenated motifs, after selective cleavage of the N–O bonds for instance.
(4+2)-/(3+2)-Domino cycloadditions involving nitroalkenes.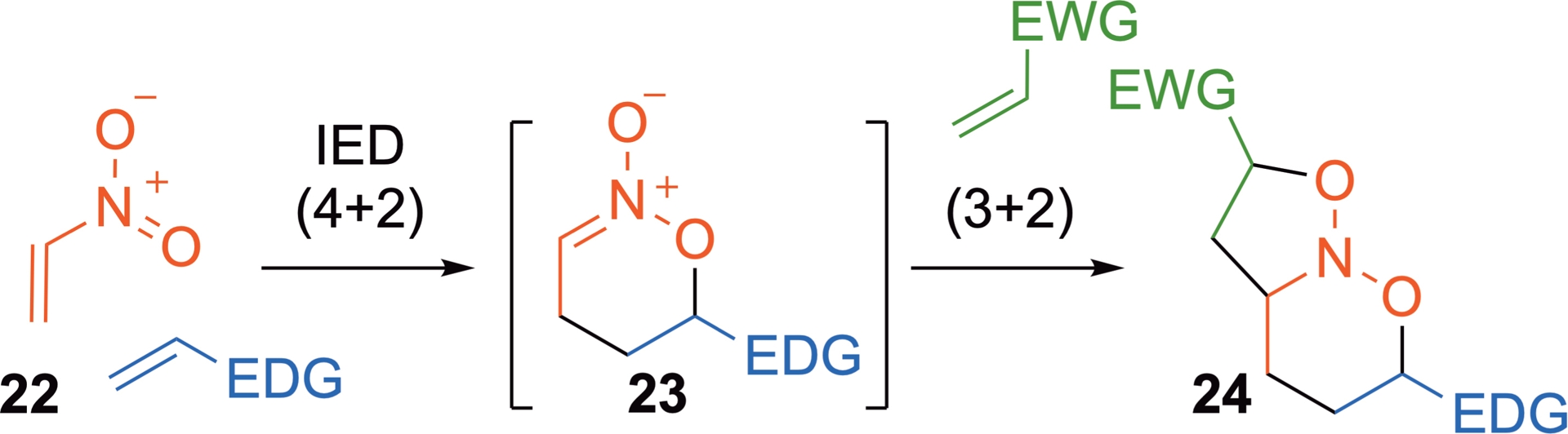
It turns out that both (4+2)- and (3+2)-cycloaddition processes are promoted under HP [4]. Hence, Scheeren et al. have demonstrated that such (4+2)-/(3+2)-reactions using nitrostyrenes as heterodienes can be efficiently promoted under HP [38]. Given the comparable electrophilic character of nitrostyrenes and 3-nitroindoles [20], we sought to explore the participation of these nitro heteroarenes in similar domino processes. If such a synthetic endeavor could be impeded by the aromaticity of the substrate, it would offer the advantage of relying on readily accessible starting materials to generate partially dearomatized polycyclic structures bearing a tetrasubstituted center at the ring junction, a feature generally difficult to control. In the first (4+2)-cycloaddition, the nitroalkene moiety is expected to react via its LUMO as an electron-deficient heterodiene, favoring interaction with an electron-rich dienophile in an inverse electron-demand (4+2)-cycloaddition. Ethyl vinyl ether was selected as a model electron-rich dienophile. In contrast, the nitronate dipole intermediate is expected to react more effectively via its HOMO with an electron-deficient dipolarophile, such as an acrylate. The complementary reactivities of these two alkene species make them excellent candidates for the design of a multicomponent domino process enabling the efficient synthesis of nitroso ketals.
Indeed, the reaction of indole 1a with ethyl vinyl ether 25 and methyl acrylate 26 in dichloromethane at r.t. successfully affords the expected nitroso ketal 27 [39]. Comparing different activation modes shows that HP is the most efficient one, leading to the expected tetracyclic adduct in good yield and with superior diastereoselectivities (Scheme 16). The reaction can also occur with thiourea- or squaramide-based organocatalysts, including chiral ones. However, the enantioselectivities observed with these catalysts are low, likely due to the competing non-catalyzed process [28].
