



 CC-BY 4.0
CC-BY 4.0
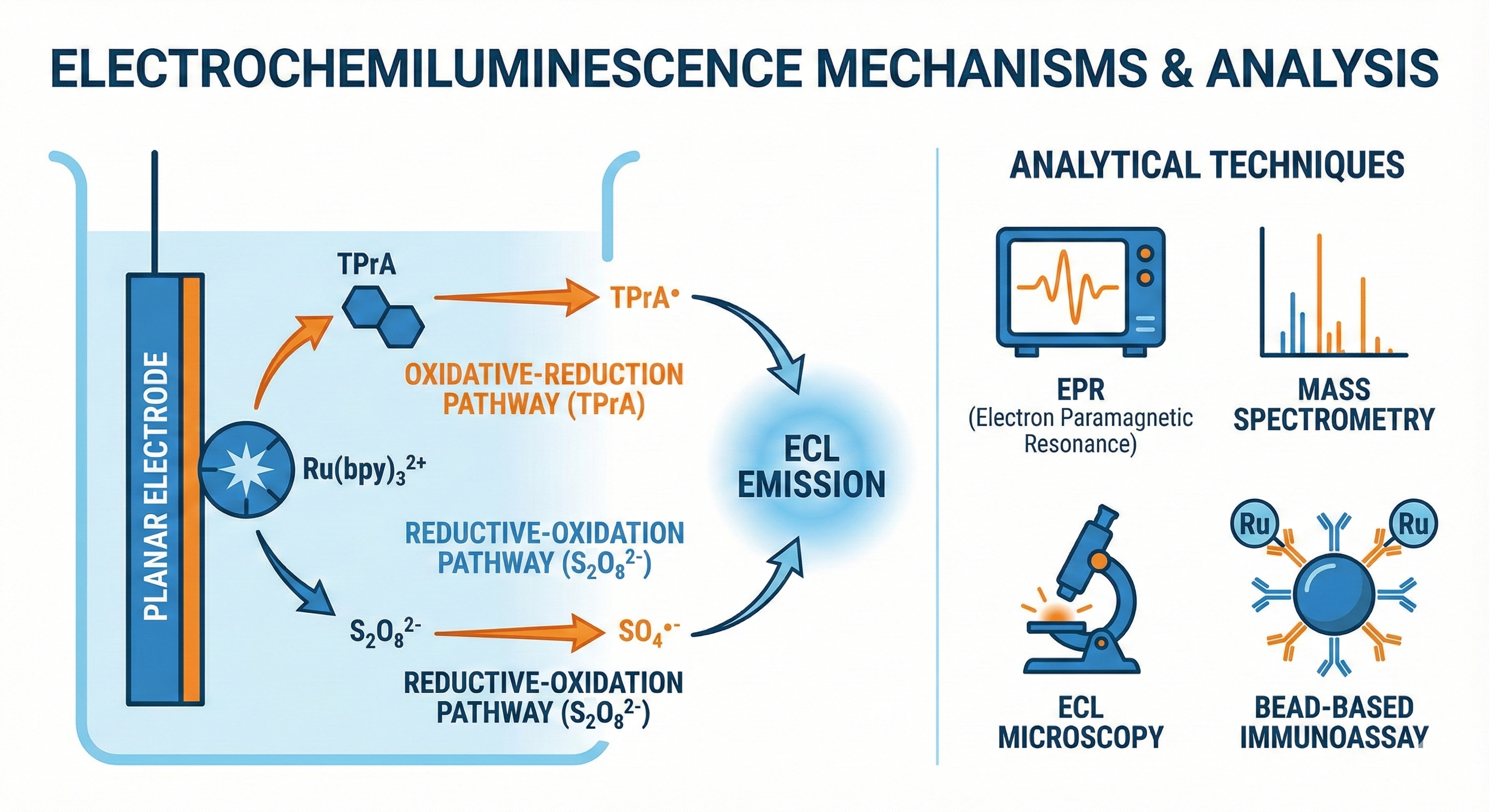
1. Introduction
Electrochemiluminescence (ECL), the production of light from electrochemically generated species, has transitioned from a laboratory curiosity to a powerful analytical technique with widespread commercial success, particularly in clinical diagnostics and bioassays [1]. This remarkable evolution was largely contingent on a pivotal mechanistic shift away from the so-called “annihilation” pathway [2]. In the annihilation mechanism, radical cations and anions of a luminophore, generated by applying alternating oxidative and reductive potentials, react to produce an excited state. While effective in aprotic organic solvents with wide potential windows, this approach is largely unfeasible in the aqueous environments required for most biological applications, where the electrochemical splitting of water severely restricts the accessible potential range [3].
The introduction of coreactants elegantly circumvented this limitation, enabling robust ECL generation from a single potential step or scan in one direction [4]. A coreactant is a species that, upon electrochemical activation, initiates a cascade of chemical reactions that culminates in the formation of the luminophore’s excited state. This innovation unlocked the full potential of ECL for bioanalysis, offering near-zero background signal, high sensitivity, and excellent spatiotemporal control [5]. At the heart of this process lie the highly reactive transient radical intermediates generated from the coreactants. The properties, reactivity, and fate of these radicals are the ultimate determinants of the efficiency, spatial distribution, and analytical performance of the entire ECL system [6].
Coreactant pathways are broadly categorized into two dominant mechanisms. The “oxidative-reduction” pathway is typified by tertiary amines, most notably TPrA. In this mechanism, the coreactant is electrochemically oxidized, and through subsequent chemical steps, generates a powerful reducing radical intermediate that drives the formation of the luminophore’s excited state [7]. Conversely, the “reductive-oxidation” pathway, exemplified by the persulfate anion (S2O82−), involves the electrochemical reduction of the coreactant to produce a potent oxidizing radical intermediate, which then reacts with the reduced form of the luminophore to generate light [8].
A deep and nuanced understanding of the generation, stability, diffusion, and reactivity of these coreactant-derived radical intermediates, such as the TPrA radical cation (TPrA•+), the α-aminoalkyl radical (TPrA•), and the sulfate radical anion (SO4•−), is therefore not merely an academic exercise. It is the fundamental prerequisite for rationally designing more efficient luminophores [9], developing novel coreactants with tailored properties, and optimizing assay conditions to push the boundaries of analytical sensitivity. This review posits that these transient species are the linchpin of coreactant ECL [10]. We will critically examine the intricate and often-debated mechanistic pathways that govern their formation, survey the sophisticated analytical strategies employed to detect and characterize these transient species, and analyze how their fundamental chemical properties directly translate into the bioanalytical performance that has made ECL an indispensable tool in modern science [11].
2. Mechanistic pathways and controversies in radical intermediate generation
The apparent simplicity of coreactant ECL, where applying a potential to a solution of luminophore and coreactant produces light, belies a sophisticated and multifaceted reaction network. The generation of radical intermediates is not a monolithic process but a series of competing and often condition-dependent pathways. This section dissects the mechanistic details and ongoing controversies surrounding the two most important coreactant systems, TPrA and persulfate, providing a critical analysis of the evidence that shapes our current understanding.
