

 CC-BY 4.0
CC-BY 4.0
1. Introduction
[4]Helicenes, called benzo[c]phenanthrenes, are organic molecules consisting of four ortho-fused benzene rings and belong to the class of polycyclic aromatic hydrocarbons (PAHs). These smallest helicenes have attracted considerable attention in view of their π-conjugated system and their twisted geometry.
The helicity of the benzo[c]phenanthrene (B[c]p) can be further reinforced through bulky substitution at 1 and 12 positions (Figure 1) of the terminal rings [1], which makes the molecule configurationally stable and can be resolved to give P- and M-enantiomers [2]. As a first example of chiral tetracyclic skeleton, Newman and co-workers [3] reported the synthesis and an optical resolution leading to the [4]helicene derivative 1. Since then, some [4]helicene derivatives have been synthesized in optically active form; for example, the lactone-type 2 [4]. Moroever, Carreño and co-workers have developed an enantioslective approach leading to the [4]helicene quinone 3 [5]. In an independent study, the [4]helicenium cation 4 was described as a chiral analog of hetero[4]helicenes [6]. This charged compound obtained after resolution has demonstrated a high configurational stability. More recently, we have reported the synthesis of the nitrile-grafted benzo[c]phenanthrene derivatives 5a–b and used them to achieve enantiopure [6]helicenes [7, 8].
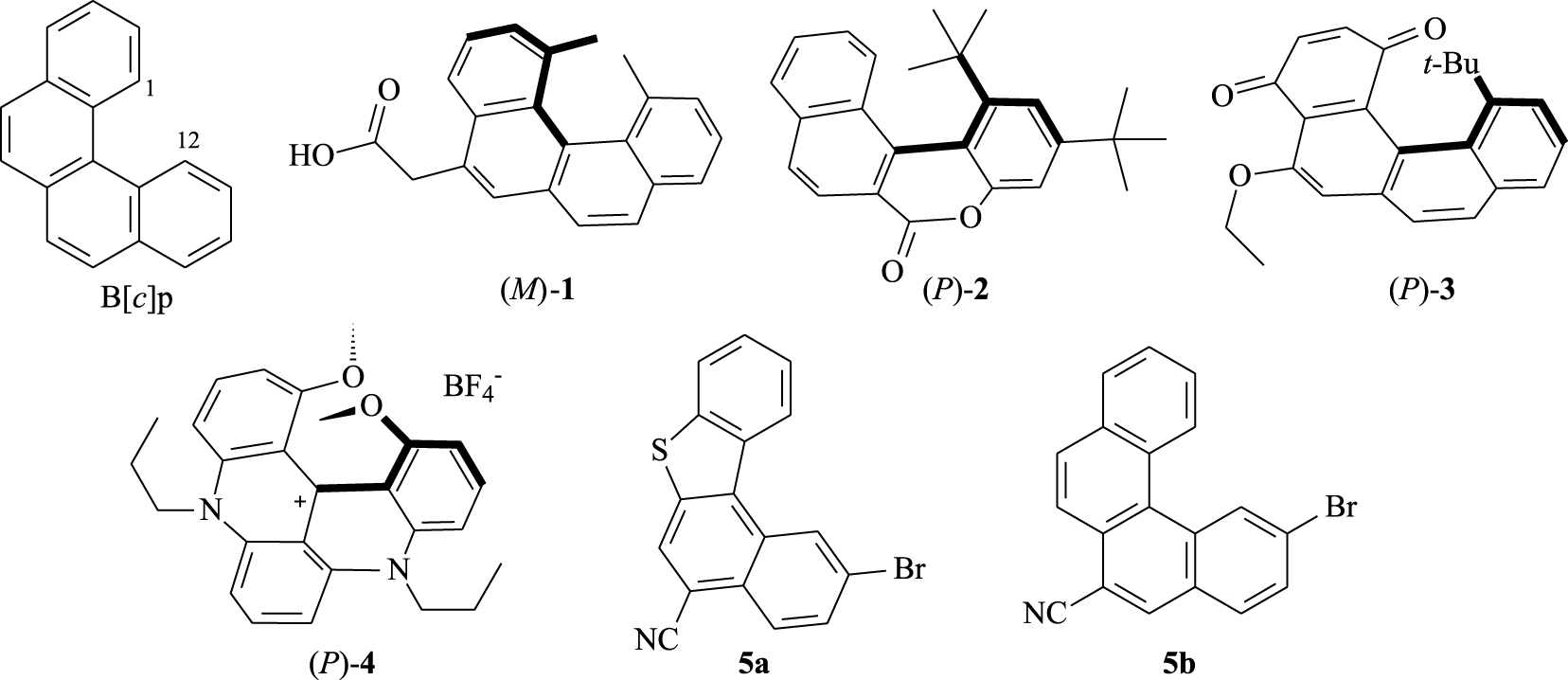
Representative examples of [4]Helicene derivatives.
[4]Helicenes have shown various applications in different areas thanks to their particular structure and their interesting properties. They were exploited in catalysis [9, 10], supramolecular assemblies [11], molecular recognition [12], and charge transfer complexation [13] and as motors [14]. Incorporation of heteroatoms into the [4]helicene skeleton has considerably amplified its electronic and photophysical properties, thus allowing its application in electronic devices [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28]. For example, the phosphorus- and boron-doped [4]helicenes 6 and 7 (Figure 2) have shown a strong green fluorescence with quantum yields of 0.83 and 0.81, respectively [29, 30]. Organometallic helicenes are also regarded as strong chiral emitters [31, 32, 33, 34, 35]. Among these conjugated systems, a cycloplatinated [6]helicene [36] has been prepared from the [4]helicene derivative 8, which reveals original electronic, chiroptical, and photophysical properties.
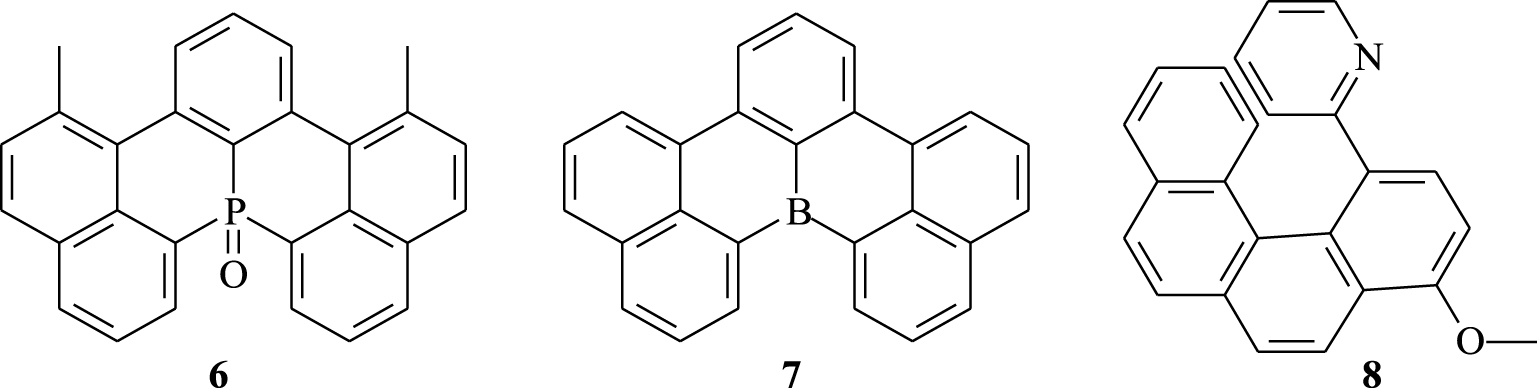
[4]Helicene derivatives as emitters.
These studies have inspired us to design and develop new [4]helicene derivatives bearing reactive functional groups by incorporating sulfur into their skeleton to evaluate their photophysical properties experimentally. Our synthetic strategy is based on the preparation of valuable α, β-unsaturated nitriles which were subjected to UV irradiation giving rise to the target [4]helicenes.
2. Experimental section
2.1. General methods
Unless otherwise noted, reagents and solvents used in this work were purchased from Sigma–Aldrich. All reactions were carried out under an argon atmosphere and were monitored by thin-layer chromatography (TLC) using commercial silica-gel plate 60 coated with a fluorescence indicator (SiliCycle Chemical division, 0.25 mm, F254). The use of UV light at 254 nm or 365 nm allowed visualization of the TLC plate. Organic compounds were purified using silica gel obtained from SiliCycle Chemical division (40–63 nm; 230–240 mesh). All mixed solvent eluents are reported as v/v solutions. Melting points were measured on a Bibby Scientific Stuart Digital, Advanced, SMP30. 1H and 13C spectra were taken on a Bruker AC 300 instrument in CDCl3 (300 MHz (1H) and 75 MHz (13C)). Signals due to the solvent served as the internal standard (CHCl3: 𝛿 7.26 for 1H, 𝛿 77.16 for 13C). Chemical shifts (𝛿) are reported in parts per million (ppm) relative to TMS. Coupling constant J is given in Hz. Photochemical reactions were performed using a falling-film photoreactor and a high-pressure Hg-vapor lamp (500 W, Helios Italquartz). UV–Vis spectra were recorded on a spectrophotometer UV-1600PC, using quartz cuvettes of 1 cm path length. Steady-state luminescence spectra were measured using a Jobin Yvon FluoroMax-2 spectrofluorometer, fitted with a red-sensitive Hamamatsu R928 photomultiplier tube. Samples for emission measurements were contained within quartz cuvettes of 1 cm path length.
2.2. General procedure for the preparation of the 𝛼,𝛽-unsaturated nitriles 9a–h
1 molar equivalent of p-substituted phenylacetonitrile was mixed with 1 molar equivalent of 2-naphtaldehyde or benzo[b]thiophene-2-carbaldehyde in dry methanol (30 mL) and the solution was stirred at 0 °C for 10 minutes. Then, 2 molar equivalents of sodium methoxide was added in small portions and the mixture was stirred for 30 minutes at 0 °C, and then for 4 hours at room temperature. The precipitate was filtered, washed with cold methanol followed by water, and dried in air.