(4+2)/(3+2)-Domino cycloadditions involving 3-nitroindole 1a, ethyl vinyl ether 25, and methylacrylate 26.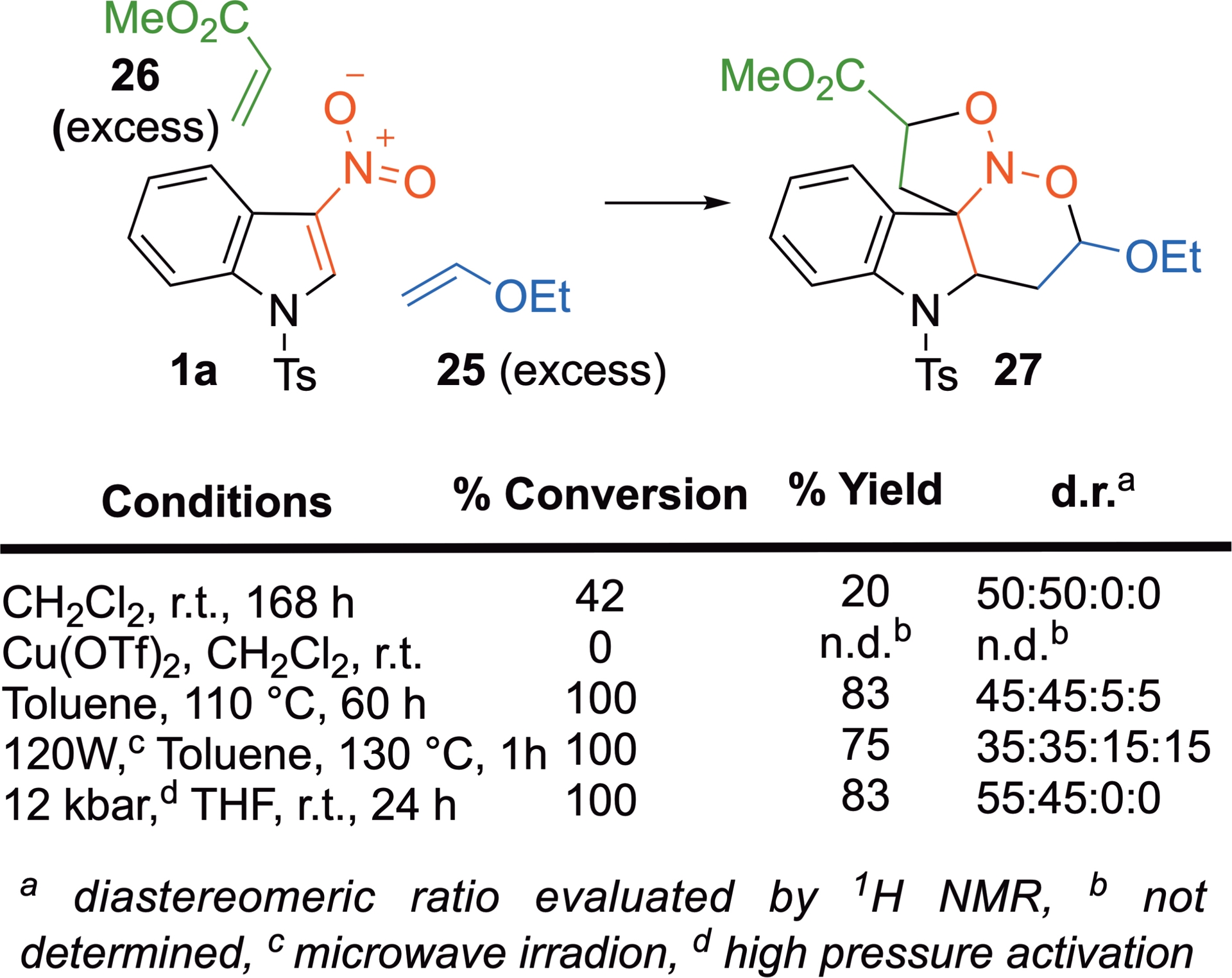
In this process, two of the eight possible diastereomer couples are formed under HP. As shown by X-ray diffraction analyses on the two separated compounds resulting from the same reaction, but involving n-butyl vinyl ether instead of ethyl vinyl ether, the difference stems from the relative stereochemistry at the methoxycarbonyl alpha position. This suggests that the initial (4+2)-cycloaddition proceeds in a purely endo mode, determining the relative configurations of both C7a at the ring junction and C6 alpha to the alkoxy group. A complete facial differentiation for the subsequent (3+2)-process, with an approach of the dipolarophile from the more accessible bottom face sets the relative stereochemistry of quaternary carbon C12b at the ring junction. This second cycloaddition, however, shows little or no endo/exo selectivity for the acrylate approach (Scheme 17) [40].