The mechanism of coreactant ECL for [Ru(bpy)3]2+ and TPrA (reproduced from Ref. [12] with permission from ACS, copyright 2002).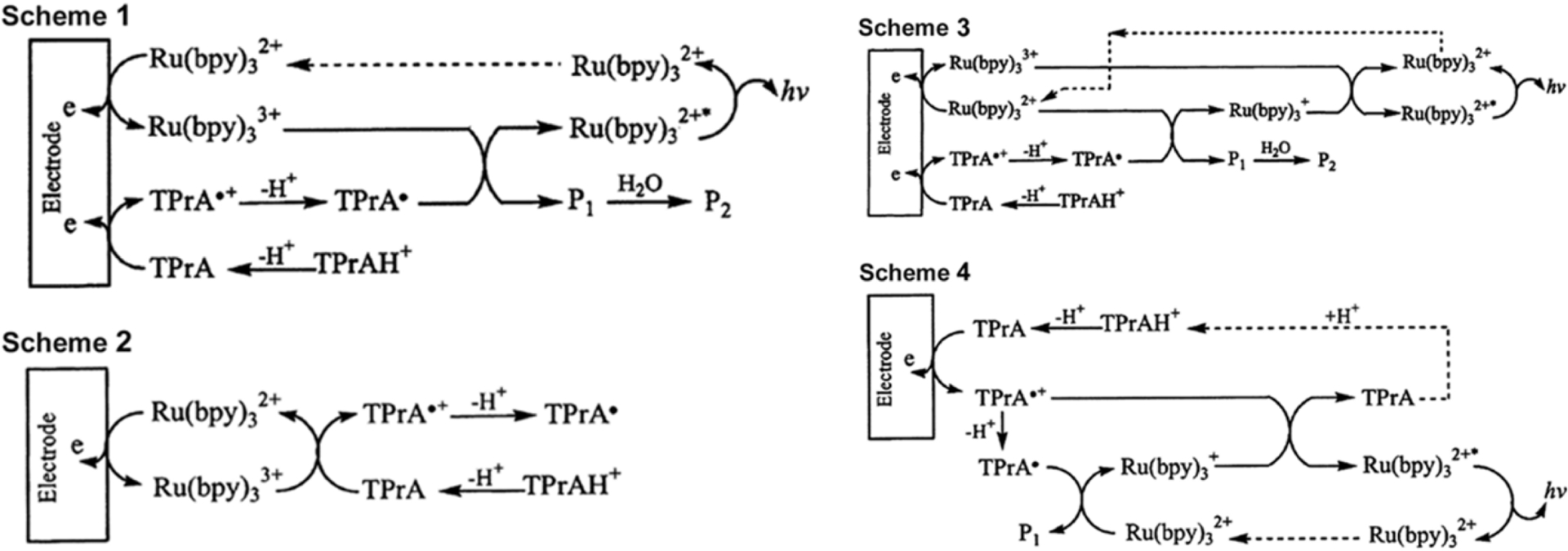
2.1. The tri-n-propylamine (TPrA) system
The tris(2,2′-bipyridine)ruthenium(II) [Ru(bpy)3]2+/TPrA system (Figure 1) is the undisputed workhorse of ECL, forming the basis of virtually all commercial immunoassay platforms. Its widespread use, however, has only intensified the scrutiny of its complex mechanism, which remains a subject of active research and debate [13]. The process begins with two foundational steps: the one-electron oxidation of TPrA at the electrode surface to form its radical cation, TPrA•+, followed by the rapid deprotonation of this species to yield the neutral, highly reducing α-aminoalkyl radical, TPrA•. The reactions are as follows:
| \begin {eqnarray} &\mathrm {TPrA} - \mathrm {e}^- {\rightarrow } \mathrm {TPrA}{\bullet }^+ \label {eq1}\end {eqnarray} | (1) |
| \begin {eqnarray} &\mathrm {TPrA}{\bullet }^+ {\rightarrow } \mathrm {TPrA}\bullet + \mathrm {H}^{+} \label {eq2} \end {eqnarray} | (2) |
The TPrA•+ intermediate is notably short-lived, with an estimated half-life of approximately 0.2 ms in neutral aqueous solution. This transient stability is a critical parameter, as it dictates that subsequent reactions involving this species are confined to a thin diffusion layer, typically on the micrometer scale, near the electrode surface [11, 14]. From this common starting point, several competing pathways can lead to the generation of the excited state.
A central and long-standing controversy in the field revolves around the initial oxidation events: the “direct oxidation” versus the “catalytic” route. The direct oxidation route, also known as the oxidative-reduction pathway, posits that at a sufficiently high anodic potential, both the luminophore and TPrA are oxidized directly at the electrode surface [15]. The resulting oxidized luminophore, then reacts with the highly reducing TPrA• radical to produce the excited state:
| \begin {eqnarray} &[\mathrm {Ru}(\mathrm {bpy})_{3}]^{2+}- \mathrm {e}^- \rightarrow [\mathrm {Ru}(\mathrm {bpy})_{3}]^{3+} \label {eq3}\end {eqnarray} | (3) |
| \begin {eqnarray} &{}[\mathrm {Ru}(\mathrm {bpy})_{3}]^{3+} + \mathrm {TPrA} \bullet \rightarrow {[\mathrm {Ru}(\mathrm {bpy})_{3}]^{2+}}^\ast + \mbox { Products} \nonumber \\ \label {eq4} \end {eqnarray} | (4) |
In contrast, the catalytic route proposes that only the luminophore is electrochemically oxidized. This species then acts as a homogeneous oxidant, diffusing into the solution to react with a neutral TPrA molecule, thereby generating the TPrA•+ radical cation in the diffusion layer, which then proceeds through deprotonation (Equation (2)) as usual:
| \begin {equation}\label {eq5} [\mathrm {Ru}(\mathrm {bpy})_{3}]^{3+} + \mathrm {TPrA} \rightarrow [\mathrm {Ru}(\mathrm {bpy})_{3}]^{2+} + \mathrm {TPrA}{\bullet }^{+} \end {equation} | (5) |
Key physicochemical properties of primary ECL radical intermediates
| Radical species | Generation method | Approximate formal/estimated redox potential | Estimated half-life/stability | Primary role in ECL | References |
|---|---|---|---|---|---|
| TPrA•+ | Electrochemical oxidation of TPrA• | E°est(TPrA•+/TPrA•) ≈− 1.7 V vs. SCE (aqueous; commonly used estimate for the reducing power of TPrA•) | ∼0.2 ms (aqueous) | Oxidant | [12, 16, 17] |
| SO4•− | Electrochemical reduction of S2O82− | E°(SO4•−/SO42−) ≈ +2.43 V vs. NHE (often quoted ∼ + 2.5–3.1 V in reviews; condition/speciation dependent) | 30–40 μs | Strong oxidant | [18, 19, 20] |
Crucially, the dominance of one pathway over the other is not an intrinsic property of the system but is dictated by the experimental conditions [21]. A wealth of evidence demonstrates that the electrode material acts as a primary “mechanistic switch”. At glassy carbon (GC) electrodes, the direct oxidation of TPrA is kinetically favored, and the direct oxidation route is generally considered to be predominant [22]. However, at noble metal electrodes like platinum (Pt) and gold (Au), the formation of passivating surface oxide layers under the positive potentials required for ECL significantly inhibits the direct heterogeneous oxidation of TPrA [23]. Under these conditions, the catalytic route becomes more significant, particularly at higher concentrations of the luminophore, which can more effectively act as a mediator for TPrA oxidation [24]. This mechanistic difference directly explains the frequently observed phenomenon of lower ECL intensities on Pt and Au electrodes compared to carbon-based electrodes for the same system [25]. Furthermore, the concentration ratio of luminophore to coreactant is a key modulator; high [[Ru(bpy)3]2+]/[TPrA] ratios strongly favor the catalytic pathway, as the abundance of electrogenerated [Ru(bpy)3]3+ provides more opportunities for homogeneous oxidation of TPrA [26]. The physicochemical characteristics of these radical intermediates are summarized in Table 1, highlighting their formation pathways, redox potentials, and stability regimes. These parameters dictate not only the efficiency but also the spatial and temporal domains of light generation in ECL [27]. For instance, the sub-millisecond lifetime of TPrA•+ confines its reactivity to the near-electrode diffusion layer, whereas the strong oxidizing potential of SO4•− in persulfate systems extends reaction zones farther into solution [28]. Comparing these radicals underscores the delicate kinetic balance between electron transfer, diffusion, and radical decay that governs overall ECL performance.