2.2.1. (Z)-2-(naphthalen-2-yl)-3-phenylacrylonitrile (9a)
Yellow solid, 81%, m.p = 117–119 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm) : 7.40–7.54 (m, 3H), 7.52–7.60 (m, 2H), 7.70 (s, 1H), 7.73–7.77 (m, 2H), 7.87–7.95 (m, 3H), 8.09 (d, J = 8.7 Hz, 1H), 8.32 (s, 1H, Hvinyl); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 111.3 (C), 117.5 (CN), 124.8 (CH), 125.5 (2CH), 126.2 (CH), 127.0 (CH), 127.2 (CH), 128.1 (CH), 128.2 (CH), 128.5 (2CH), 128.6 (CH), 129.8 (CH), 130.8 (C), 132.6 (C), 133.6 (C), 134.2 (C), 141.6 (CH); HRMS (MALDI-TOF) Cald. For (C19H13NNa) [M+Na]+: 278.0945. Found: 278.0941; Anal. Calcd for C19H13N: C, 89.38; H, 5.13. Found: C, 89.32; H, 5.11.
2.2.2. (Z)-2-(4-methylphenyl)-3-(naphthalen-2-yl)acrylonitrile (9b)
White solid, 81%, m.p = 128–130 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 2.41 (s, 3H, CH3), 7.26 (d, J = 8.1 Hz, 2H), 7.50–7.57 (m, 2H), 7.61–7.64 (m, 3H), 7.84–7.92 (m, 3H), 8.05 (d, J = 8.7 Hz, 1H), 8.28 (s, 1H, Hvinyl); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 20.6 (CH3), 111.3 (C), 117.6 (CN), 124.8 (CH), 125.4 (2CH), 126.2 (CH), 126.9 (CH), 127.2 (CH), 128.1 (CH), 128.2 (CH), 129.2 (2CH), 129.6 (CH), 130.9 (C), 131.4 (C), 132.7 (C), 133.5 (C), 138.8 (C), 140.5 (CH); HRMS (MALDI-TOF) Cald. For (C20H15NNa) [M+Na]+: 292.1102. Found: 292.1098; Anal. Calcd for C20H15N: C, 89.19; H, 5.61. Found: C, 89.11; H, 5.59.
2.2.3. (Z)-2-(4-fluorophenyl)-3-(naphthalen-2-yl) acrylonitrile (9c)
White solid, 73%, m.p = 148–150 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.18 (t, J = 8.4 Hz, 2H), 7.54–7.59 (m, 2H), 7.63 (s, 1H), 7.69–7.74 (m, 2H), 7.87–7.95 (m, 3H), 8.07 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H, Hvinyl); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 110.2 (C), 115.4 (d, JC-F = 21.9 Hz, 2CH), 117.4 (CN), 124.6 (CH), 126.3 (CH), 127.1 (CH), 127.2 (CH), 127.3 (CH), 127.4 (CH), 128.2 (2CH), 129.8 (CH), 130.3 (C), 130.6 (C), 132.6 (C), 133.7 (C), 141.5 (CH), 161.1 (d, JC-F = 248 Hz, C–F).; 19F NMR, 282 MHz, CDCl3): 𝛿 (ppm): − 108.84; HRMS (MALDI-TOF) Cald. For (C19H12FNNa) [M+Na]+: 296.0851. Found: 296.0847; Anal. Calcd for C19H12FN: C 83.50; H 4.43. Found: C 83.40; H 4.45.
2.2.4. (Z)-2-(4-chlorophenyl)-3-(naphthalen-2-yl) acrylonitrile (9d)
White solid, 80%, m.p = 167–169 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.43 (d, J = 8.4 Hz, 2H), 7.53–7.60 (m, 2H), 7.65–7.68 (m, 3H), 7.87–7.95 (m, 3H), 8.08 (dd, J1 = 1.5 Hz, J2 = 7.5 Hz, 1H), 8.31 (s, 1H, Hvinyl); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 110.6 (C), 117.7 (CN), 125.1 (CH), 126.8 (CH), 127.2 (2CH), 127.7 (2CH), 128.7 (2CH), 129.2 (2CH); 130.5 (CH), 131.0 (C), 133.1 (C), 133.2 (C), 134.2 (C), 135.2 (C), 142.4 (CH); HRMS (MALDI-TOF) Cald. For (C19H12ClNNa) [M+Na]+: 312.0555. Found: 312.0551; Anal. Calcd for C19H12ClN: C 78.76; H 4.17. Found: C 78.81; H 4.11.
2.2.5. (Z)-3-(benzo[b]thiophen-2-yl)-2-phenylacrylonitrile (9e)
Yellow solid, 87%, m.p = 128–130 °C; 1H NMR (300 MHz, CDCl3) : 𝛿 (ppm): 7.37–7.48 (m, 5H), 7.68–7.71 (m, 3H), 7.81–7.87 (m, 3H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 110.4 (C), 117.2 (CN), 121.9 (CH), 124.0 (CH), 124.5 (CH), 125.4 (2CH), 125.9 (CH), 128.6 (2CH), 128.8 (CH), 129.0 (CH), 133.4 (C), 134.0 (CH), 137.1 (C), 138.2 (C), 140.6 (C); HRMS (MALDI-TOF) Cald. For (C17H11NSNa) [M+Na]+: 284.0509. Found: 284.0505; Anal. Calcd for C17H11NS: C 78.13; H 4.24. Found: C 78.03; H 4.31.
2.2.6. (Z)-3-(benzo[b]thiophen-2-yl)-2-(p-tolyl)acrylonitrile (9f)
Yellow solid, 90%, m.p = 158–160 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 2.40 (s, 3H, CH3), 7.24 (d, J = 8.1 Hz, 2H), 7.36–7.43 (m, 2H), 7.56 (d, J = 8.1 Hz, 2H), 7.66 (s, 1H, Hvinyl), 7.81–7.86 (m, 3H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 20.6 (CH3), 110.4 (C), 117.3 (CN), 121.8 (CH), 123.9 (CH), 124.4 (CH), 125.3 (2CH), 125.8 (CH), 128.5 (CH), 129.3 (2CH), 130.6 (C), 133.0 (CH), 137.3 (C); 138.3 (C), 139.1 (C), 140.5 (C); HRMS (MALDI-TOF) Cald. For (C18H13NSNa) [M+Na]+: 298.0666. Found: 298.0662; Anal. Calcd for C18H13NS: C 78.51; H 4.76. Found: C 78.63; H 4.81.
2.2.7. (Z)-3-(benzo[b]thiophen-2-yl)-2- (p-fluorophenyl)acrylonitrile (9g)
Yellow solid, 79%, m.p = 178–180 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.11–7.16 (m, 2H), 7.37–7.45 (m, 2H), 7.62–7.67 (m, 3H), 7.80–7.86 (m, 3H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 109.0 (C), 115.6 (d, JC-F = 22 Hz, 2CH), 117.2 (CN), 121.9 (CH), 124.1 (CH), 124.6 (CH), 126.1 (CH), 127.2 (d, JF-C = 8.2 Hz, CH), 129.3 (d, JF-C = 8 Hz, CH), 129.5 (C), 134.04 (CH), 134.07 (CH), 136.9 (C), 138.1 (C), 140.5 (C), 161.1 (d, JF-C = 249.2 Hz, C–F); 19F NMR, 282 MHz, CDCl3): 𝛿 (ppm): −111.18; HRMS (MALDI-TOF) Cald. For (C17H10FNSNa) [M+Na]+: 302.0415. Found: 302.0411; Anal. Calcd for C17H10FNS: C 73.10; H 3.61. Found: C 73.03; H 3.58.
2.2.8. (Z)-3-(benzo[b]thiophen-2-yl)-2- (p-chlorophenyl)acrylonitrile (9h)
Yellow solid, 72%, m.p = 203–205 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.40–7.44 (m, 2H), 7.44–7.47 (m, 2H), 7.60 (d, J = 6.9 Hz, 2H), 7.70 (s, 1H), 7.83–7.88 (m, 3H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 109.0 (C), 116.9 (CN), 121.9 (CH), 124.1 (CH), 124.6 (CH), 126.1 (CH), 126.5 (2CH), 128.8 (2CH), 129.5 (CH), 131.8 (C), 134.3 (CH), 134.9 (C), 136.8 (C), 138.1 (C), 140.7 (C); HRMS (MALDI-TOF) Cald. For (C17H10ClNSNa) [M+Na]+: 318.0120. Found: 318.0116; Anal. Calcd for C17H10ClNS: C 69.03; H 3.41. Found: C 69.07; H 3.44.
2.3. General procedure for the photocyclization reaction
1-liter solution of toluene containing 600 mg of α, β-unsaturated nitrile 9 and 1.1 molar equivalents of iodine was degassed for 10–15 minutes. Then, 50 molar equivalents of propylene oxide were added and the mixture was irradiated for 2–3 hours using a falling-film photoreactor and a high-pressure Hg-vapor lamp (500 W, Helios Italquartz). After completion of the reaction (TLC), the solvent was removed under reduced pressure and the crude residue was purified by silica-gel column chromatography with cyclohexane/EtOAc (80:20) as the eluent. Spectroscopic data of [4]helicene derivatives are given subsequently.