Proposed pathways for the (4+2)-/(3+2)-sequence accounting for the observed diastereoselectivity.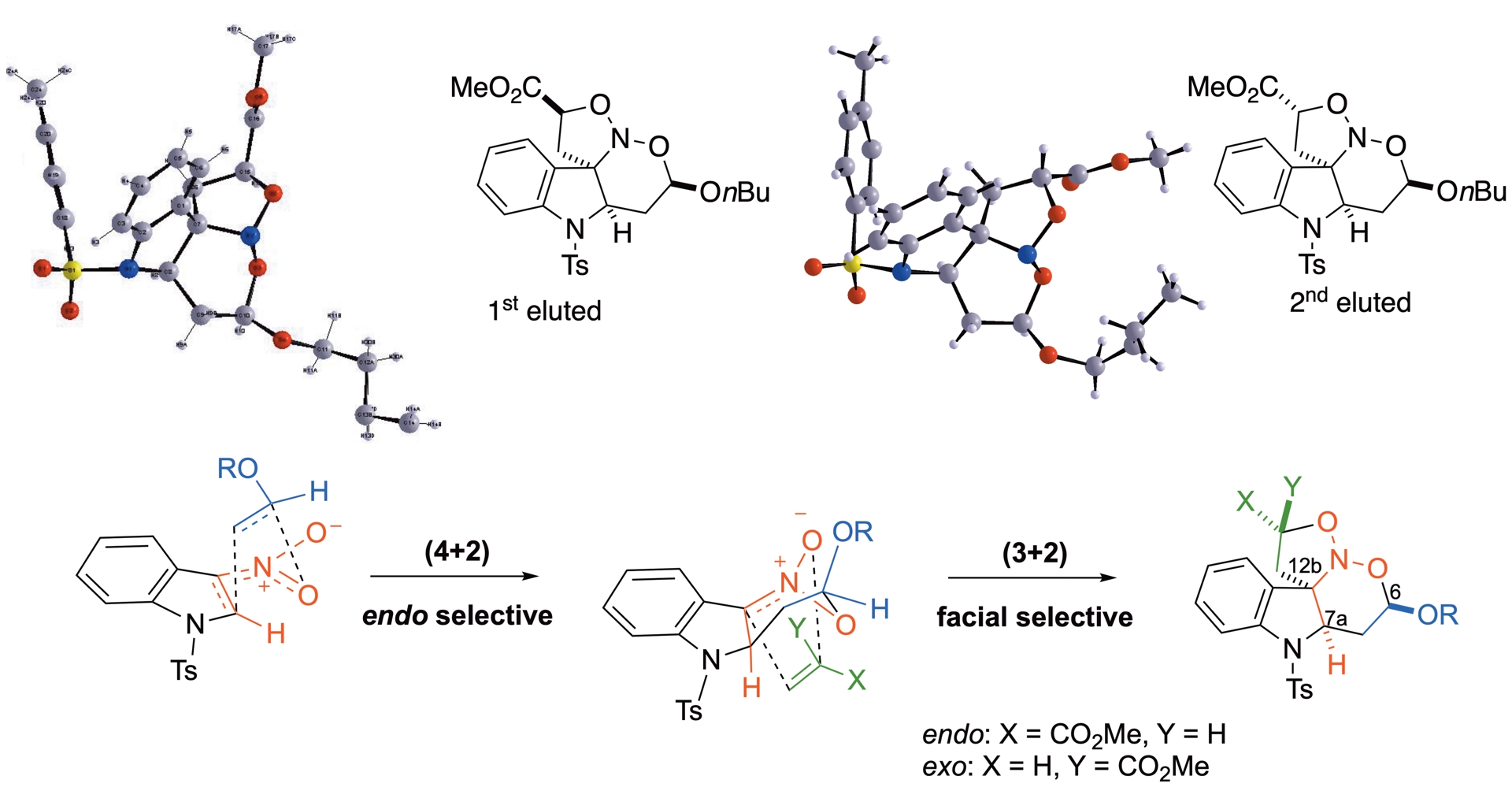
The diastereoselectivity is improved when other dipolarophiles are employed, notably when they bear an aromatic moiety (Scheme 18) [39].
