

1 Introduction
In recent years, phosphines where the phosphorus atom is included into a cyclic moiety have been developed as chiral ligands for applications in enantioselective, transition-metal-promoted catalysis. Representative examples include DuPHOS 1 [1], BPE 2 [2], TangPHOS 3 [3], RoPHOS 4 [4], PennPHOS 5 [5], phosphinane 6 [6], and the ferrocenylphosphine 7 [7] (Fig. 1).
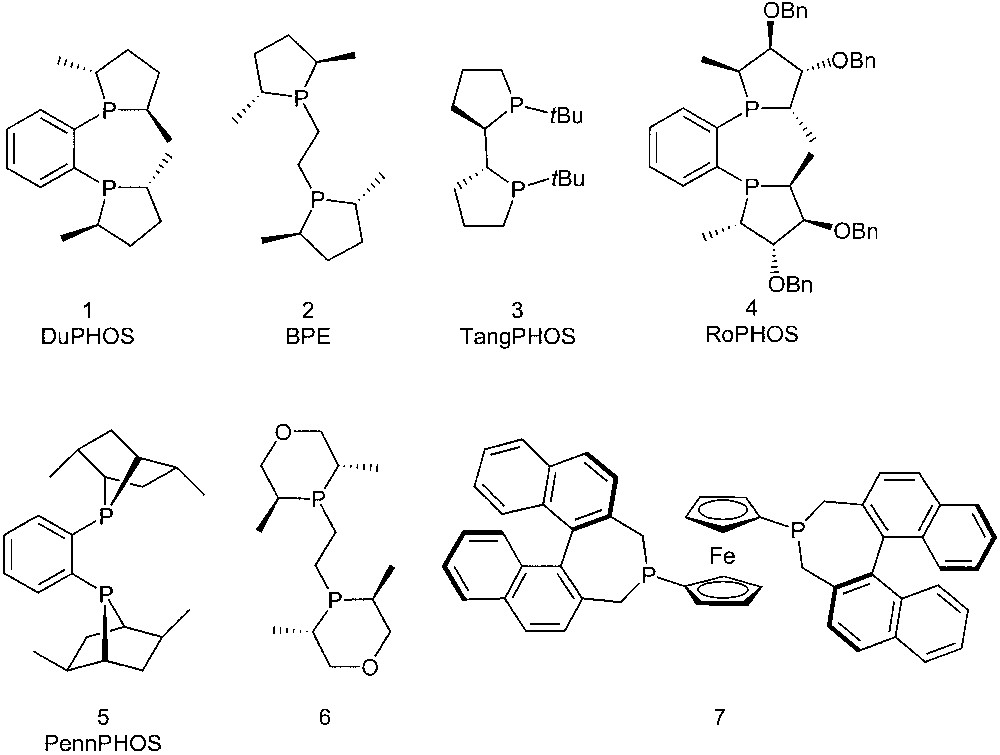
Mainly five-membered rings, but also a few examples of six- and seven-membered cyclic phosphines appear in this series. Their catalytic behaviour is mainly determined by the C2 symmetry, the strong electron-donating power, as well as by the conformational constraints induced by the cyclic structure. All the above ligands are very efficient chiral auxiliaries in a number of catalytic reactions.
Thus, it appears that heterocyclic phosphines are emerging at present as an especially interesting class of chiral ligands, with peculiar catalytic properties. In this context, phosphetane rings also have been used as chiral synthons for ligand design.
This short review summarizes the most recent developments in the synthesis of chiral phosphetanes and their applications in enantioselective organometallic catalysis.
2 Background
The systematic preparation of chiral phosphetanes, with catalytic purposes, started in the early 90’s8 with the development of a large series of mono- and bidentate derivatives of general formula 8 (Fig. 2).