2.3.1. Benzo[c]phenanthrene-5-carbonitrile (10a)
Yellow solid, 71%, m.p = 125–127 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.72–7.75 (m, 2H, H9,11), 7.78–7.81 (m, 3H, H2,3,7 or 8), 7.95 (d, J = 8.4 Hz, 1H, H8 or H7), 8.04–8.07 (m, 1H, H10), 8.29 (s, 1H, H6), 8.44–8.49 (m, 1H, H4), 9.05–9.08 (m, 1H, H12), 9.11–9.14 (m, 1H, H1); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 108.8 (C), 117.2 (CN), 125.3 (CH), 125.5 (CH), 126.3 (CH), 126.9 (CH), 127.0 (CH), 127.1 (CH), 127.8 (CH), 127.9 (CH), 128.1 (CH), 128.2 (CH), 128.5 (C), 129.0 (C), 129.5 (C), 129.8 (C), 130.3 (C), 134.0 (CH), 134.2 (C); HRMS (MALDI-TOF) Cald. For (C19H11N) [M]+: 253.0891. Found: 253.0887; Anal. Calcd for C19H11N: C 90.09, H 4.38. Found: C 90.17, H 4.41; FT-IR: ν (cm−1): 534, 567, 607, 662, 735, 807, 903, 1063, 1160, 1240, 1367, 1424, 1496, 1592, 1728, 1929, 2210, 3059.
2.3.2. 2-Methylbenzo[c]phenanthrene-5-carbonitrile (10b)
Yellow solid, 81%, m.p = 185–187 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 2.68 (s, 3H, CH3), 7.64 (d, J = 8.1 Hz, 1H, H3), 7.71–7.78 (m, 3H, H9,11,7 or 8), 7.92 (d, J = 8.7 Hz, 1H, H8 or H7), 8.03–8.06 (m, 1H, H10), 8.20 (s, 1H, H6), 8.32 (d, J = 8.1 Hz, 1H, H4), 8.90 (s, 1H, H1), 9.05 (d, J = 9 Hz, 1H, H12); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 21.2 (CH3), 108.6 (C), 117.4 (CN), 125.1 (CH), 125.6 (CH), 126.2 (CH), 126.7 (CH), 127.4 (CH), 127.7 (CH), 127.9 (CH), 128.1 (CH), 128.4 (C), 128.7 (C), 128.8 (CH), 129.1 (C), 129.4 (C), 129.7 (C), 133.0 (CH), 134.1 (C), 137.1 (CH); HRMS (MALDI-TOF) Cald. For (C20H13N) [M]+: 267.1047. Found: 267.1043; Anal. Calcd for C20H13N: C 89.86, H 4.90. Found: C 89.96, H 4.94; FT-IR: ν (cm−1): 519, 664, 747, 811, 901, 1047, 1156, 1238, 1384, 1439, 1502, 1611, 2221, 2940, 3067.
2.3.3. 2-Fluorobenzo[c]phenanthrene-5-carbonitrile (10c)
Yellow solid, 82%, m.p = 225–227 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.53–7.60 (m, 1H, H3), 7.74–7.82 (m, 3H, H9,11,7 or 8), 7.98 (d, J = 8.4 Hz, 1H, H8 or H7), 8.06 (d, J = 8.1 Hz, 1H, H10), 8.26 (s, 1H, H6), 8.42 (dd, JH-H = 9 Hz, JH-F = 6 Hz, 1H, H4), 8.68 (dd, JH-H = 1.8 Hz, JH-F = 11.7 Hz 1H, H1), 8.9 (d, J = 8.1 Hz, 1H, H12); 13C NMR (101 MHz, CDCl3): 𝛿 (ppm): 108.9 (C), 113.2 (d, JC-F = 24.03 Hz, CH), 116.8 (d, C-F = 24.4 Hz, CH), 117.7 (CN), 126.0 (CH), 127.3 (CH), 127.45 (C), 127.46 (C), 127.5 (CH), 127.7 (CH), 128.1 (d, JC-F = 9.1 Hz, CH), 128.9 (CH), 129.3 (CH), 129.4 (C), 129.5 (C), 131.3 (d, JF-C = 8.9 Hz, C), 133.9 (CH), 134.5 (C), 160.8 (d, JF-C = 248.6 Hz, C-F); 19F NMR, 282 MHz, CDCl3): 𝛿 (ppm): −109.6; HRMS (MALDI-TOF) Cald. For (C19H10NF) [M]+: 271.0797. Found: 271.0793; Anal. Calcd for C19H10NF: C 84.12, H 3.72. Found: C 84.03, H 3.70; FT-IR: ν (cm−1): 542, 647, 670, 711, 743, 807, 895, 1183, 1416, 1504, 1616, 1720, 2218, 3059.
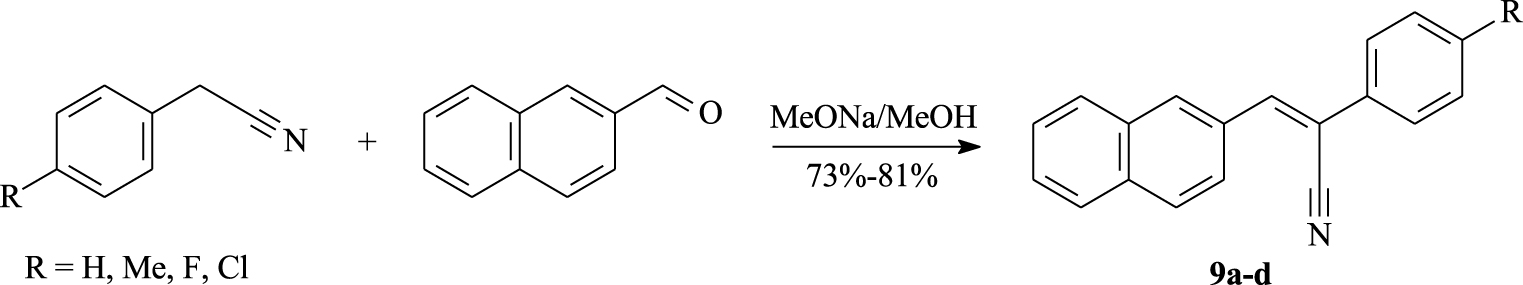
Synthesis of α,β-unsaturated nitriles 9a–d from 2-naphtaldehyde.
2.3.4. 2-Chlorobenzo[c]phenanthrene-5-carbonitrile (10d)
Yellow solid, 85%, m.p = 137–139 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.61–7.69 (m, 4H, H3,9,11,7 or 8), 7.86 (d, J = 8.7 Hz, 1H, H8 or H7), 7.93 (dd, J1 = 2.4 Hz, J2 = 7.2 Hz, 1H, H10), 8.16 (s, 1H, H6), 8.23 (d, = 8.7 Hz, 1H, H4), 8.84 (d, J = 8.1 Hz, H12), 8.96 (d, J = 1.8 Hz, 1H, H1); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 108.8 (C), 117.5 (CN), 125.9 (CH), 127.2 (CH), 127.4 (CH), 127.5 (CH), 127.7 (CH), 127.8 (CH), 128.1 (CH), 128.9 (CH), 129.0 (C), 129.1 (C), 129.2 (C), 129.3 (CH), 129.5 (C), 130.8 (C), 134.1 (C), 134.6 (C), 134.7 (CH); HRMS (MALDI-TOF) Cald. For (C19H10NCl) [M]+: 287.0501. Found: 287.0497; Anal. Calcd for C19H10NCl: C 79.31, H 3.50. Found: C 79.41, H 3.55; FT-IR: ν(cm−1): 506, 638, 677, 748, 802, 904, 1013, 1052, 1091, 1154, 1240, 1357, 1388, 1419, 1489, 1591, 1692, 2223, 3059.
2.3.5. Benzo[b]naphtho[2,1-d]thiophene- 7-carbonitrile (10e)
Yellow solid, 80%, m.p = 172–174 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.51–7.67 (m, 2H, H2,3), 7.71–7.84 (m, 2H, H9,10), 7.99 (dd, J1 = 2.1 Hz, J2 = 7.2 Hz, 1H, H4), 8.27 (s, 1H, H6), 8.38 (dd, J1 = 1.5 Hz, J2 = 8.1 Hz, 1H, H8), 8.79 (dd, J1 = 1.8 Hz, J2 = 7.5 Hz 1H, H1), 8.96 (d, J = 8.1 Hz, 1H, H11); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 108.7 (C), 117.1 (CN), 122.8 (CH), 123.1 (CH), 124.9 (CH), 125.0 (CH), 126.1 (CH), 126.3 (CH), 126.4 (CH), 127.4 (CH), 127.9 (CH), 129.6 (C), 129.7 (C), 132.3 (C), 135.0 (C), 136.1 (C), 140.7 (C); HRMS (MALDI-TOF) Cald. For (C17H9NS) [M]+: 259.0455. Found: 259.0451; Anal. Calcd for C17H9NS: C 78.74, H 3.50. Found: C 78.65, H 3.47; FT-IR: ν (cm−1): 476, 539, 610, 657, 713, 744, 775, 870, 1012, 1115, 1154, 1201, 1241, 1320, 1367, 1430, 1509, 1580, 2219.
2.3.6. 10-Methylbenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (10f)
Yellow solid, 80%, m.p = 220–222 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 2.68 (s, 3H, CH3), 7.52–7.65 (m, 3H, H2,3,9), 7.97 (d, J = 7.8 Hz, 1H, H4), 8.17 (s, 1H, H6), 8.24 (d, J = 8.4 Hz, 1H, H8), 8.72 (s, 1H, H11), 8.77 (d, J = 8.1 Hz, 1H, H1); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 21.8 (CH3), 108.5 (C), 117.2 (CN), 122.6 (CH), 122.8 (CH); 124.7 (CH), 124.9 (CH), 125.9 (CH), 126.1 (CH), 126.3 (CH), 127.8 (C), 128.3 (CH), 129.8 (C), 131.8 (C), 135.1 (C), 136.1 (C), 138.1 (C), 140.6 (C); HRMS (MALDI-TOF) Cald. For (C18H11NS) [M]+: 273.0612. Found: 273.0608; Anal. Calcd for C18H11NS: C 79.09, H, 4.06. Found: C 79.19, H 4.01; FT-IR: ν (cm−1): 542, 623, 655, 711, 751, 807, 879, 927, 1031, 1071, 1160, 1183, 1240, 1312, 1360, 1384, 1432, 1521, 1616, 1704, 2210, 2923, 3059.