The mechanistic picture was further refined by a seminal 2002 paper from Bard and coworkers, which reconciled several anomalous observations by proposing a “new route”. This pathway involves the reaction between the TPrA radical cation, TPrA•+, and the reduced form of the luminophore, [Ru(bpy)3]+. The reduced luminophore is generated via a competing reaction pathway where the potent TPrA• radical reduces a ground-state [Ru(bpy)3]2+ molecule:
| \begin {eqnarray} &[\mathrm {Ru}(\mathrm {bpy})_{3}]^{2+} + \mathrm {TPrA}\bullet \rightarrow [\mathrm {Ru}(\mathrm {bpy})_{3}]^{+} + \mbox {Products}\qquad \label {eq6}\end {eqnarray} | (6) |
| \begin {eqnarray} &[\mathrm {Ru}(\mathrm {bpy})_{3}]^{+} + \mathrm {TPrA}{\bullet }^+ \rightarrow [\mathrm {Ru}(\mathrm {bpy})_{3}]^{2+*} + \mathrm {TPrA}\qquad \label {eq7} \end {eqnarray} | (7) |
This mechanism is particularly vital to explain the oxidative (TPrA•+) excitation ECL phenomenon, where light is generated at potentials positive enough to oxidize TPrA but insufficient to directly oxidize [Ru(bpy)3]2+ (typically <1.0 V vs. saturated calomel electrode [SCE]) [17]. In this scenario, direct generation of [Ru(bpy)3]3+ at the electrode is impossible. Instead, only TPrA is oxidized, creating a flux of both TPrA•+ and TPrA• radicals diffusing away from the electrode. The subsequent homogeneous reactions (6) and (7) are then solely responsible for generating the excited state. This oxidative (TPrA•+) excitation mechanism is of paramount importance in the context of modern bioassays, especially bead-based immunoassays [29]. In these formats, the luminophore is conjugated to an antibody and immobilized on the surface of a microbead, which is spatially separated from the electrode surface. The ability of the electrogenerated TPrA radicals to diffuse over micrometer-scale distances to reach and react with the luminophore labels on the bead is the fundamental principle that enables these highly sensitive assays to function [30].
Comparison of mechanistic pathways in the [Ru(bpy)3]2+/TPrA system
| Pathway name | Required electrode potential (Eapp) | Key reactants at electrode | Critical homogeneous reactions | Dominant conditions/applications |
|---|---|---|---|---|
| Direct oxidation | Eapp > E°(Ru2+/3+) | [Ru(bpy)3]2+, TPrA | [Ru(bpy)3]3++ TPrA•→[Ru(bpy)3]2+∗ | GC electrodes; Low [[Ru(bpy)3]2+]/[TPrA] ratio [31] |
| Catalytic | Eapp > E°(Ru2+/3+) | [Ru(bpy)3]2+ | [Ru(bpy)3]3++ TPrA → [Ru(bpy)3]2++ TPrA•+ | Pt, Au electrodes; High [[Ru(bpy)3]2+]/[TPrA] ratio [32, 33, 34] |
| Oxidative (TPrA•+) excitation | E°(TPrA/TPrA•+) < Eapp < E°(Ru2+/3+) | TPrA only | [Ru(bpy)3]++ TPrA•+→ [Ru(bpy)3]2+∗ | Bead-based immunoassays; Spatially separated luminophore [35] |
The comparative features of the three principal mechanistic routes are summarized in Table 2, which delineates their operative potentials, key reactants, and dominant experimental regimes. This tabulation underscores that the boundary between “direct”, “catalytic”, and “oxidative (TPrA•+) excitation” mechanisms is not rigid but dynamically dictated by the electrode surface chemistry and the redox landscape [36]. For instance, while both the direct and catalytic pathways operate above the Ru2+/3+ oxidation potential, they diverge sharply in the identity of the electroactive species and in the locus of radical generation (heterogeneous vs. homogeneous) [37]. The oxidative (TPrA•+) excitation regime, operating at potentials below the Ru oxidation potential, introduces a spatial decoupling between charge injection and light emission, enabling photon generation far from the electrode interface [38]. This mechanistic diversity provides an essential framework for rational assay design: selecting electrode materials that favor TPrA oxidation enhances ECL efficiency, while exploiting the oxidative (TPrA•+) excitation pathway is indispensable for bead- and nanoparticle-based formats where diffusion-mediated radical transfer drives excitation [39].
2.2. The persulfate (S2O82−) system: radical vs. non-radical pathways
Persulfate, specifically the peroxydisulfate anion (S2O82−), is the archetypal coreactant for the reductive-oxidation pathway [40]. This pathway is particularly useful for luminophores that are more readily reduced than oxidized and has found extensive use not only in ECL but also in the broader field of advanced oxidation processes (AOPs) for environmental remediation, providing a rich literature from which to draw mechanistic insights.
The canonical mechanism involves the one-electron electrochemical reduction of S2O82− at the cathode. This reaction cleaves the peroxide bond to generate the highly oxidizing sulfate radical anion (SO4•−) and a stable sulfate ion SO42−.
| \begin {equation}\label {eq8} \mathrm {S}_{2}\mathrm {O}_{8}^{2-} + \mathrm {e}^- \rightarrow \mathrm {SO}_{4}^{2-} + \mathrm {SO}_{4}{\bullet }^- \end {equation} | (8) |
The SO4•− radical is a powerful one-electron oxidant, with a standard redox potential reported in the range of +2.5 to +3.1 V vs. normal hydrogen electrode (NHE), comparable to that of the hydroxyl radical [18]. In the context of ECL, this potent radical reacts with the electrochemically generated reduced form of the luminophore (L•−) in a highly exergonic electron transfer reaction to produce the light-emitting excited state, L∗:
| \begin {eqnarray} &\mathrm {L} + \mathrm {e}^- \rightarrow \mathrm {L}{\bullet }^- \label {eq9}\end {eqnarray} | (9) |
| \begin {eqnarray} &\mathrm {L}{\bullet }^- + \mathrm {SO}_{4}{\bullet }^- \rightarrow \mathrm {L}^* + \mathrm {SO}_{4}^{2-} \label {eq10} \end {eqnarray} | (10) |
An important distinction from the TPrA system, highlighted by the work of Paolucci and Valenti, is that a heterogeneous ECL mechanism analogous to the oxidative (TPrA•+) excitation route is not feasible with persulfate [41]. Because the SO4•− radical acts as an oxidant, the luminophore must first be reduced to L•− for the light-producing reaction to occur [42]. Therefore, in persulfate systems, both the luminophore and the coreactant must be able to interact with the electrode surface, unlike in bead-based TPrA assays where the luminophore can be spatially separated from the electrode.
While the SO4•−-mediated pathway is well-established, a significant controversy has emerged, largely from the AOPs field, regarding the potential role of non-radical pathways in persulfate activation. Proponents of non-radical mechanisms suggest that oxidation can occur via pathways that do not involve free sulfate radicals [43]. These proposed mechanisms include direct electron transfer from a substrate to the persulfate molecule, often mediated by a catalyst surface, or the generation of other reactive oxygen species such as singlet oxygen (1O2) [44]. Evidence for these pathways often relies on chemical quenching experiments, where the addition of common radical scavengers like methanol or tert-butanol (which react rapidly with SO4•− and •OH) fails to completely inhibit the degradation of a target pollutant.
The focus of this controversy lies in the difficulty of unequivocally proving the absence of radicals and the potential for misinterpretation of indirect evidence. The boundary between radical and non-radical processes can be ambiguous, and it is likely that multiple pathways coexist, with their relative contributions being highly dependent on factors such as pH, the nature of the catalyst, and the electronic properties of the substrate [45]. For example, at high pH, SO4•− can react with OH− to generate the hydroxyl radical (•OH), adding another layer of complexity to the system. While this debate is most active in the context of pollutant degradation, its implications for persulfate-based ECL are significant [46]. If non-radical pathways or alternative radical species are major contributors to the oxidative chemistry, they could represent untapped avenues for designing new and more efficient ECL systems.