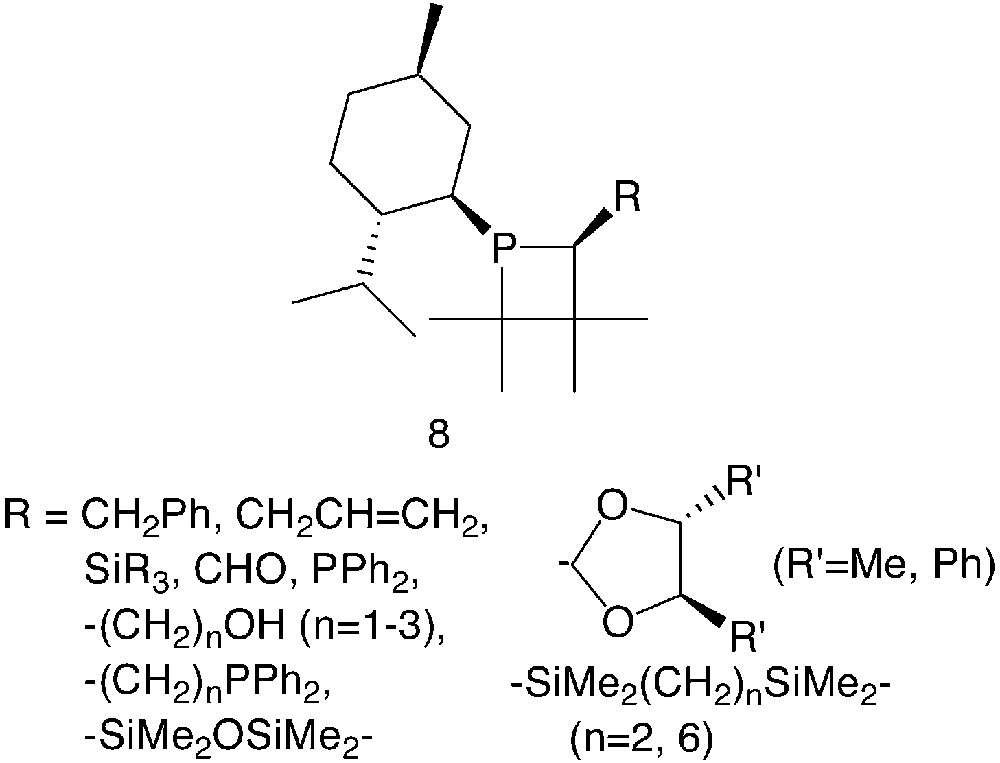
In these compounds, the phosphorus atom itself represents a stereogenic centre. For R ≠ H, the α-carbon is chiral as well. The menthyl substituent served as chiral auxiliary to produce the optically pure phosphetanes through fractional crystallisation of diastereomeric mixtures of the parent phosphetane oxide (R = H). A number of α-substituents, R, were introduced either directly via diastereoselective metalation–alkylation reactions of the parent phosphetane oxide (R = CH2Ph [8], CH2CH=CH2 [8], CHO [9], SiR3 [10], PPh2 [11]) or by subsequent modifications of the R group (R = –(CH2)nOH [12], –(CH2)nPPh2 [12], and 4,5-disubstituted-1,3-dioxolan-1-yl groups [10, 13]). Monodentate phosphetanes of this series were evaluated mainly in palladium-catalysed olefin hydrosilylation reactions [9, 14] and allylic nucleophilic substitutions [13] on model substrates.
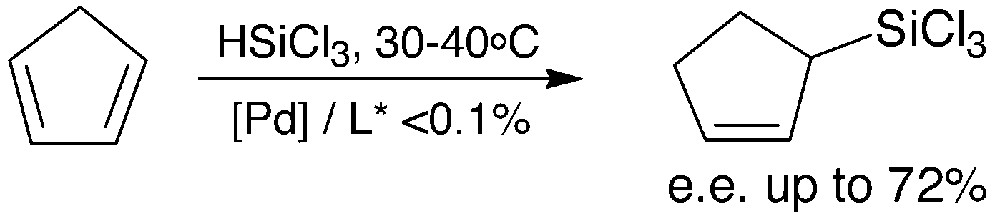
Notably, high catalytic activity and moderate to high enantioselectivities were obtained in the catalytic hydrosilylation of cyclopentadiene (Fig. 3).