Influence of the dipolarophile on the diastereoselectivity of the (4+2)-/(3+2)-process.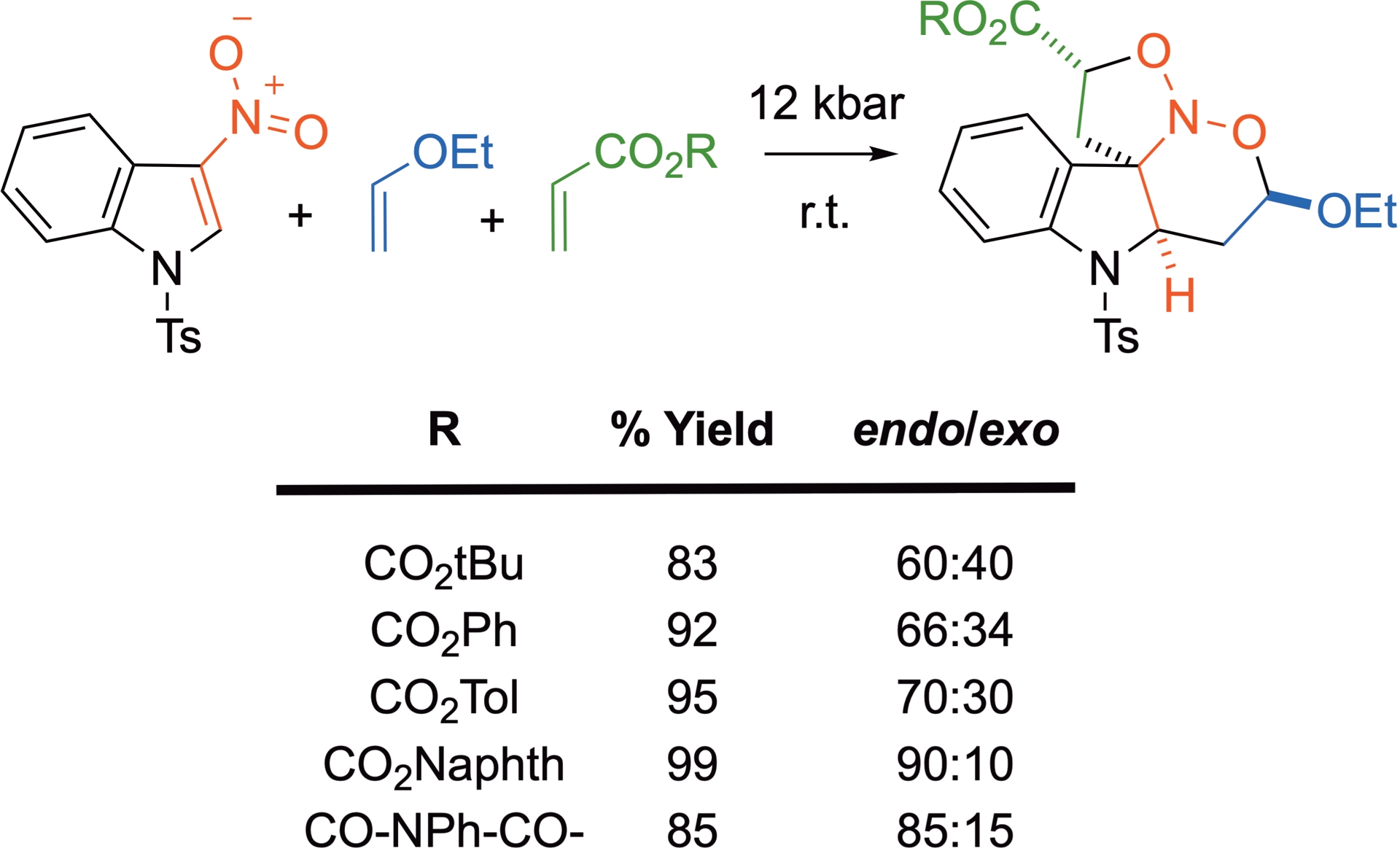
One of the key strengths of this method is the straightforward and efficient conversion of the resulting nitroso ketals. Therefore, the simple catalytic reduction of endo-27 under 15 bar hydrogen using Raney nickel® for 48 h results in the formation of pyrrolizidone 28, isolated in 98% yield, with the original characteristics of the stereogenic centers completely retained (Scheme 19). Most likely, this product results from the sequential cleavage of two N–O bonds, formation, and subsequent reduction of an imine intermediate to construct ring D, followed by lactamization to complete ring C. Similarly, reduction of the diastereomer exo-27 led to the formation of the corresponding diastereomeric tetracyclic lactam.