2.3. A broader perspective: alternative and bio-derived coreactants
The search for coreactants beyond TPrA and persulfate is a vibrant area of research, driven by the desire for lower toxicity, improved biocompatibility, higher efficiency, and novel mechanistic pathways. A variety of aliphatic amines and related compounds have been explored as alternatives to TPrA. For example, 2-(dibutylamino)ethanol (DBAE) was introduced as a less toxic and less volatile alternative. Biological buffers containing tertiary amine moieties, such as HEPES and BIS-TRIS, have also been shown to function as effective coreactants, offering the potential for simplified assay formulations where the buffer, electrolyte, and coreactant are the same molecule [47]. Comparative studies of these analogs reveal a complex interplay between the coreactant’s structure, the stability of its radical intermediates, and the resulting ECL efficiency [48]. Wang and coworkers demonstrated that the ECL distance (related to radical lifetime) and reactivity of the coreactant co-determine the sensitivity of bead-based immunoassays, and found that BIS-TRIS offered a superior balance of these properties compared to TPrA, leading to a significant enhancement in sensitivity [49]. In addition, reduced nicotinamide adenine dinucleotide (NADH), a ubiquitous endogenous redox cofactor, should be considered a biocompatible coreactant in [Ru(bpy)3]2+-based ECL: it is recognized among established [Ru(bpy)3]2+ coreactants, can be generated enzymatically in situ from NAD+ in dehydrogenase-coupled assays to drive ECL without adding exogenous amine reagents, and has recently been exploited as an efficient endogenous coreactant for ECL sensing and imaging in biological systems [50, 51, 52].
Reaction mechanism of electrochemiluminescence generation from [Ru(bpy)3]2+ on BDD electrode (platinum spiral and Ag/AgCl as working and counter/reference electrodes) with sulfate ions (reproduced from Ref. [53] with permission from ACS, copyright 2016).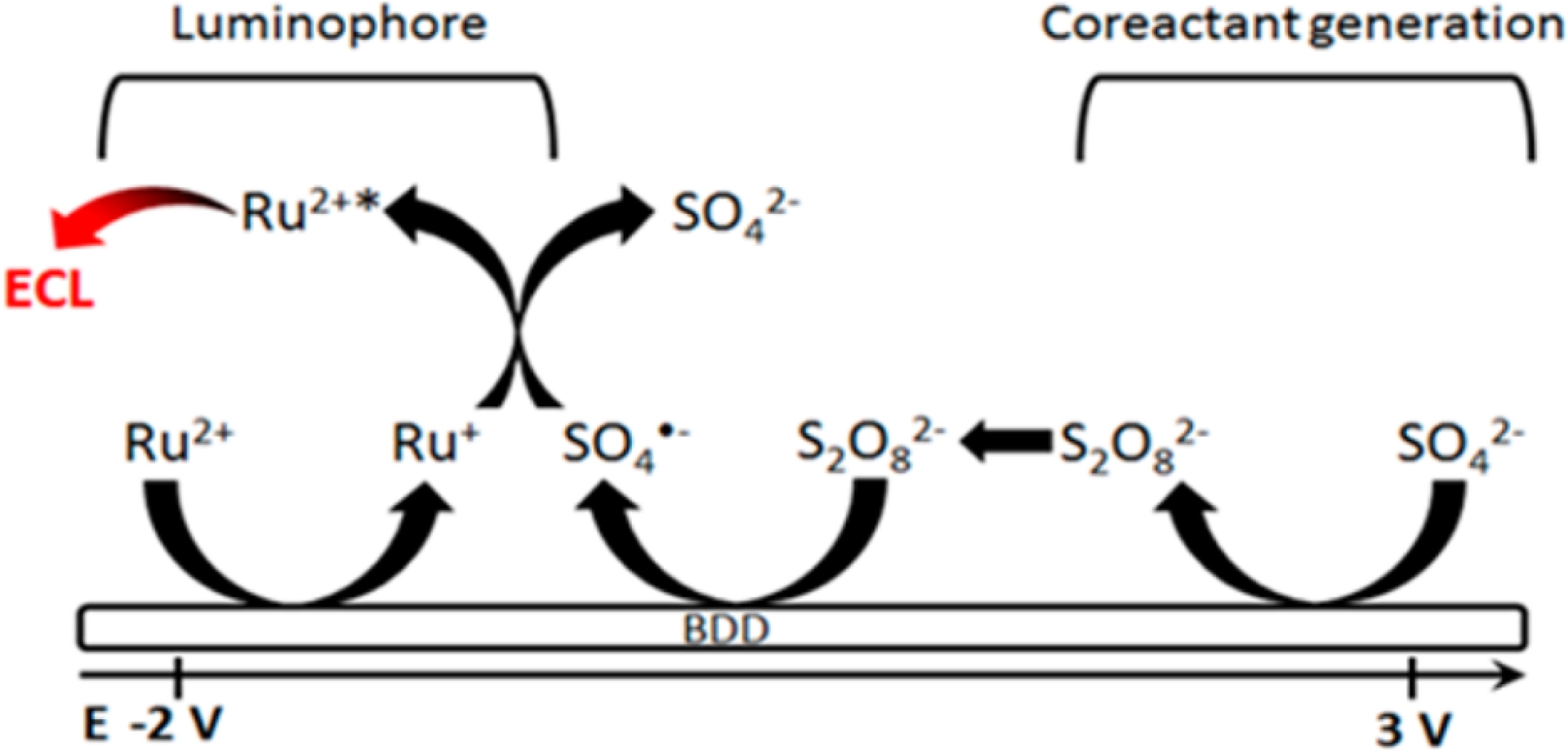
Selected alternative coreactants and their radical intermediates
| Coreactant name | Type | Key radical(s) generated | Relative ECL efficiency (vs. TPrA) | Notable advantages/disadvantages | References |
|---|---|---|---|---|---|
| 2-(dibutylamino) ethanol (DBAE) | Oxidative-reduction | Amine radical cation, α-aminoalkyl radical | Variable; can be higher at low concentrations | Lower toxicity and volatility than TPrA | [54] |
| BIS-TRIS | Oxidative-reduction | Amine radical cation, α-aminoalkyl radical | Can be >200% higher in bead assays | Biocompatible buffer; balances reactivity and diffusion | [55] |
| Oxalate (C2O42−) | Oxidative-reduction | CO2•− | Generally lower than TPrA | Classic coreactant; less efficient in many systems | [55, 56] |
| In-situ-generated S2O82− | Reductive-oxidation | SO4•− | Potentially high | “Coreactant-free” system; requires BDD electrode | [57] |
| Carbohydrazide | Intramolecular | CON4H6• | High in intramolecular systems | Covalently linked to luminophore; improves efficiency | [58] |
A particularly innovative strategy involves generating the coreactant in situ from an inert and benign precursor present in the bulk solution. A prime example is the work by Irkham and colleagues, who utilized the unique properties of boron-doped diamond (BDD) electrodes [53]. BDD electrodes can withstand extremely high anodic potentials in aqueous solution without significant water oxidation. This allows for the direct oxidation of inert sulfate anions (SO42−), a common component of many buffers, into the highly reactive peroxydisulfate (S2O82−) coreactant directly at the electrode surface. This in-situ generated S2O82− then participates in a conventional reductive-oxidation ECL mechanism with [Ru(bpy)3]2+ (Figure 2). This “coreactant-free” approach is highly attractive for bioanalysis as it eliminates the need to add potentially interfering or toxic coreactants to the sample, thereby improving the biocompatibility and simplifying the assay design. Other novel approaches include the use of metal–organic gels (MOGs) with amino-rich groups as solid-state coreactants, which can enhance ECL through both chemical functionality and electrocatalytic effects [59]. The representative examples summarized in Table 3 highlight how structural diversity among alternative and bio-derived coreactants translates into distinct radical intermediates and performance outcomes. The contrast between oxidative-reduction and reductive-oxidation types emphasizes how radical lifetime, diffusion distance, and redox balance jointly dictate ECL efficiency. Particularly, the high activity of BIS-TRIS and in-situ-generated S2O82− illustrates two emerging paradigms: exploiting biocompatible multifunctional buffers and electrode-driven radical generation. These findings collectively redefine the design space for next-generation, sustainable, and low-toxicity ECL systems.