Moderate catalytic activities were observed in the allylic substitutions promoted by the palladium–phosphetane complexes. The enantioselectivity was highly dependent on the nature of the R substituent as well as on the relative configuration of the various chiral centres of the phosphetane ligands. The best enantiomeric excesses were obtained with phosphetane 8a, bearing a 4,5-diphenyl-1,3-dioxolan-2-yl substituent of R,R-configuration, associated with an R-configured phosphorus (Fig. 4).
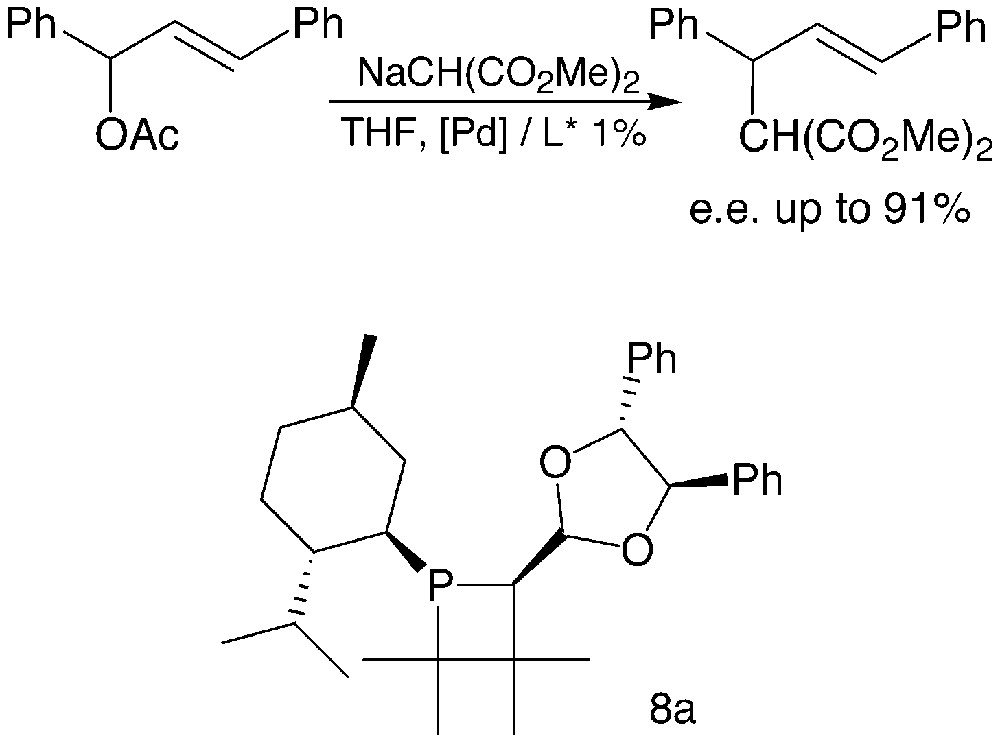
The synthesis and catalytic applications of phosphetanes 8 have been summarised in a recent review [15].
Generally speaking, studies on phosphetanes 8 indicated that phosphetane rings are suitable structural units for building chiral ligands for selected applications in asymmetric catalysis. Thus, following the pioneering work above, a new family of phosphetane-based chiral ligands has been developed recently. These phosphetanes fall into the general formula 9, where the symmetrically substituted α-carbons are the only stereogenic centres (Fig. 5).