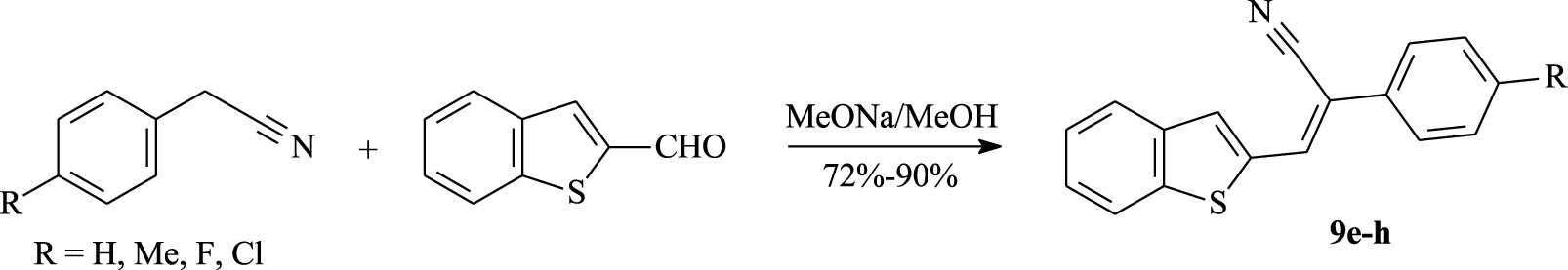
Synthesis of α,β-unsaturated nitriles 9e–h from benzo[b]thiophene-2-carbaldehyde.
Photocyclization of the α,β-unsaturated nitriles 9a–d into [4]helicenes 10a–d.
2.3.7. 10-Fluorobenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (10g)
Yellow solid, 84%, m.p = 235–237 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.53 (td, J1 = 2.1 Hz, J2 = 8.1 Hz, 1H, H3), 7.60–7.71 (m, 2H, H2,9), 8.04 (d, J = 8.1 Hz, 1H, H8), 8.30 (s, 1H, H6), 8.41 (dd, JH-H = 9 Hz, JH-F = 5.7 Hz, 1H, H4), 8.62 (dd, JH-H = 2.4 Hz, JH-F = 11.1 Hz, 1H, H11), 8.73 (d, J = 7.8 Hz, 1H, H1); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 108.5 (d, JF-C = 23.3 Hz, CH), 109.0 (C), 116.5 (d, JF-C = 24.6 Hz, CH), 117.5 (CN), 123.5 (CH), 125.0 (CH), 125.7 (CH), 127.0 (C), 127.1 (CH), 127.2 (CH), 129.1 (d, JF-C = 9.4 Hz, CH), 131.0 (C), 132.2 (C), 135.2 (C), 137.7 (C), 141.1 (C), 160.6 (d, JF-C = 246.6 Hz, CF); 19F NMR (282 MHz, CDCl3): 𝛿 (ppm): −109.17; HRMS (MALDI-TOF) Cald. For (C17H8FNS) [M]+: 277.0361. Found: 277.0357; Anal. Calcd for C17H8FNS: C 73.63, H, 2.91. Found: C 73.71, H, 2.97; ν (cm−1) = 584, 630, 711, 747, 820, 892, 973, 1037, 1154, 1181, 1236, 1326, 1353, 1426, 1516, 1616, 2222, 3063.
2.3.8. 10-Chlorobenzo[b]naphtho[2,1-d]thiophene-7-carbonitrile (10h)
White solid, 96%, m.p = 212–214 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 7.58–7.67 (m, 3H, H2,3,9), 7.99 (d, J = 7.5 Hz, 1H, H4), 8.22 (s, 1H, H6), 8.25 (d, J = 9 Hz, 1H, H8), 8.62 (d, J = 8.4 Hz, 1H, H1), 8.83 (d, J = 1.5 Hz, 1H, H11); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 108.3 (C), 116.7 (CN), 122.4 (CH), 122.9 (CH), 124.5 (CH), 125.2 (CH), 126.6 (CH), 127.1 (CH), 127.43 (CH), 127.47 (CH), 127.7 (C), 129.9 (C), 131.1 (C), 134.3 (C), 134.4 (C), 137.0 (C), 140.6 (C); HRMS (MALDI-TOF) Cald. For (C17H8ClNS) [M]+: 293.0065. Found: 293.0061; Anal. Calcd for C17H8ClNS: C 69.51, H 2.74. Found: C 69.62, H 2.78; FT-IR: ν (cm−1): 485, 560, 609, 667, 717, 741, 783, 808, 883, 907, 948, 1089, 1130, 1155, 1238, 1312, 1354, 1429, 1611, 2222, 3058.
Chemical yields of the α,β-unsaturated nitriles 9a–h
| α,β-unsaturated nitrile | R | Yield (%)a |
|---|---|---|
| 9a | H | 81 |
| 9b | Me | 80 |
| 9c | F | 73 |
| 9d | Cl | 80 |
| 9e | H | 87 |
| 9f | Me | 90 |
| 9g | F | 79 |
| 9h | Cl | 72 |
aIsolated yields.
3. Results and discussion
In the first step, we prepared different α,β-unsaturated nitriles, as precursors for the [4]helicenes, through Knoevenagel reaction. We sought to functionalize our olefin using various p-substituted phenylacetonitriles, which were condensed with 2-naphtaldehyde affording nitriles 9a–d in 73%–81% yields (Scheme 1, Table 1). A wide variety of literature conditions for the Knoevenagel condensation can be performed [37, 38]. Herein, we report an economic and rapid synthesis, using sodium methoxide as a base and dry methanol as a solvent at room temperature to provide the desired diarylethenes, with Z-configuration, which are recovered by simple filtration.
Chemical yields of [4]helicenes 10e–h
| [4]helicene | R | Yield (%)a |
|---|---|---|
| 10a | H | 71 |
| 10b | Me | 81 |
| 10c | F | 82 |
| 10d | Cl | 85 |
| 10e | H | 80 |
| 10f | Me | 80 |
| 10g | F | 84 |
| 10h | Cl | 96 |
aIsolated yields.
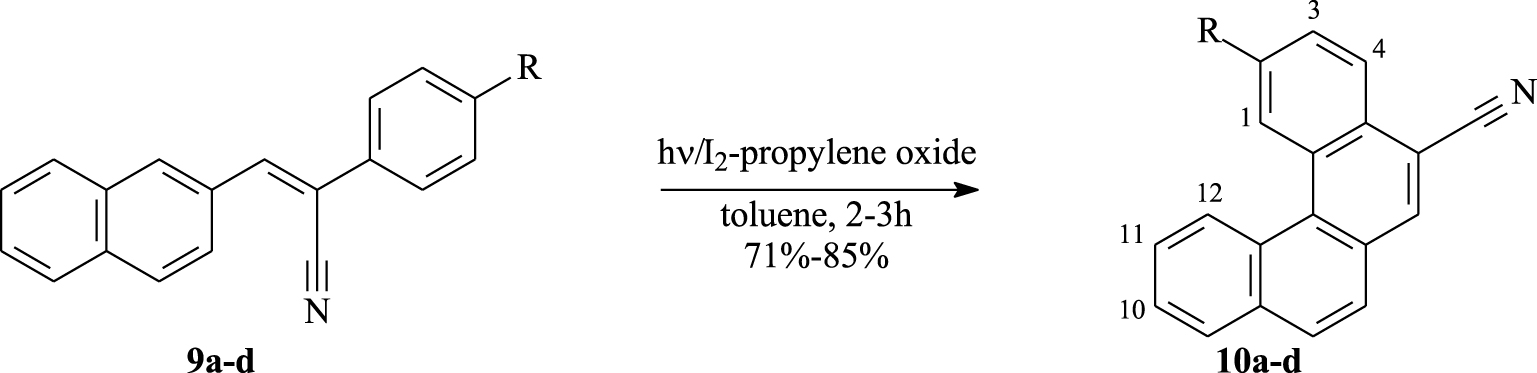
Expansion between 7.4 and 9.2 ppm of 1H–1H COSY NMR spectra (CDCl3, 300 MHz, 298 K) of compounds 10b (top) and 10c (down).
According to the same synthetic approach, we have carried out the Knoevenagel reaction between the p-substituted arylacetonitriles and benzo[b]thiophene-2-carbaldehyde which provides a series of (hetero)functionalized α, β-unsaturated nitriles 9e–h in 72%–90% yields (Scheme 2, Table 1).

Photocyclization of the α,β-unsaturated nitriles 9e–h into [4]helicene 10e–h.
The resulting α,β-unsaturated nitriles 9a–h were then subjected to photocyclization providing new tetracyclic systems using an immersion 500 W Hg-vapor lamp (Scheme 3). The irradiation was carried out using 600 mg of each α,β-unsaturated nitrile in 1 L of toluene, in the presence of iodine and propylene oxide. The photocyclization reaction requires high dilution to avoid intramolecular couplings [39] or dimerization of the substrate [40]. Thus, diarylethenes 9a–d were photoconverted into the benzo[c]phenanthrene-5-carbonitrile derivatives 10a–d in 71%–85% yields (Table 2).
On the other hand, similar experimental conditions were used for the photolysis of α,β-unsaturated nitriles 9e–h. This allows the formation of the expected benzo[b]naphto[2,1-d]thiophene-7-carbonitrile derivatives 10e–h in 80%–96% yields (Scheme 4, Table 2).
The structural assignment of [4]helicenes 10a–h were deduced from their 1H, 13C, and 1H–1H COSY NMR spectra. Their selected 1H NMR data and chemical shifts are gathered in Tables 3 and 4. For example, protons H1 and H12 for compounds 10a–d undergo a large deshielding effect compared to the other aromatic protons owing to the magnetic anisotropic effect in the vicinity of the terminal benzene rings, representing the characteristic signals of a [4]helicene [41]. A singlet around 𝛿 = 8.29, 8.20, 8.26, and 8.16 ppm is mainly attributed to proton H6 for compounds 10a, 10b, 10c, and 10d, respectively. The assignment of the other signals can be determined based on the 1H–1H COSY NMR.