3. Advanced methodologies for the detection and characterization of radical intermediates
The transient nature and low concentration of coreactant radical intermediates make their detection and characterization a formidable analytical challenge. Elucidating the complex mechanisms discussed in the previous section has only been possible through the development and application of a suite of highly specialized, sensitive, and often coupled analytical techniques. No single method provides a complete picture; rather, robust mechanistic conclusions are built upon a convergence of evidence from complementary approaches that probe different aspects of the radical’s identity, reactivity, and spatial behavior.
3.1. Spectroscopic and spectroelectrochemical detection
Spectroscopic methods provide some of the most direct and unambiguous evidence for the existence and structure of radical intermediates. Among these, electron paramagnetic resonance (EPR) spectroscopy stands out as the definitive technique for studying species with unpaired electrons. EPR is analogous to NMR but probes the spins of electrons rather than nuclei, yielding spectra with characteristic hyperfine splitting patterns that serve as a “fingerprint” for a specific radical. A landmark achievement in ECL mechanism research was the direct EPR detection of the TPrA radical cation. By using a flow-cell setup to rapidly transport the electrochemically generated species from the electrode into the EPR cavity before it could decompose, they captured its spectrum providing incontrovertible proof of its formation as a key intermediate in the ECL pathway.
For radicals that are too short-lived or exist at concentrations below the limit of direct EPR detection, the technique of spin trapping is indispensable [60]. In this method, a diamagnetic “spin trap” molecule, such as 5,5-dimethyl-1-pyrroline N-oxide (DMPO), is added to the system. The trap reacts rapidly with the transient radical to form a much more stable and persistent nitroxide radical adduct, which accumulates to a concentration sufficient for EPR detection [61]. The hyperfine splitting constants of the resulting adduct’s spectrum are characteristic of the trapped radical, allowing its identification. Spin trapping has been crucial for identifying the formation of both sulfate radicals and hydroxyl radicals in persulfate-based systems [62]. However, careful control experiments are essential, as it has been shown that spin traps can sometimes react directly with the persulfate precursor, leading to the formation of artifactual signals that could be misinterpreted as evidence for a specific radical pathway.
Schematic of the RT-Triplex setup for electricity-luminescence-mass synchronization. Insets show the sideview schematic diagrams of the EC reaction occurring at the two ends of the capillary (reproduced from ref [63] with permission from RSC, copyright 2022).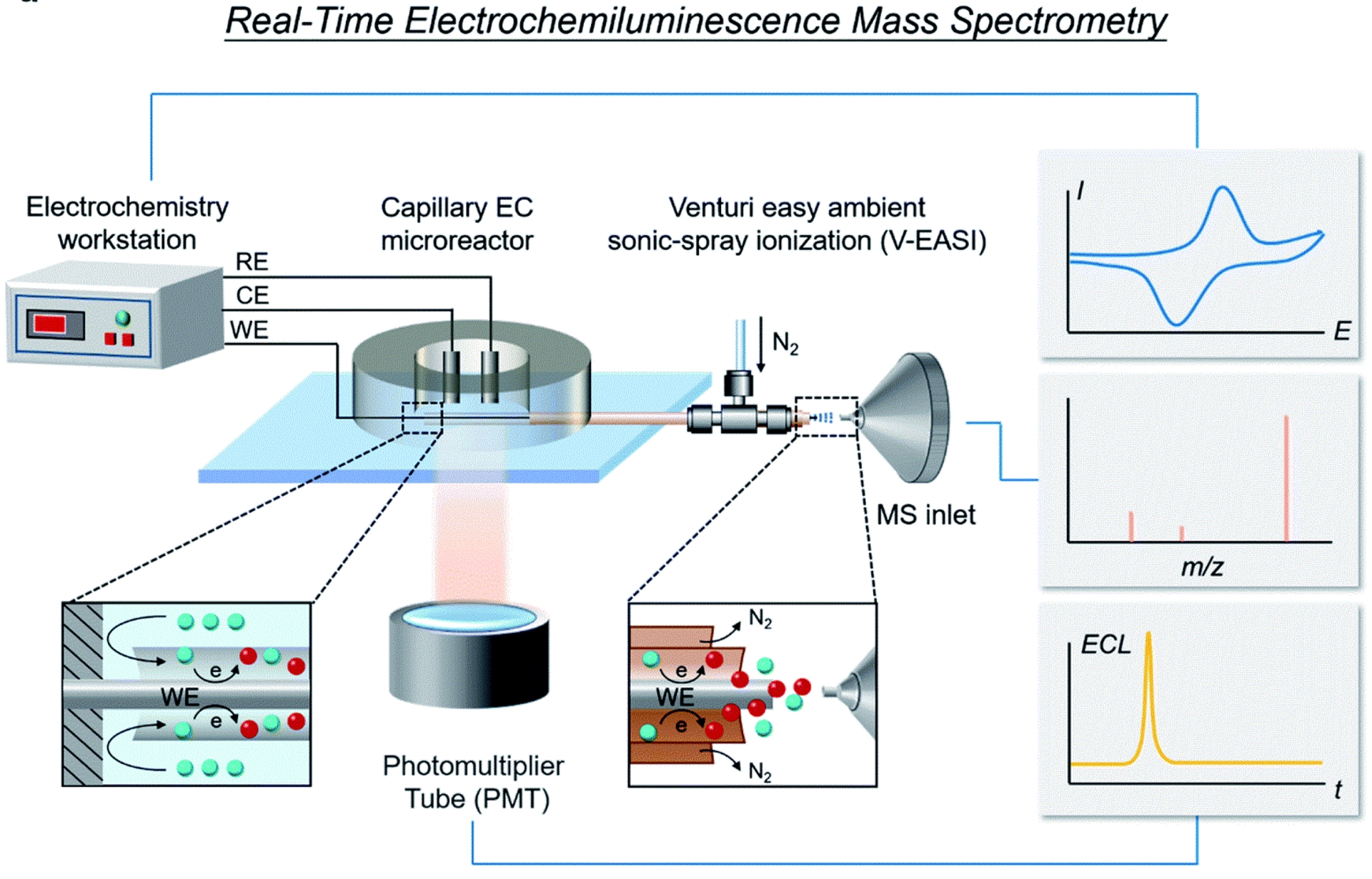
Spectroelectrochemistry (SEC) provides a powerful complementary view by coupling an electrochemical experiment with a spectroscopic measurement, typically UV–Visible absorption spectroscopy, in the same cell [64]. By monitoring the absorbance spectrum at the electrode surface as a function of the applied potential, SEC can track the depletion of reactants and the formation of stable or semi-stable products and intermediates in real time [65]. While it generally lacks the specificity to identify transient radicals directly, it provides invaluable kinetic data and helps to correlate electrochemical events (e.g., current peaks) with specific chemical transformations, thereby helping to piece together the overall reaction sequence [66]. To illustrate a quantitative-use case that dovetails with coreactant ECL chemistry, thin-layer UV–Vis SEC has been applied to the [Ru(bpy)3]2+ system under oxidative bias to follow the disappearance of the MLCT band (∼450–460 nm) and concomitant growth of the oxidized species’ signatures in real time [67]. In practice, stepping the potential to the RuII/III region produces an immediate approximately linear drop of A450 within the first hundreds of milliseconds [68, 69, 70], followed by a slower exponential phase; fitting the time-resolved A(t) traces yields pseudo-first-order rate constants for consumption of [Ru(bpy)3]2+ that increase with overpotential, and the absorbance change scales with the electrolysis charge in a Nernstian manner. These SEC-derived kinetics have been used side by side with ECL readouts in the canonical [Ru(bpy)3]2+/amine coreactant platform to rationalize how the build-up of [Ru(bpy)3]3+ during the anodic half-cycle gates the downstream radical chemistry of the amine; e.g., studies that combine SEC with mechanistic probes on the [Ru(bpy)3]2+/TPrA couple demonstrate that the spectroscopic depletion of RuII (ΔA450 ≳ 10–30% in thin cells) coincides with the potential window where ECL turns on, providing a quantitative bridge between the electrochemical generation of RuIII and the chemical step that yields the emissive *RuII state [71].