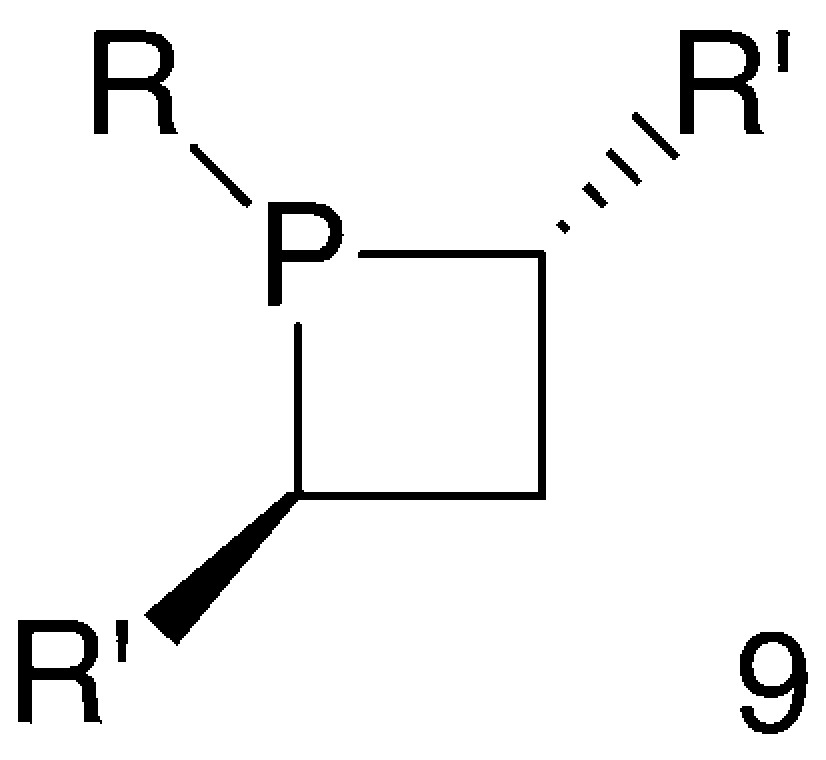
The synthesis and properties of this new family of chiral phosphetanes are presented hereafter.
3 Recent developments
The first examples of phosphetanes of the general formula 9 were described in 1997 [16] and, since then, many others have been prepared. Their easy availability has been, undoubtedly, a key point for their development as enantiomerically pure auxiliaries in catalytic reactions.
The synthetic approach to phosphetanes 9 involves the reaction between a primary phosphine and a chiral 1,3-dielectrophile such as the bis-mesylate or the cyclic sulphate of an enantiomerically pure 1,3-diol, in the presence of bases, as shown in Fig. 6.
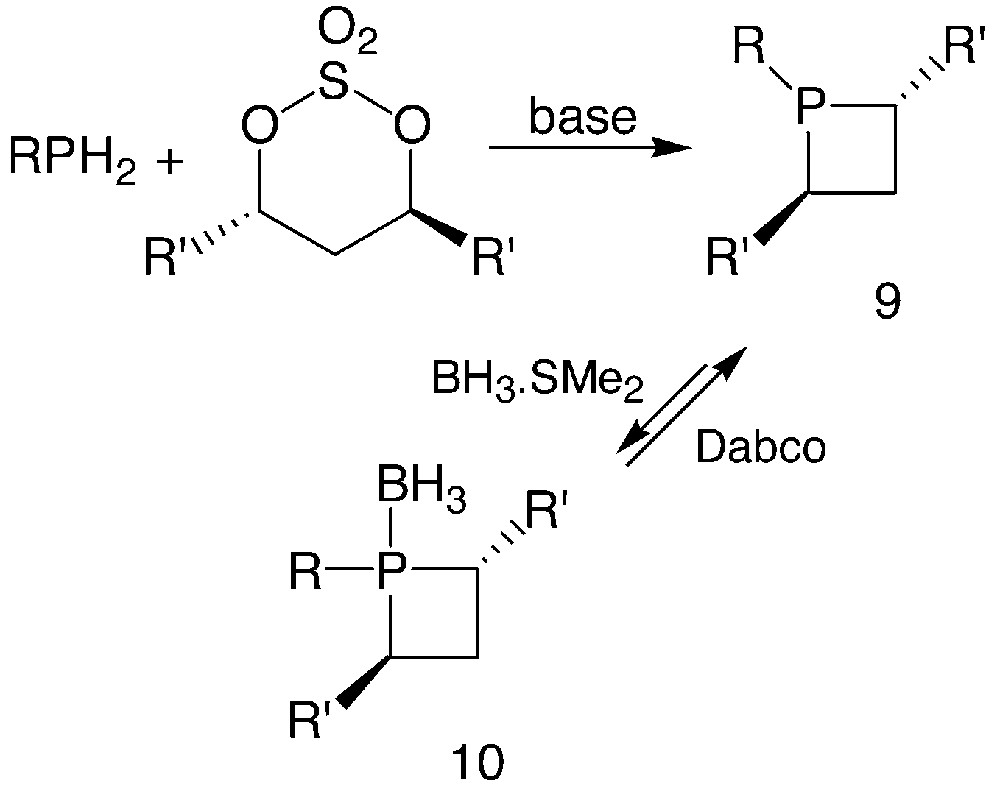
Usually, n-BuLi or s-BuLi are used as the base, in experimental conditions that depend on the nature of the primary phosphines. Starting from PhPH2, the dilithiated phosphine is formed and reacted then with the cyclic sulphate between –40 °C and room temperature [16]. In most cases, the dilithiated phosphine being hardly available, a two-step reaction must be performed: the monoanion is reacted with the cyclic sulphate, then a second equivalent of base is added to achieve the cyclisation [16, 17]. An alternative procedure involves slow addition of two equivalents of base to a solution containing the primary phosphine and the cyclic sulphate in a 1:1 ratio [18]. For the synthesis of