Starting with compound 10b, the 1H NMR spectrum displayed the characteristic methyl group at 2.68 ppm and eight signals relative to a total of ten aromatic protons. A singlet at 8.90 ppm and a doublet at 9.05 ppm (J = 9 Hz) correspond to protons H1 and H12, respectively. On the 1H–1H COSY NMR spectrum (Figure 3), proton H12 shows a cross peak with a proton among three others appearing as a multiplet at 7.71–7.78 ppm. This latter proton shows another cross peak with the signal proton (m, 1H) at 8.03–8.06 ppm. Consequently, H11 belongs to the multiplet at 7.71–7.78 ppm and the signal which appears as a multiplet within the range 8.03–8.06 ppm is therefore relative to proton H10. A correlation is observed between the doublet at 𝛿 = 7.64 ppm (J = 8.1 Hz) and the doublet at 𝛿 = 8.32 ppm (J = 8.1 Hz), which allows us to assign these two doublets to protons H3 and H4, respectively. The difference in NMR chemical shifts between them is closely explained by the effect of substituents at C2 (Me) and C5 (CN). H4 is more deshielded than H3 owing to the proximity of cyano group. Also, the doublet at 𝛿 = 7.92 ppm (J = 8.7 Hz) shows a cross peak with the multiplet (3H) within the range 7.71–7.78 ppm. So, this doublet could be attributed to proton H7 or H8 and the multiplet was assigned to protons H9, H11, and H7 or H8.
Selected characteristic 1H NMR (300 MHz) data (𝛿 in ppm) for compounds 10a–d
| Compound | 𝛿 (H1) | 𝛿 (H6) | 𝛿 (H4) | 𝛿 (H12) |
|---|---|---|---|---|
 | 9.11–9.14 (m) | 8.44–8.49 (m) | 8.29 (s) | 9.05–9.08 (m) |
 | 8.90 (s) | 8.32 (d) | 8.20 (s) | 9.05 (d) |
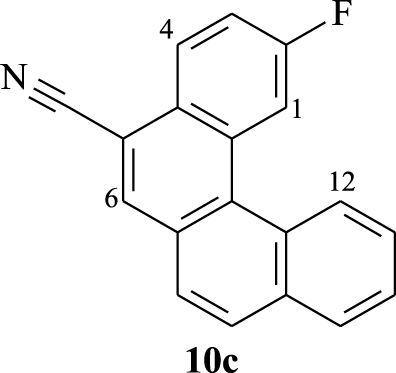 | 8.68 (dd) | 8.42 (dd) | 8.26 (s) | 8.9 (d) |
 | 8.96 (d) | 8.23 (d) | 8.16 (s) | 8.84 (d) |
s, singlet; d, doublet; dd, doublet of doublet; m, multiplet.
According to the structure of compound 10c, it is easy to assign the doublet of doublet at 𝛿 = 8.68 ppm (JH-H = 1.8 Hz, JH-F = 11.7 Hz) to proton H1 that couples with fluorine and proton H3. As a proton inside the crown, H12 reveals as a doublet with characteristic downfield shifts at 𝛿 = 8.9 ppm (J = 8.1 Hz). The assignment of the other signals could be elaborated thanks to COSY experiment (Figure 3). For instance, the correlation between the doublet of H12 and the multiplet at 7.74–7.82 ppm, which is relative to 3 protons, confirms that one of these protons is H11. That proton (H11) shows a correlation with the signal appearing at 8.06 ppm, which is clearly attributed to proton H10. The multiplet at 7.53–7.60 ppm corresponds to H3 since it shows two correlations: one with H1 and a second with the doublet of doublet at 8.42 ppm (JH-H = 9 Hz, JH-F = 6 Hz) corresponding to H4. When observing the correlations between the multiplet at 7.74–7.82 ppm and the doublet appearing at 𝛿 = 7.98 ppm (J = 8.4 Hz), we can assign this last signal to proton H7 or H8 and the multiplet to protons H9, H11, and H8 or H7.
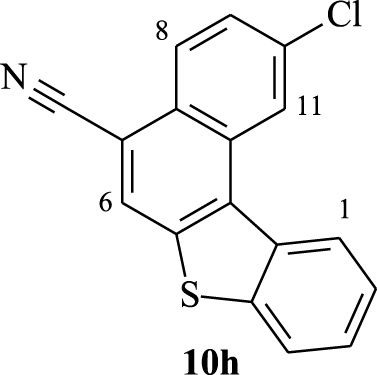
Furthermore, the 1H NMR data of compounds 10e–h show the same characteristics of a [4]helicene: the two most deshielded signals are assigned to protons H1 and H11 which are inside the crown. A singlet and a doublet or doublet of doublet corresponding to protons H6 and H8, respectively, are deshielded owing to the electron-withdrawing effect of the nitrile group and also represent characteristic protons of this family of benzo[b]naphtho[2,1-d]thiophene-7-carbonitrile. As previously, the assignment of the other signals was based on 1H–1H COSY NMR spectral analysis (Figure 4). In the case of compound 10f, protons H11 and H8 appear as a singlet at 𝛿 = 8.72 ppm and a doublet at 𝛿 = 8.24 ppm (J = 8.4 Hz), respectively. Each one of them displays a cross peak with the same proton signal belonging to the multiplet (3H) at 7.52–7.65 ppm, which is clearly attributed to H9. We noted a cross peak between the doublet of H1 (𝛿 = 8.77 ppm) and the last multiplet (3H) and a cross peak between the doublet at 𝛿 = 7.97 ppm (J = 7.8 Hz) and the same multiplet. These correlations clearly proved that the multiplet around 7.52–7.65 ppm corresponds to protons H2 and H3, while the doublet at 𝛿 = 7.97 ppm is attributed to H4.
Compound 10h exhibits very similar 1H NMR data to that of 10f within the range of 7.20–9.00 ppm. Indeed, the doublet at 𝛿 = 8.62 ppm (J = 8.4 Hz) and the doublet at 𝛿 = 8.83 ppm (J = 1.5 Hz) could be assigned to H1 and H11, respectively. The 1H–1H COSY NMR spectrum of 10h (Figure 4) shows the same four correlations as that of 10f between H1 and H2, H11 and H9, H8 and H9, and H4 and H3. This allows us to conclude that the multiplet around 7.58–7.67 ppm corresponds to protons H2, H3, and H9.
Selected characteristic 1H NMR (300 MHz) data (𝛿 in ppm) for compounds 10e–h
| Compound | 𝛿 (H1) | 𝛿 (H6) | 𝛿 (H8) | 𝛿 (H11) |
|---|---|---|---|---|
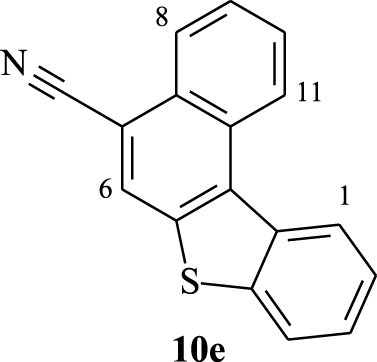 | 8.79 (dd) | 8.27 (s) | 8.38 (dd) | 8.96 (d) |
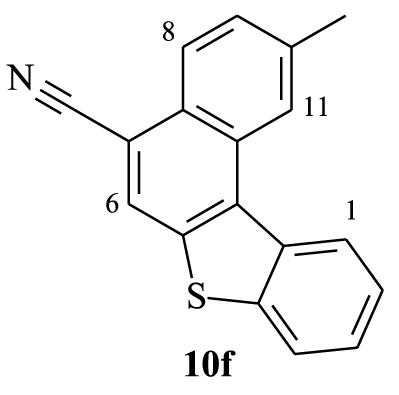 | 8.77 (d) | 8.17 (s) | 8.24 (d) | 8.72 (s) |
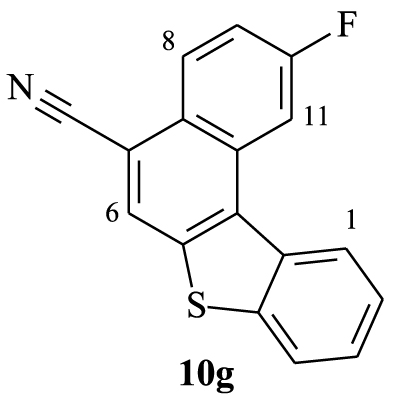 | 8.73 (d) | 8.30 (s) | 8.04 (d) | 8.62 (dd) |
 | 8.62 (d) | 8.22 (s) | 8.25 (d) | 8.83 (d) |
s: singlet, d: doublet, dd: doublet of doublet, m: multiplet.