Computed relative free energy profile for the electrocatalytic oxidation of TPrA (reproduced from Ref. [72] with permission from Wiley, copyright 2025).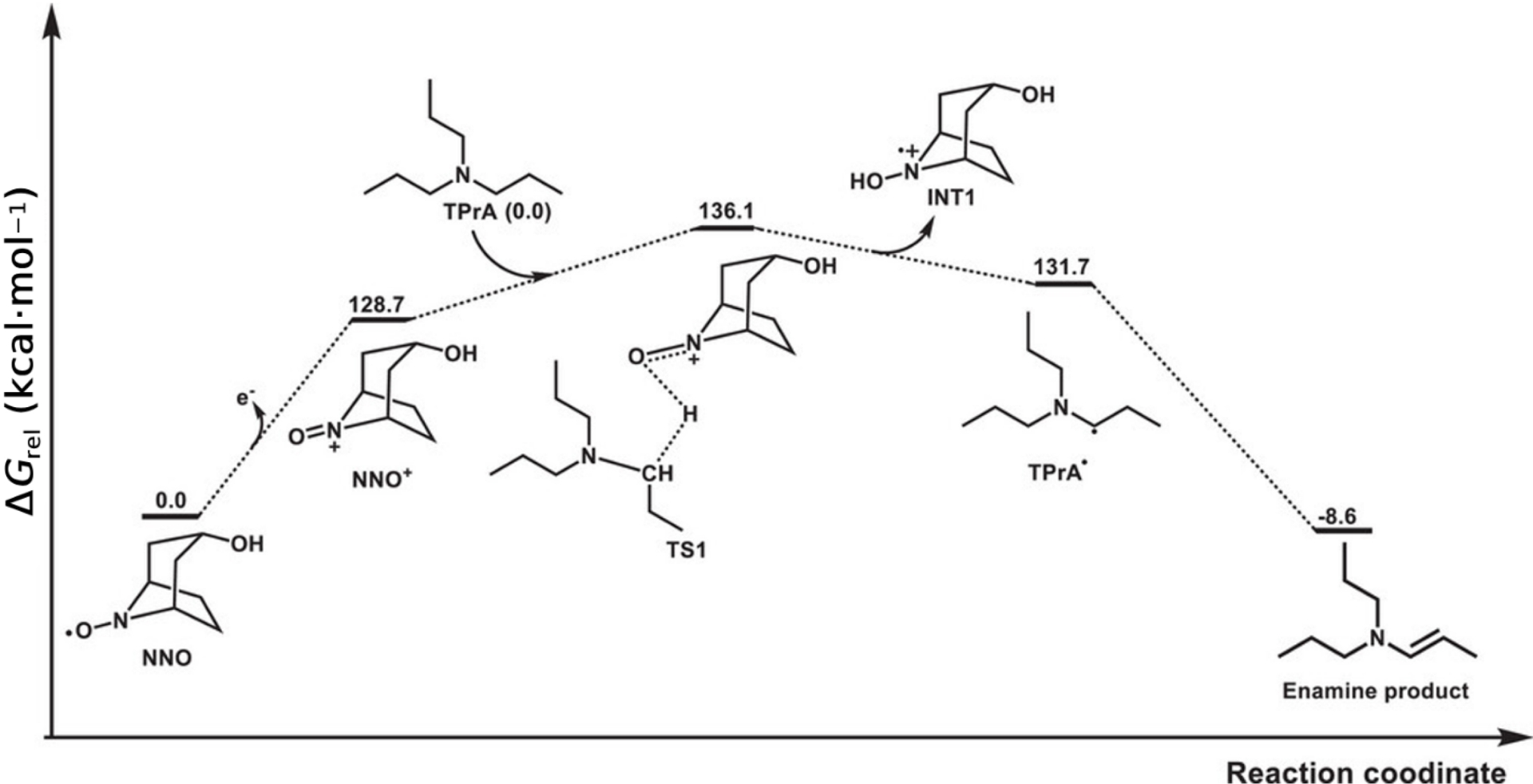
3.2. Mass spectrometry and computational approaches
The direct identification of intermediates by their mass-to-charge ratio offers an orthogonal and highly specific detection method. Electrochemical mass spectrometry (EC–MS) achieves this by interfacing an electrochemical flow cell with a mass spectrometer, allowing the continuous, real-time analysis of species generated at the electrode. Recent innovations in ambient ionization techniques have led to the development of platforms capable of capturing even very short-lived intermediates. To illustrate the quantitative power of EC–MS in ECL studies, Zhang and coworkers [63] built a real-time, triply synchronized platform (RT-Triplex) that couples a capillary electrochemical microreactor directly to a Venturi easy ambient sonic-spray MS while recording the photomultiplier tube (PMT) signal (Figure 3). In this geometry, fleeting intermediates with sub-millisecond lifetimes were transferred from the electrode–electrolyte interface to the MS inlet at a volumetric flow of 152 μL⋅min−1 (linear velocity ≈ 0.108 m⋅s−1), enabling time-aligned molecular and optical readouts. Using a BODIPY/TPrA ECL system, they detected the iminium ion [Pr2N = CHEt]+ (m/z 142.1592) from TPrA electrooxidation and observed that its extracted-ion chronogram turned on at 0.76 V, essentially coincident with the ECL onset at 0.79 V measured by the PMT. Moreover, post-electrolysis infusion experiments showed that introducing TPrA collapsed the “tailing” of the BODIPY⋅+ (m/z 318.2081) signal to ∼4 s, quantitatively linking coreactant availability to radical persistence. Together with direct capture of characteristic m/z features for other key intermediates (e.g., luminol-derived L–OOH at m/z 208.0358 under alkaline conditions, concomitant with intensified ECL), these measurements provide potential-resolved, number-anchored evidence that ties specific radical/proton-transfer steps to the light output in real time [73]. Such synchronized datasets—onset potentials, transport rates, and decay times—move EC–MS beyond qualitative assignment and into kinetic discrimination of competing ECL pathways at the electrode interface [74].