the most air-sensitive phosphetanes, the borane-complexation technique is conveniently used to protect the phosphorus atom: an excess of the BH3.SMe2 complex is added to the crude reaction mixture to form the borane-phosphetane complex 10. After purification, phosphetanes can be easily displaced from their borane complexes by heating with an excess of 1,8-diazabicyclo[2.2.2]octane (Dabco) [16, 17].
Given that a great number of primary phosphines are either commercially available or easily prepared through known methods, the broad scope of the synthetic approach above is ensured. On the other hand, its practical utility relies mainly on the availability of symmetrically substituted chiral 1,3-diols. Both enantiomers of 2,4-pentanediol are commercially available and many other diols are obtained, in large amounts, by enantioselective hydrogenation of the corresponding 1,3-diketones promoted by ruthenium complexes. Atropoisomeric chiral ligands such as BINAP and MeO–BIPHEP afford very high diastereomeric and enantiomeric excesses in these hydrogenations [19–21] (Fig. 7).
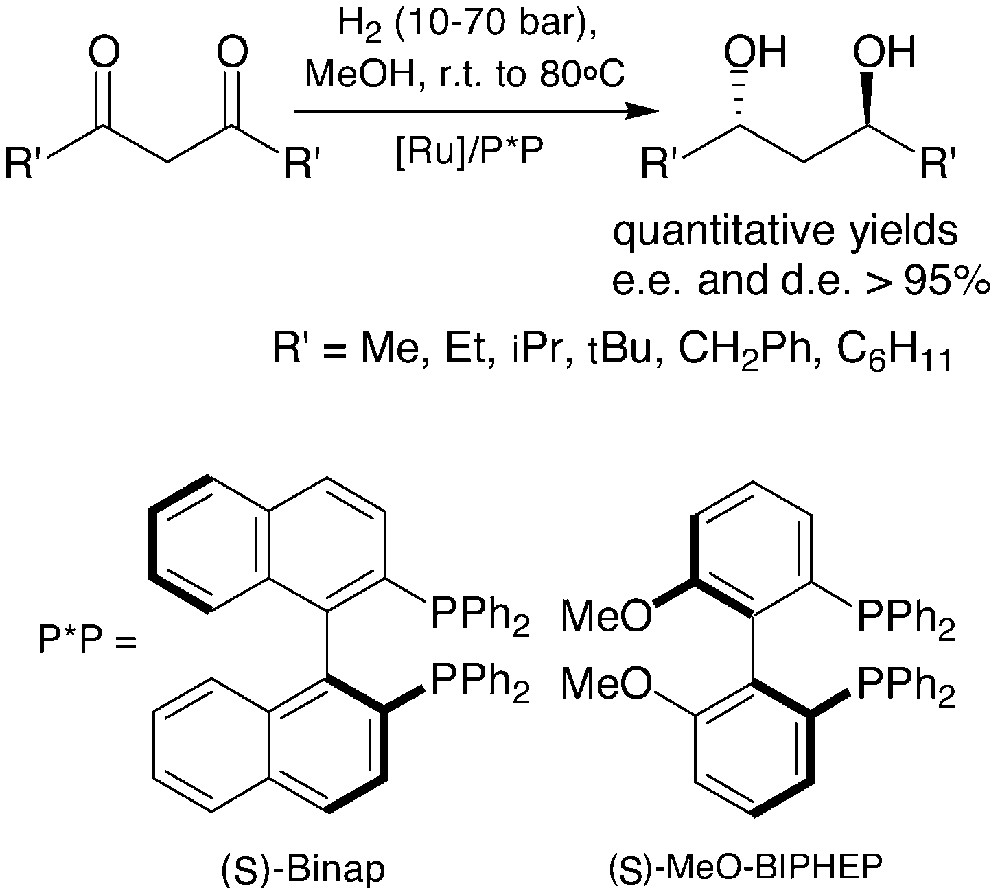
The cyclic sulphates of the anti-1,3-diols shown in Fig. 7 have been used for the synthesis of a number of monodentate and bidentate phosphetanes, according to the general method in Fig. 6. Selected examples of thus obtained, monodentate phosphetanes are shown hereafter (Fig. 8) [16, 17, 22, 23].