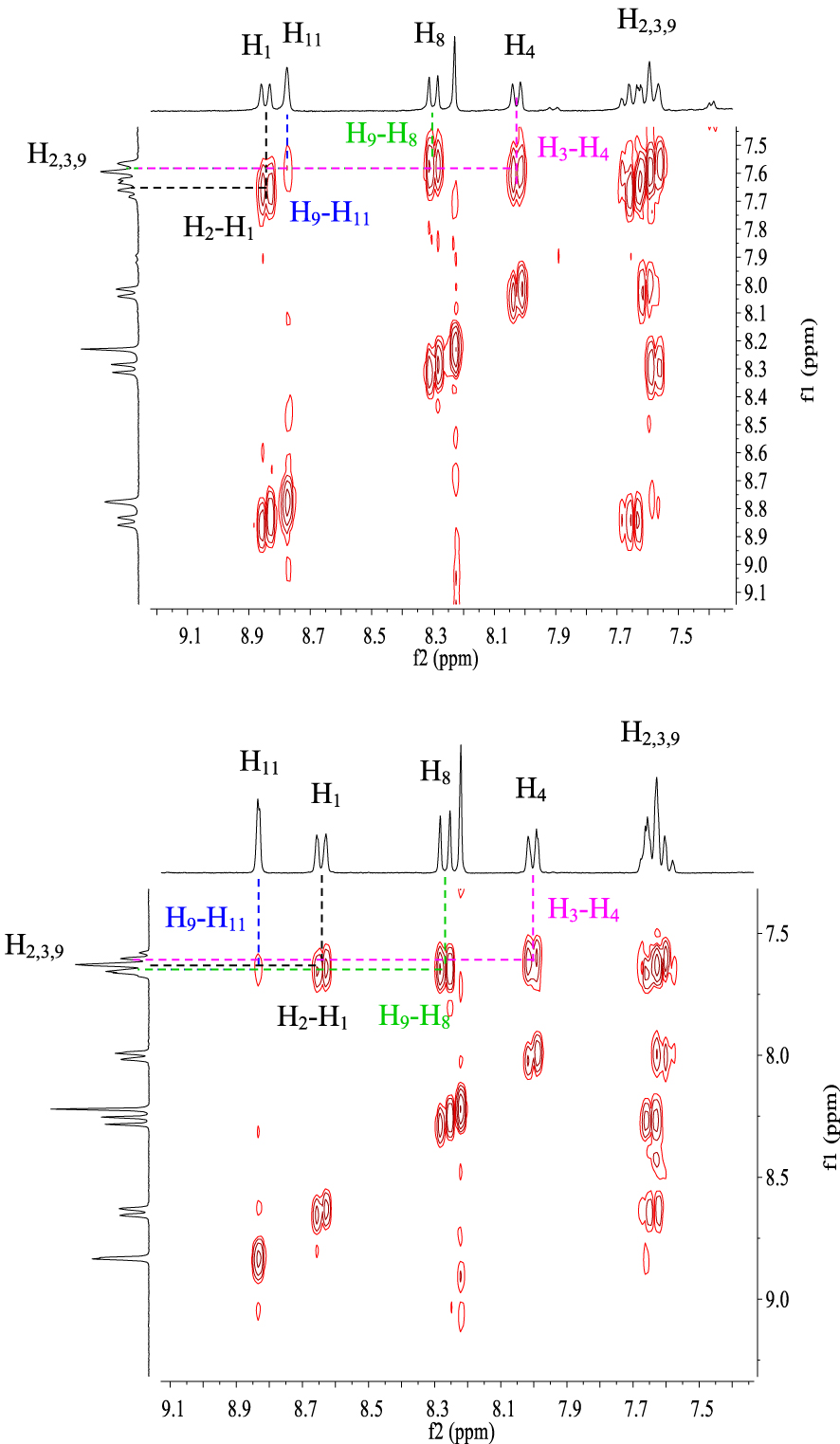
Expansion between 7.4 and 9.1 ppm of 1H–1H COSY NMR spectra (CDCl3, 300 MHz, 298 K) of compounds 10f (top) and 10h (down).
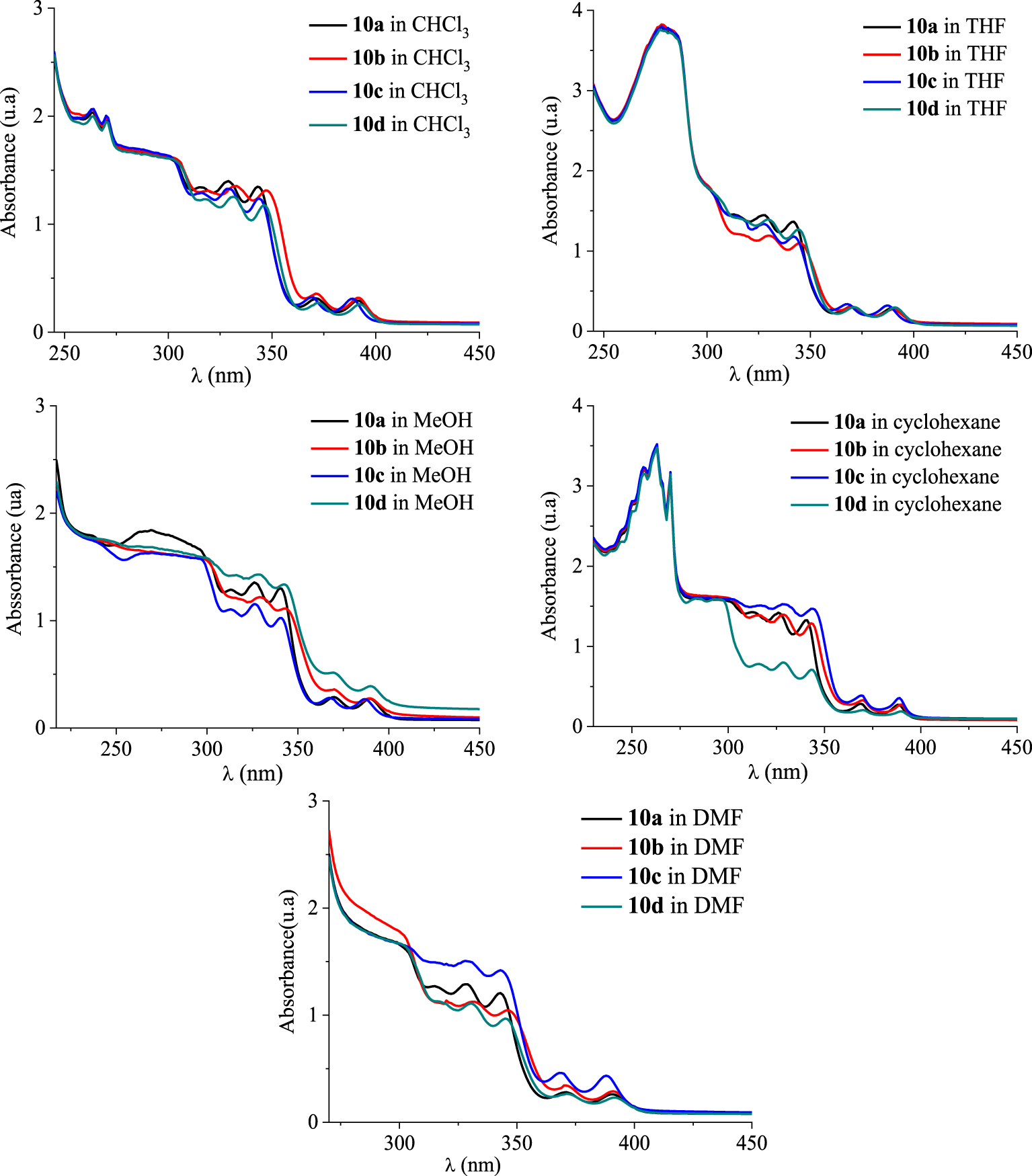
UV–Vis absorption spectra of [4]helicenes 10a–d in CHCl3, THF, MeOH, cyclohexane, and DMF in dilute solutions (c ≈ 1.5 × 10−5 mol⋅L−1).
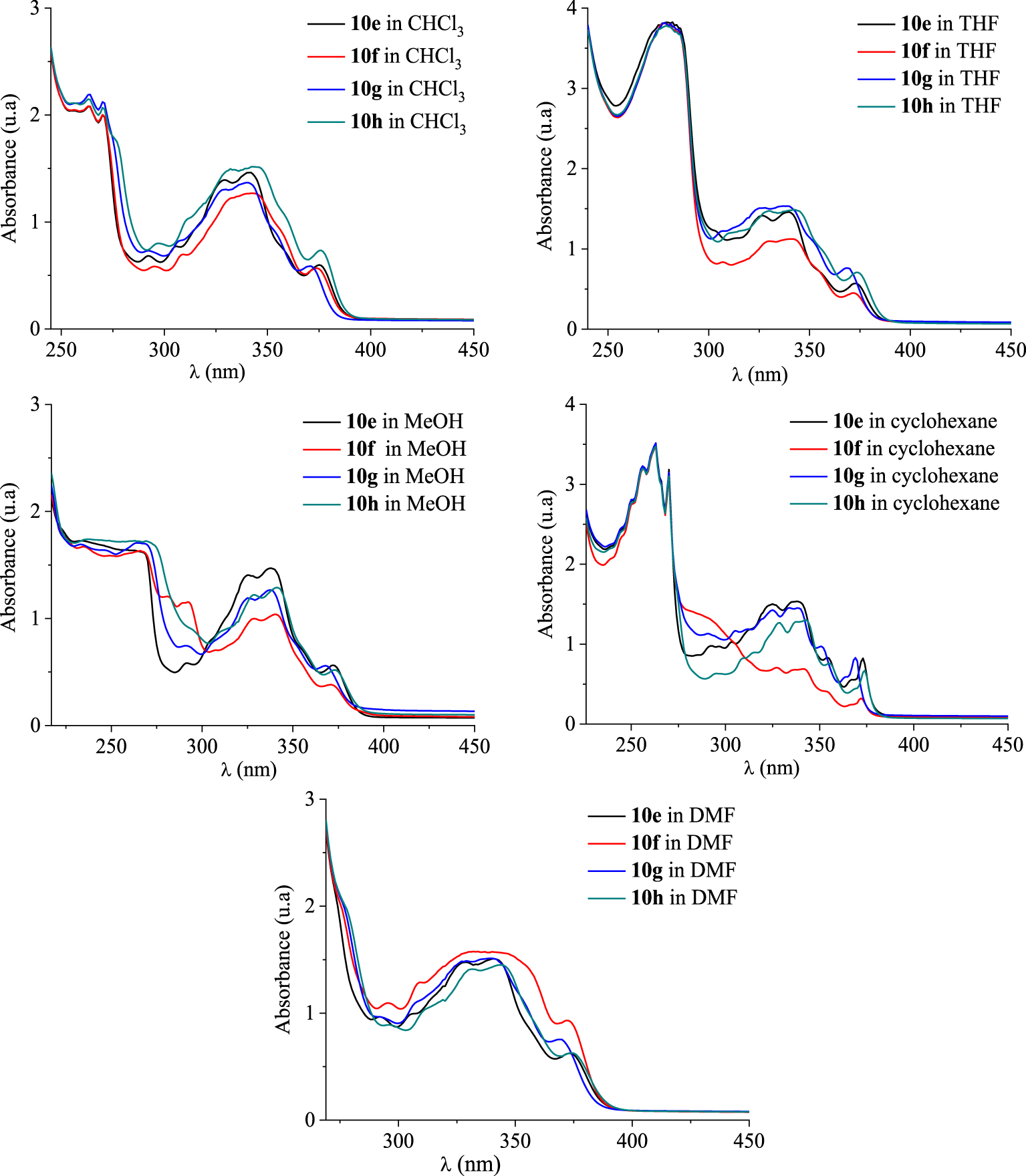
UV–Vis absorption spectra of [4]helicenes 10e–h in CHCl3, THF, MeOH, cyclohexane, and DMF in dilute solutions (c ≈ 1.5 × 10−5 mol⋅L−1).