ECL images of a single bead labeled with [Ru(bpy)3]2+ in a 0.3 M phosphate buffer with 180 mM TPrA (Ru@Beads) (a) without and (b) with 100 μM of [Ir(dfppy)2(pt-TEG)]+. (c) Comparison between the single-bead ECL intensity profiles of Ru@Beads (grey line) and Ru@Beads/[Ir(dfppy)2(pt-TEG)]+ (red line). Inset: histogram of the comparison between the respective averaged maximum values of ECL intensity (reproduced from Ref. [75] with permission from RSC, copyright 2024).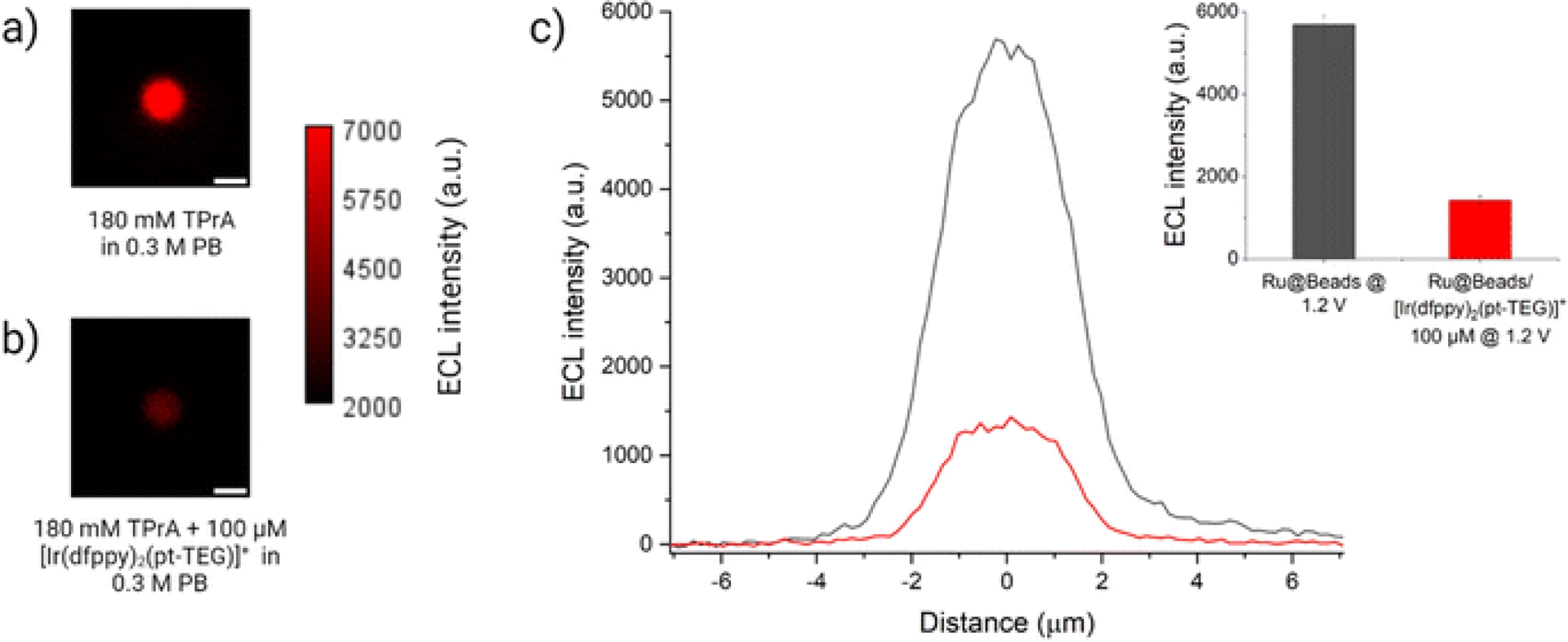
Computational chemistry, particularly density functional theory (DFT), has become an essential tool for augmenting and interpreting experimental findings [76]. DFT calculations can provide invaluable insights into the electronic structure, geometry, and energetic properties of proposed radical intermediates [77]. For example, theoretical calculations can be used to predict the standard redox potentials of radical couples, which are often difficult to measure experimentally but are critical for assessing the thermodynamic feasibility of a proposed ECL pathway [78]. Furthermore, DFT can be used to simulate EPR spectra, helping to confirm the assignment of an experimentally observed signal to a specific radical structure [79]. By modeling reaction pathways and calculating activation energies for transition states, computational approaches can help to rationalize kinetic observations and explain why certain coreactants or pathways are more efficient than others [80]. A recent study illustrates how such calculations deliver quantitative, mechanism-level insight for coreactant ECL [72]. Using nortropine N-oxyl (NNO) as an organic redox mediator in the archetypal [Ru(bpy)3]2+/amine system, DFT was employed to map the free-energy profile of the key oxidation step that generates the reducing amine-derived species (Figure 4) [81]. The computed activation free energy for hydride transfer from TPrA to the oxidized mediator (NNO+) is only ΔG = 7.4 kcal⋅mol−1 in acetonitrile, implying very fast kinetics and, consequently, efficient population of the coreactant radical manifold under mild anodic bias. This low barrier rationalizes the experimentally observed facilitation of ECL at lower oxidation overpotentials when NNO is present and provides a thermodynamic/kinetic basis for the enhanced emission efficiency reported for the redox-mediated route. By quantitatively connecting a specific transition state to macroscopic observables (onset and brightness), the DFT analysis does not simply “support” the mechanism but predicts its feasibility and impact in numerical terms, thereby guiding the design of alternative mediators and coreactants with comparably accessible barriers.
3.3. Microscopy and spatially resolved techniques
While the aforementioned techniques identify what the intermediates are, ECL microscopy (ECLM) reveals where they react. By replacing the conventional photomultiplier tube (PMT) with a sensitive camera (e.g., an EMCCD), ECLM captures a two-dimensional image of the light emission originating from the electrode surface. Although ECLM does not detect the radicals directly, it provides powerful, spatially resolved information about the reaction zone, which is intimately linked to the diffusion and lifetime of the radical intermediates. The thickness of the ECL emission layer [82], for instance, can be measured and correlated with the diffusion length of the key radicals, offering a method to distinguish between different mechanistic pathways [83].
Fracassa et al. [75] using ECLM to study bead-based assays has been particularly illuminating. By imaging single luminophore-labeled microbeads on an electrode surface, they observed that ECL emission originates from the entire surface of the bead, even regions several micrometers away from the electrode (Figure 5). This observation provides compelling visual evidence for the oxidative (TPrA•+) excitation mechanism, demonstrating that TPrA-derived radicals are indeed generated at the electrode and must diffuse through the solution to reach and react with the distal luminophore labels. This ability to map reactivity in space makes ECLM an indispensable tool for understanding the mechanisms of heterogeneous and spatially confined ECL systems, which are the most relevant for modern bioanalytical applications [84]. The analytical landscape summarized in Table 4 highlights how diverse techniques collectively unravel the fleeting existence of coreactant radicals. Each method contributes a distinct layer of mechanistic insight—EPR offering structural fingerprints, SEC providing kinetic correlations, EC–MS yielding molecular confirmation, and ECLM visualizing spatial dynamics. Together, these complementary tools bridge temporal and spatial scales from nanoseconds to micrometers. Notably, the integration of in-situ and operando approaches, combining spectroscopic and imaging modalities, is transforming ECL research toward quantitative mapping of radical generation, transport, and reaction efficiency in real time.
Advanced analytical techniques for radical intermediate detection
| Technique | Principle | Information obtained | Key advantages | Key limitations |
|---|---|---|---|---|
| EPR—direct detection | Absorption of microwave radiation by unpaired electrons in a magnetic field. | Unambiguous radical structure; Concentration | Unsurpassed specificity for radicals | Requires relatively stable radicals at sufficient concentration |
| EPR—spin trapping | Chemical reaction of a transient radical with a “trap” to form a persistent radical adduct | Identification of ultra-short-lived or low-concentration radicals | Greatly extends the range of detectable radicals | Susceptible to artifacts; indirect detection [85] |
| SEC | Simultaneous electrochemical control and UV–Vis spectroscopic monitoring at the electrode | Concentration profiles of chromophore species vs. potential; Reaction kinetics | Provides real-time kinetic data | Generally not specific for transient radical species |
| EC–MS | Coupling of an electrochemical cell to a mass spectrometer for real-time analysis | Molecular weight of intermediates and products; Reaction pathways | High sensitivity and molecular specificity | Can be complex to implement; ionization efficiency varies |
| ECLM | Imaging of light emission from the electrode surface with a camera | Spatial distribution of ECL reaction; Thickness of the emission layer | Provides powerful spatial information on reaction zones | Indirect detection of radicals; provides information on light emission only |
4. Bioanalytical performance: linking radical properties to assay sensitivity and selectivity
The profound commercial and scientific success of ECL, particularly in the form of electrochemiluminescence immunoassays (ECLIA), stems from its exceptional analytical performance, including high sensitivity, wide dynamic range, and robustness. This performance is not an emergent property of the system as a whole but is a direct consequence of the fundamental physicochemical properties of the coreactant radical intermediates [86]. Bridging the gap between the mechanistic details discussed previously and their practical impact on bioassay function is crucial for the rational improvement of existing technologies and the development of new ones.