Reduction of nitroso ketal 27.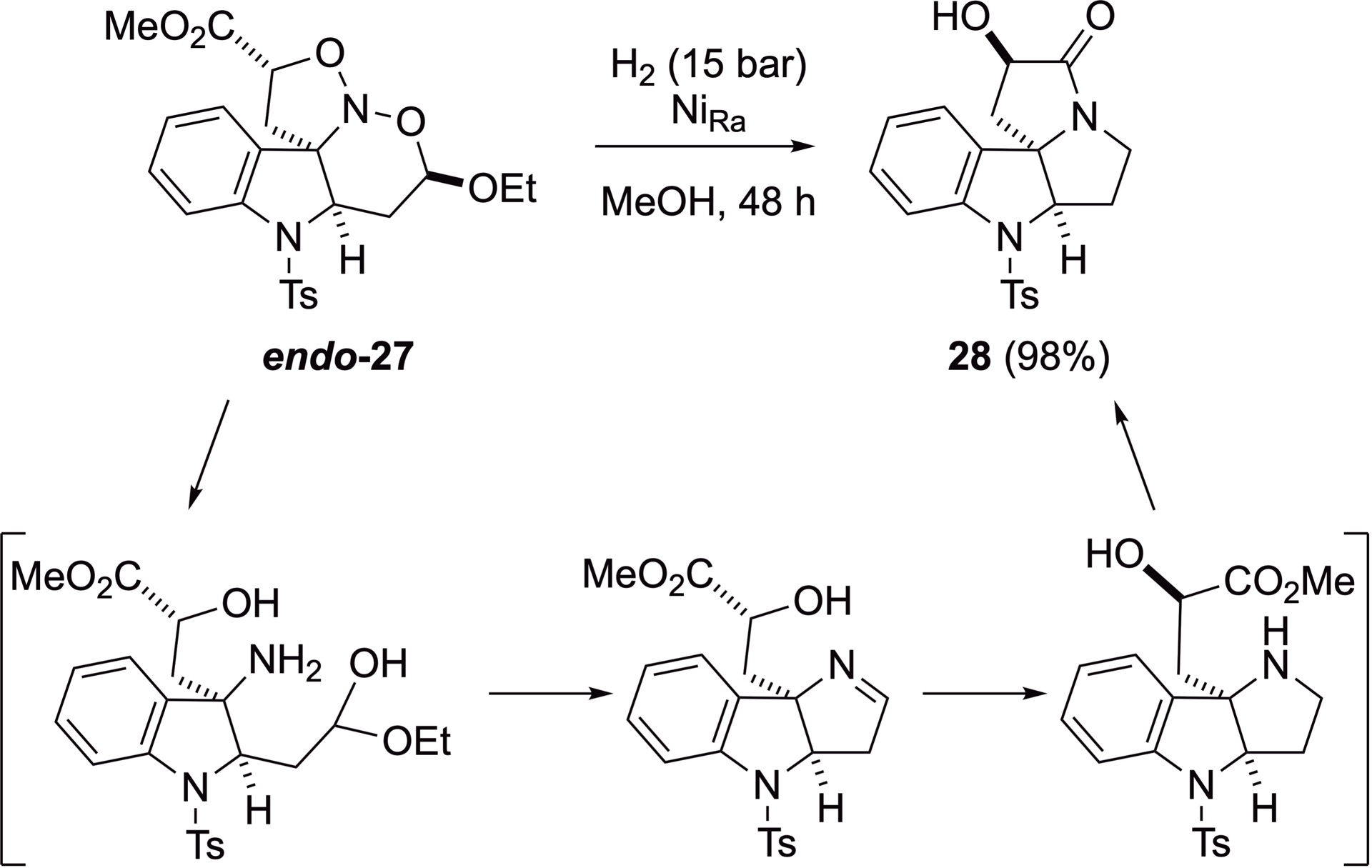
Extension of this process to other ring systems has been achieved with 3-nitropyrrole (Scheme 20). As expected, this more aromatic substrate requires a higher pressure of 16 kbar and heating to 50 °C to react. It leads to the corresponding nitroso ketal obtained in a 25:75 endo/exo ratio and isolated in an overall 81% yield. Note that this smaller heterocycle leads to a stereoselectivity different from that above. Here again, the nitroso ketal can be efficiently reduced into the corresponding tricyclic compound under similar hydrogenation conditions and the tricyclic amine is obtained in 68% isolated yield.