UV–Vis absorption properties of 10a–h in CHCl3, THF, MeOH, and cyclohexane
| CHCl3 | THF | ||||||
|---|---|---|---|---|---|---|---|
| Compound | 𝜆absa (nm) | 𝜆onset (nm) | Eg-opb (eV) | 𝜆absa (nm) | 𝜆onset (nm) | Eg-opb (eV) | |
| 10a | 232, 263, 269, 315, 328, 343, 371, 391 | 402 | 3.08 | 279, 303, 313, 327, 341, 369, 389 | 400 | 3.10 | |
| 10b | 263, 270, 299, 318, 332, 347, 370, 391 | 401 | 3.09 | 279, 303, 314, 328, 342, 367, 387 | 398 | 3.11 | |
| 10c | 263, 270, 299, 315, 328, 343, 368, 388 | 397 | 3.12 | 279, 303, 316, 329, 344, 371, 390 | 397 | 3.12 | |
| 10d | 263, 270, 299, 318, 331, 346, 372, 392 | 402 | 3.08 | 279, 303, 316, 330, 345, 370, 391 | 400 | 3.10 | |
| 10e | 263, 270, 292, 306, 336, 370 | 389 | 3.18 | 279, 303, 325, 340, 354, 372 | 386 | 3.21 | |
| 10f | 263, 270, 294, 306, 341, 374 | 388 | 3.19 | 279, 303, 329, 340, 354, 372 | 387 | 3.20 | |
| 10g | 263, 270, 292, 306, 336, 370 | 384 | 3.22 | 279, 303, 325, 338, 354, 368 | 383 | 3.23 | |
| 10h | 263, 270, 296, 309, 331, 344, 375 | 390 | 3.17 | 279, 311, 329, 343, 355, 374 | 388 | 3.19 | |
| MeOH | Cyclohexane | ||||||
| 𝜆absa (nm) | 𝜆onset (nm) | Eg-opb (eV) | 𝜆absa (nm) | 𝜆onset (nm) | Eg-opb (eV) | ||
| 10a | 235, 268, 313, 326, 340, 370, 389 | 400 | 3.10 | 243, 249, 256, 262, 270, 297, 312, 326, 340, 368, 388 | 397 | 3.12 | |
| 10b | 279, 315, 329, 343, 369, 389 | 403 | 3.07 | 243, 249, 256, 262, 270, 299, 316, 328, 343, 369, 388 | 396 | 3.13 | |
| 10c | 237, 279, 313, 326, 341, 367, 387 | 398 | 3.11 | 243, 249, 256, 262, 270, 297, 315, 328, 343, 369, 389 | 397 | 3.12 | |
| 10d | 242, 279, 316, 327, 342, 370, 389 | 401 | 3.09 | 243, 249, 256, 262, 270, 283 295, 315, 328, 343, 369, 389 | 398 | 3.11 | |
| 10e | 235, 263, 290, 301, 324, 338, 372 | 388 | 3.19 | 243, 249, 256, 262, 270, 291 302, 311, 324, 337, 354, 366, 373 | 383 | 3.23 | |
| 10f | 235, 266, 281, 291, 327, 340, 371 | 386 | 3.21 | 243, 249, 256, 262, 270, 292 305, 327, 340, 353, 366, 371 | 382 | 3.24 | |
| 10g | 233, 245, 266, 291, 307, 323, 337, 368 | 383 | 3.23 | 243, 249, 256, 262, 270, 290 305, 311, 324, 333, 338, 350, 362, 370 | 378 | 3.28 | |
| 10h | 236, 268, 294, 310, 327, 341, 373 | 386 | 3.21 | 243, 249, 256, 262, 270, 294 308, 313, 328, 342, 355, 367, 373 | 383 | 3.23 | |
aAbsorption maxima measured in solution (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature.
b The optical gap (Eg-op) was estimated from the onset point of the absorption spectrum: Eg-op = 1240∕𝜆onset.
Photophysical properties of the novel compounds 10a–h have been performed using UV–Vis absorption and photoluminescence (PL) spectroscopies at room temperature. The absorption spectra were measured in different solvents such as chloroform (CHCl3), methanol (MeOH), tetrahydrofuran (THF), N,N-dimethylformamide (DMF), and cyclohexane and the resulting optical parameters are gathered in Tables 5 and 6. In dilute chloroform solutions (c ≈ 1.5 × 10−5 mol⋅L−1), the absorption spectra of compounds 10a–d exhibited similar features including several bands, with very close absorption coefficients, that are characteristic of π–π* and n–π* transitions (Figure 5). In the high-energy region, two thin and intense absorption bands are observed at 263 nm (ε ∼ 132 000–138 000 M−1⋅cm−1) and 270 nm (ε ∼ 128 000–134 000 M−1⋅cm−1). By examining the low-energy region (300–350 nm), three absorption bands are clearly identified around 315, 328, and 343 nm for compounds 10a, 10c, and 10d, respectively, and a bathochromic shift of about 5 nm was noted for the absorption maxima of 10b. At longer wavelengths (>350 nm), two weaker absorption bands are located between 350 nm and 400 nm with intensities within the range 17 900–24 800 M−1⋅cm−1 revealing a hypsochromic shift of about 3–4 nm for the absorption maxima of 10c.
The absorption spectra of 10a–d in the other solvents showed markedly different behaviors especially below 300 nm. In cyclohexane and THF, the tetracyclic systems 10a–d absorbed strongly within the range 250–300 nm. In THF solutions, their absorption spectra displayed a strong absorption at 279 nm (ε ≈ 253 000 M−1⋅cm−1) and a shoulder peak located at 298 nm. In MeOH, only a broad band was observed with a maximum peak (111 000–122 000 M−1⋅cm−1) around 268 nm. By changing CHCl3, MeOH, and THF to cyclohexane, the UV–Vis spectra showed a vibronic absorption. When using DMF, a weak absorption at 296–301 nm could be found.
We noticed a remarkable difference regarding the intensities of the absorption bands for compounds 10a–d in cyclohexane, methanol, and DMF within the range 300–400 nm. Compared to 10a–c, compound 10d exhibited five less intense absorption bands (𝜆 = 316, 328, 343, 369, and 389 nm) in cyclohexane, CHCl3, and DMF, but the most intense ones in MeOH. On the contrary, the intensities of the bands for compound 10c were found to be the highest in DMF and cyclohexane, but low in MeOH. The other compounds exhibited similar bands with comparable intensities.
As depicted in Figure 6, the absorption spectra of compounds 10e–h in CHCl3, THF, MeOH, cyclohexane, and DMF at room temperature exhibit similar features as compounds 10a–d below 278 nm, but they absorbed differently in the highest wavelengths. In the low-energy region (>300 nm), the absorption spectra of the benzo[b]naphto[2,1-d]thiophene-7-carbonitriles 10e–h show several and remarkably larger bands, compared to those of 10a–d. This resulting behavior may be explained by the elongation of the conjugated structure due to the presence of a thiophene nucleus in 10e–f. It is interesting to notice that compound 10h shows a bathochromic shift of about 3–4 nm for the low-energy maxima at 374 nm in THF. Moreover, in cyclohexane the low-energy maxima at 373 nm for compounds 10e,h are slightly shifted to the highest wavelengths.