Influence of electrode material on TPrA oxidation and ECL emission
| Electrode material | TPrA oxidation behavior | Dominant ECL pathway | Relative ECL intensity | Implications for bioanalysis | References |
|---|---|---|---|---|---|
| GC | Easy, broad irreversible oxidation peak starting at ∼0.6 V vs. SCE | Direct oxidation | High | Preferred for fundamental studies and disposable sensors due to high efficiency | [31, 87] |
| Au | Inhibited by surface oxide formation; catalytic oxidation at low potentials | Catalytic (at high potentials) or oxidative (TPrA•+) excitation | Moderate to low | Requires surface modification or additives to improve performance; widely used in reusable sensors | [31, 32, 88] |
| Pt | Strongly inhibited by surface oxide formation | Catalytic | Low | Generally yields weak ECL with TPrA, limiting its use in sensitive assays | [31, 87] |
| BDD | Sluggish kinetics for TPrA oxidation | Direct oxidation | Moderate | Can enhance ECL of labeled beads; enables in-situ coreactant generation | [57, 89] |
4.1. The direct impact of radical intermediates on biosensor function
The stability, diffusion distance, and reactivity of these intermediates are the key parameters that govern this process [90]. A critical insight is the operation of a “Goldilocks Principle” for radical stability in the context of heterogeneous bioassays, such as the widely used magnetic bead-based format. In these systems, the luminophore is immobilized on a microbead surface, physically separated from the electrode. For an ECL signal to be generated, the coreactant radicals must be stable enough to survive diffusion across the micrometer-scale gap from the electrode to the bead surface [91]. However, they must also remain highly reactive to participate in an efficient, thermodynamically favorable electron-transfer reaction upon arrival to generate the luminophore’s excited state. A radical that is too reactive might decompose before reaching the bead, while a radical that is too stable may lack the necessary thermodynamic driving force for efficient light generation [92]. The remarkable success of TPrA in these assays is largely because its radical intermediates, particularly TPrA•+, possess a near-optimal balance: its half-life of ∼0.2 ms allows diffusion over the required length scale, while the resulting TPrA• remains a potent enough reductant to drive the ECL reaction efficiently.
The intrinsic reactivity of the radical, governed by its standard redox potential, directly determines the ECL efficiency and, by extension, the analytical sensitivity of a biosensor. The highly negative redox potential of the TPrA• radical (ca. −1.7 V vs. SCE) ensures that its reaction with oxidized or ground-state luminophores is highly exergonic, leading to a high probability of forming the excited state and thus of a strong light signal [93]. Any factor that increases the effective concentration or generation rate of these key radicals, such as using an electrode material that facilitates TPrA oxidation (Table 5), tends to directly increase the ECL signal and improve the limit of detection (LOD) [94].
However, the high reactivity that makes these radicals effective can also be a source of limitations. Radical intermediates can engage in unwanted side reactions with components of the biological sample matrix, such as proteins or endogenous antioxidants. These “dark” pathways consume the radicals without producing light, effectively quenching the ECL signal and potentially compromising assay accuracy. Furthermore, the stability of the radicals themselves can be influenced by the sample matrix; for instance, pH changes can alter the rate of the critical deprotonation step of TPrA•+ [95]. Therefore, the robustness of an ECL immunoassay depends not only on the specificity of the antibody–antigen interaction but also on the stability and predictable reactivity of the coreactant radical intermediates within complex biological fluids like serum or plasma [96].
4.2. Rational design of coreactants and assay formats
A deep mechanistic understanding of radical intermediates enables the rational design of superior ECL systems. Instead of relying on empirical screening, researchers can now tailor coreactant structures and assay configurations to optimize radical generation and reactivity. One promising strategy is to overcome the limitations of intermolecular diffusion by physically linking the coreactant and luminophore. For example, covalently integrating coreactant moieties into the structure of a porous emitter, such as a covalent organic framework (COF), has been shown to enhance ECL intensity by over three orders of magnitude [97]. This dramatic improvement is attributed to the facilitation of rapid intra-framework charge transfer between the luminophore and coreactant radicals, maximizing the efficiency of the light-generation step.
Another advanced approach involves the use of synergistic coreactant systems or “co-reaction accelerators”. In these systems, a second chemical species is added to catalyze the generation of the primary coreactant radicals. For instance, Ding and Su [89] demonstrated that adding a small amount of triethanolamine (TEOA), a relatively weak ECL coreactant, to the classic [Ru(bpy)3]2+/TPrA system resulted in a more than tenfold amplification of the ECL signal. They proposed a “chemical oxidation mechanism” where the electrochemically generated TEOA radical cation acts as a homogeneous oxidant for TPrA in the solution phase [98]. This opens an additional, highly efficient channel for the generation of TPrA radicals, effectively accelerating the overall reaction kinetics and boosting the light output [99]. These examples illustrate a clear trend in the field: moving from simply using coreactants to actively engineering the generation, environment, and reactivity of their radical intermediates to achieve unprecedented analytical performance.
5. Conclusions and future perspectives
The study of coreactant radical intermediates has been central to the transformation of ECL from a physical phenomenon into a dominant bioanalytical technology. This review has traced the critical role of these transient species, from their generation at the electrode surface to their ultimate impact on assay performance. It is clear that the chemical identity, redox properties, stability, and transport dynamics of radicals derived from coreactants like TPrA and persulfate are the fundamental parameters that dictate the efficiency and applicability of ECL systems. The intricate web of competing mechanistic pathways, such as the direct oxidation, catalytic, and oxidative (TPrA•+) excitation routes in the TPrA system, is not merely of academic interest but has profound practical consequences, determining which electrode materials and assay formats yield optimal results. Similarly, the ongoing investigation into radical versus non-radical pathways in persulfate chemistry highlights a frontier where fundamental understanding continues to evolve. The development of a sophisticated analytical arsenal, including EPR spectroscopy, real-time mass spectrometry, and ECL microscopy, has been indispensable in providing the experimental evidence needed to construct and validate these complex mechanistic models.
Despite significant progress, several key questions and challenges remain, representing fertile ground for future research. The precise nature and kinetics of “dark” side reactions involving coreactant radicals, especially within complex biological matrices, are still not fully understood and represent a major factor in assay stability and reproducibility. While the dominant conditions for different TPrA pathways are qualitatively known, a comprehensive, quantitative model that can predict the relative contributions of each pathway under any given set of experimental parameters has yet to be developed. Furthermore, the role of non-radical pathways in persulfate-based ECL remains an open and intriguing question that warrants further investigation.
Looking forward, the field is poised for exciting advancements driven by a continued focus on the chemistry of radical intermediates. A major direction will be the rational design of novel coreactants. This will move beyond simple structural analogs to “smart” molecules with tailored radical stabilities, enhanced biocompatibility, or even built-in responsiveness to biological analytes. The integration of coreactant functionalities into nanostructured materials, such as MOFs (metal–organic frameworks) and COFs, represents a powerful strategy to control the local radical environment and enhance charge-transfer efficiency. Progress will also be heavily reliant on the development of advanced operando characterization techniques. The future lies in multi-modal platforms that can simultaneously capture electrochemical, spectroscopic, and spatial information in real time to provide a complete, dynamic picture of the reaction layer. Finally, computational and machine learning approaches will play an increasingly vital role. High-throughput DFT screening could rapidly identify promising new coreactant structures, while machine learning models could be trained on multi-parameter experimental data to predict optimal assay conditions and even assist in deciphering complex mechanistic data. By continuing to focus on the fundamental chemistry of these pivotal radical intermediates, the scientific community can ensure that ECL remains at the forefront of analytical and bioanalytical innovation.
CRediT authorship contribution statement
Yifan He: Conceptualization, Investigation, Formal analysis, Writing—original draft.
Yanfei Lv: Formal analysis, Visualization.
Li Fu: Supervision, Writing—review & editing.
Shichao Zhao: Conceptualization, Writing—review & editing.
Hassan Karimi-Maleh: Supervision, Writing—review & editing.
Acknowledgements
This work was supported by the Hassan Research Initiation Fund Grant Number KYQD2024-046 and Theoretical and Experimental Development of an MXene/GoldAmplified Electrochemical Biosensor for Monitoring Lung Cancer Drugs: Adagrasib and Afatinib Dimaleate Grant Number 2025K064, The Quzhou Affiliated Hospital of Wenzhou Medical University.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.