(4+2)/(3+2)-Domino cycloadditions involving 3-nitropyrrole 11, ethyl vinyl ether 25, and methyl acrylate 26 and subsequent reduction of nitroso ketal 29.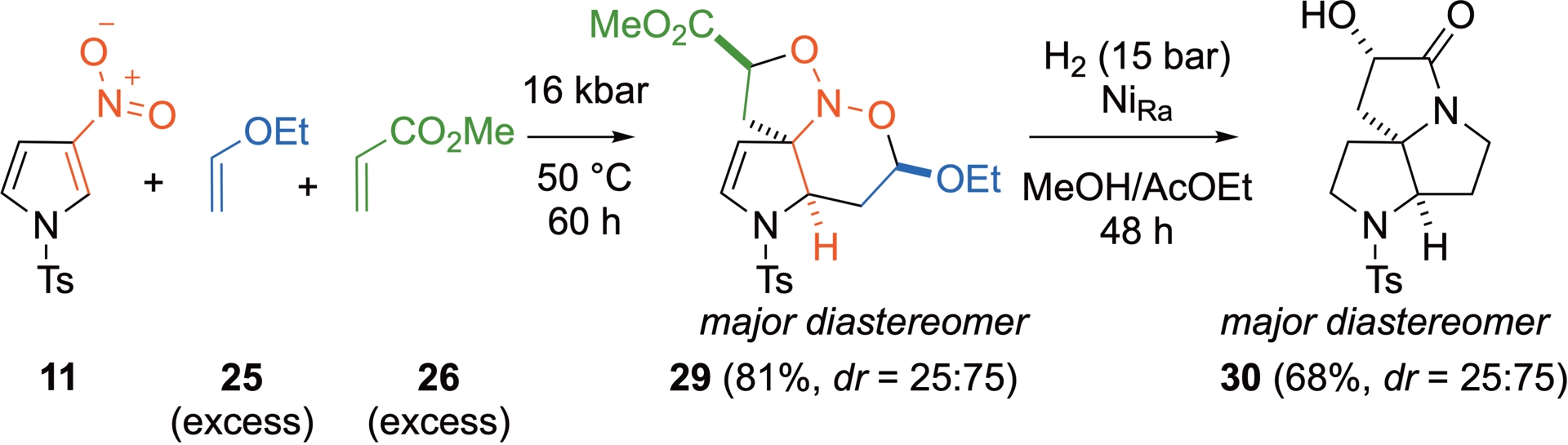
Although these tetra- or tricyclic structures are not commonly found in natural products, they certainly exhibit functional and three-dimensional topological characteristics that could contribute to the development of novel pharmacological scaffolds.
4. Conclusion
In conclusion, dearomative cycloadditions involving electron-deficient (hetero)arenes are strongly favored by high-pressure conditions. These easily available aromatic compounds can act as dienophiles or heterodienes, in reaction with electron-rich dienes or dienophiles, providing an efficient access to the corresponding three-dimensional polycyclic adducts. High-pressure conditions primarily promote these reactions by overcoming the high energy barrier required to disrupt aromaticity, thereby enabling more efficient and selective transformations. High pressure favors forward cycloadditions over retroreactions, which is a key point since dearomatization is often endergonic and reversible under normal thermal conditions. High pressure can indeed shift equilibria toward product formation since cycloadditions are characterized by highly negative activation volumes, associated to a decrease in volume from reactants to the transition state and products. They also stabilize tightly packed transition states and intermediates. High pressure (HP) simultaneously disfavors the retrocycloadditions involving an elimination reaction, which are characterized by a positive activation volume. Because these dearomative cycloadditions proceed at lower temperatures under HP, the side reactions and decomposition processes are limited. In many cases, HP conditions also help improving selectivity and stereocontrol as the pressurized confined environment can favor specific diastereomers or regioisomers, thus improving the stereochemical outcome of cycloadditions. In summary, acting as a physical promoter, HP facilitates cycloadditions that disrupt aromaticity and enables the access to strained or unusual polycyclic structures. In doing so, HP helps in building complex three-dimensional polycyclic scaffolds with a high sp3 character that are important in pharmaceutics and materials science.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization from this article, and have declared no affiliations other than their research organizations.