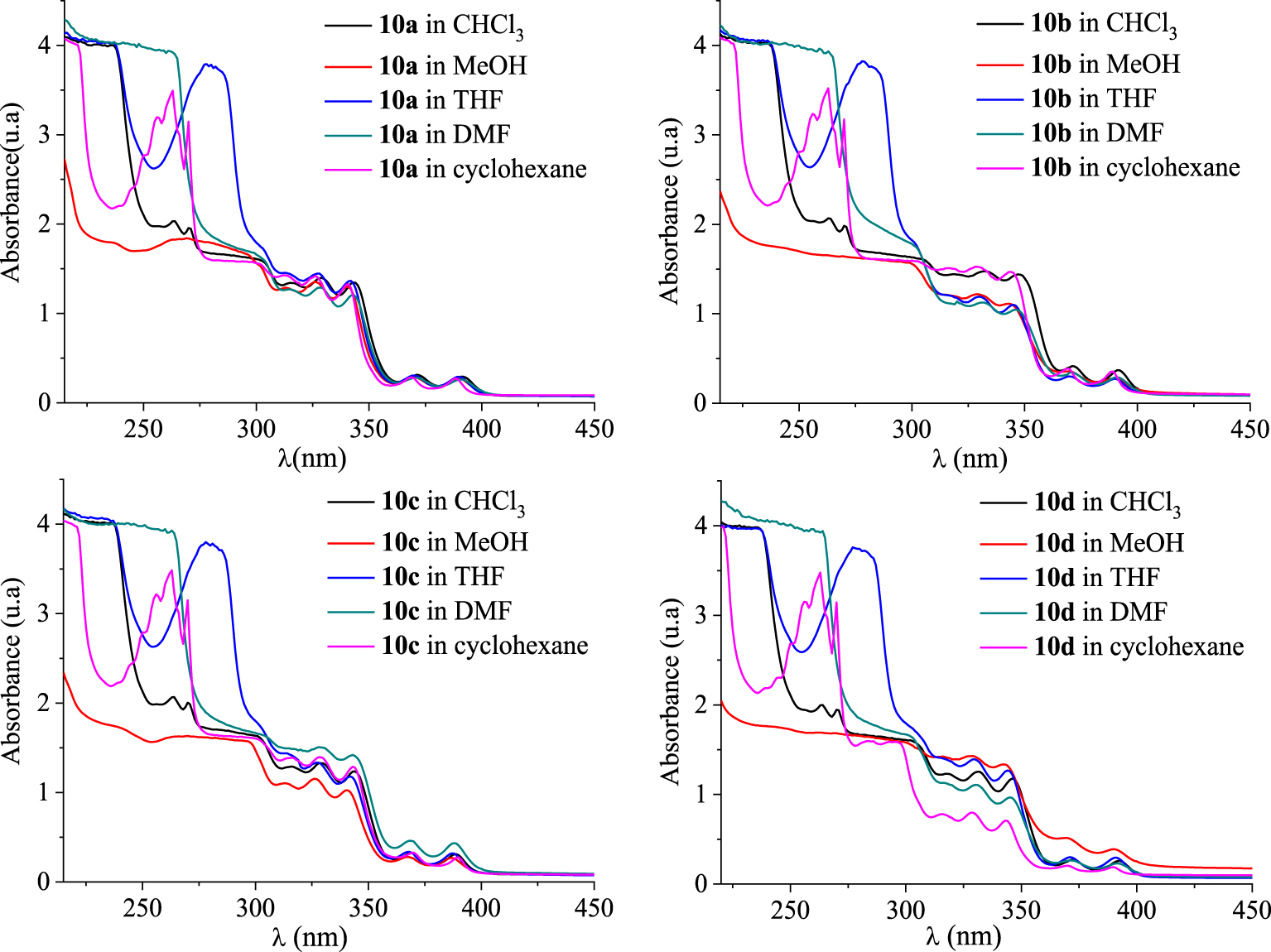
UV–Vis absorption spectra of [4]helicenes 10a–d in increasing polarity of the tested solvents (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature.
UV–Vis absorption properties of 10a–h in DMF
| Compound | 𝜆absa (nm) | 𝜆onset (nm) | Eg-opb (eV) |
|---|---|---|---|
| 10a | 296, 314, 328, 343, 370, 390 | 403 | 3.07 |
| 10b | 299, 320, 331, 346, 370, 390 | 403 | 3.07 |
| 10c | 301, 316, 329, 343, 368, 387 | 401 | 3.09 |
| 10d | 297, 317, 330, 345, 371, 391 | 403 | 3.07 |
| 10e | 292, 304, 326, 341, 373 | 389 | 3.18 |
| 10f | 294, 308, 326, 341, 373 | 388 | 3.19 |
| 10g | 292, 306, 325, 340, 371 | 386 | 3.21 |
| 10h | 296, 312, 329, 344, 374 | 391 | 3.17 |
aAbsorption maxima measured in solution (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature.
bThe optical gap (Eg-op) was estimated from the onset point of the absorption spectrum: Eg-op = 1240∕𝜆onset.

UV–Vis absorption spectra of [4]helicenes 10e–h in increasing polarity of the tested solvents (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature.
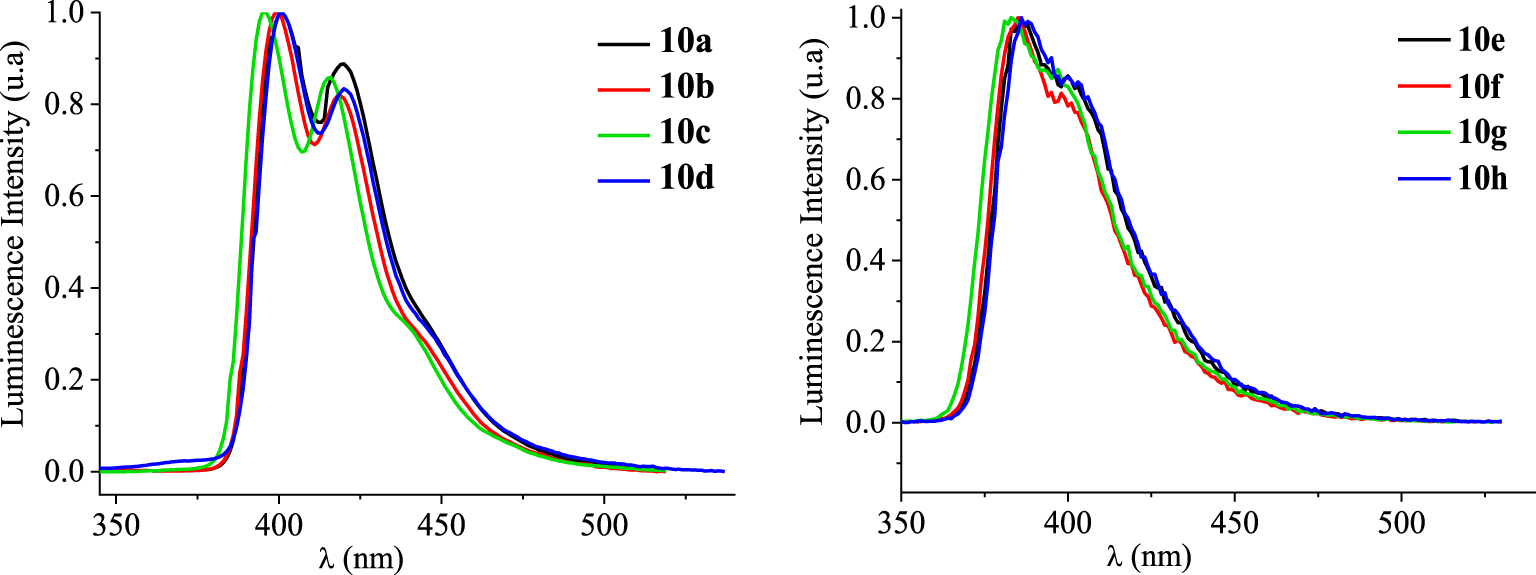
Normalized emission spectra (𝜆exc = 340 nm) of 10a–h in dilute chloroform solutions (c ≈ 1.5 × 10−5 mol⋅L−1).
Photoluminescence properties of [4]helicenes 10a–h in chloroform. Stokes shifts are calculated in wavelength and wavenumber units
| Compound | Photoluminescence | Stokes shift | |||
|---|---|---|---|---|---|
| FWHMd (nm) | 𝛥ν (cm−1) | ||||
| 10a | 400b, 419, 441c | 43 | 9 | 575 | |
| 10b | 399b, 419, 438c | 41 | 8 | 512 | |
| 10c | 395b, 415, 435c | 39 | 7 | 456 | |
| 10d | 400b, 420, 440c | 41 | 8 | 510 | |
| 10e | 386b, 395c | 41 | 16 | 1120 | |
| 10f | 385b, 396c | 38 | 11 | 763 | |
| 10g | 388b, 395c | 40 | 13 | 917 | |
| 10h | 387b, 397c | 40 | 12 | 826 | |
a Emission measured in chloroform (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature; fluorescence excitation at 340 nm.
b Emission maximum (
c Shoulder peak.
d Spectrum full width at half maximum.
As shown in Figures 7 and 8, a strong absorbance accompanied by a large bathochromic shift of about 15 nm within the range 250–300 nm is observed in dilute THF solutions. Compounds 10c, 10f, 10g, and 10h showed less absorption bands when changing the solvent from CHCl3, cyclohexane, DMF, or THF to MeOH. The change of solvent does not considerably affect the photophysical features of compounds 10a,e at longer wavelengths ranging from 320 nm to 400 nm. The absorption bands above 350 nm are less intense than the other observed ones and could be ascribed to the intramolecular charge transfer (ICT) from the [4]helicene unit to the electron-withdrawing cyano group. The optical gap energy (Eg-opt) of each [4]helicene was calculated from the absorption onset and was found to be 3.07 to 3.31 eV (Tables 5 and 6). Their UV profiles resembled those of some tetracyclic systems having interesting photophysical properties [42, 43, 44].
The photoluminescence (PL) spectra of [4]helicenes 10a–h have been recorded in chloroform solutions (c ≈ 1.5 × 10−5 mol⋅L−1) at room temperature showing a blue light emission (Figure 9). Compounds 10a–d present structured emissions displaying two bands with different intensities followed by one shoulder peak (Table 7). The emission maximum of the fluorinated derivative 10c (𝜆ems = 395 nm) is shifted to the lower wavelengths by 4–5 nm, in comparison to those of 10a,b,d (𝜆ems = 399–400 nm). Unlike the previous compounds, the emission spectra of the benzo[c]phenanthrene-like derivatives 10e–h show one slightly wider main emission band (𝜆ems = 385–388 nm) incorporating a shoulder peak around 395 nm.
4. Conclusion
In conclusion, we have achieved the synthesis of various α,β-unsaturated nitriles bearing reactive functional groups, which are precursors of new [4]helicenes. Our synthetic strategy involves only two steps and offers a simple and direct pathway toward a series of functionalized tetracyclic π-conjugated systems in good overall yields. The UV–Vis absorption and the photoluminescence properties of the target benzo[c]phenanthrene and benzo[b]naphto[2,1-d]thiophene derivatives have been experimentally evaluated in solutions and an emission in the blue region of the visible spectrum was noted. The obtained results seem encouraging for the examination of such compounds as promising materials for optoelectronic applications.