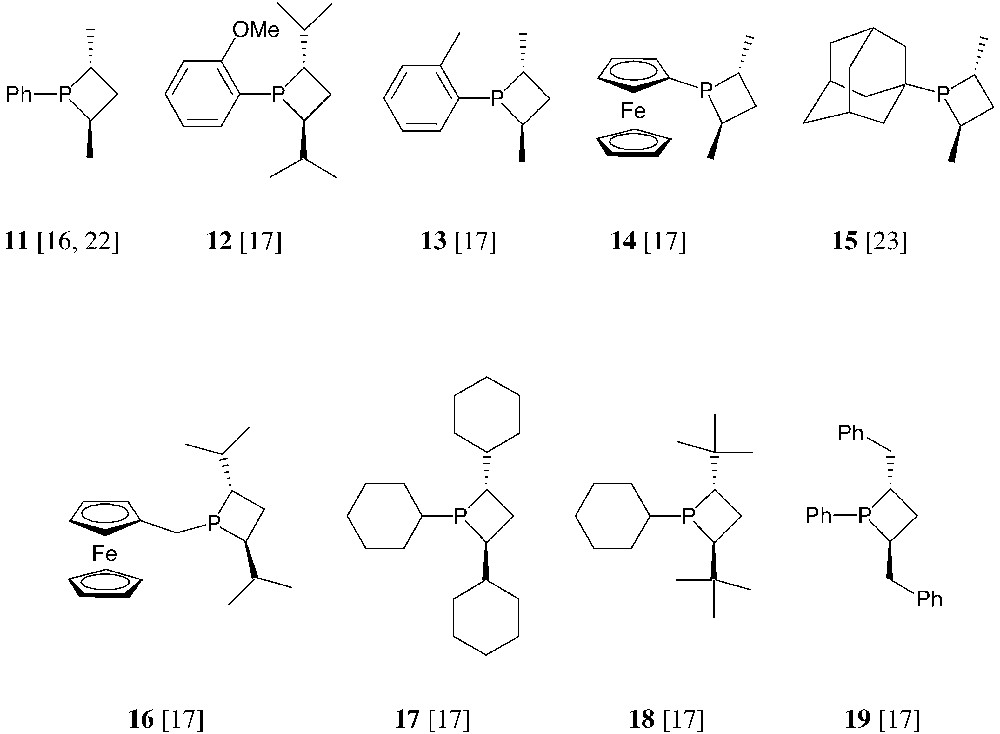
This overview shows that the structural features of monodentate phosphetanes are easily modulated by the suitable choice of the starting materials. Also, their properties and behaviour as chiral ligands should be finely tuned by these structural variations. Thus, for instance, P-alkyl-substituted phosphetanes such as 15, 16 and 18, should behave as better electron-donors toward transition metals then the analogous P-aryl derivatives 11 or 12. Moreover, the sterical hindrance of the ring substituents varies drastically when going, for instance, from phosphetanes 11 to 15, 17 or 18. The sterical hindrance, and especially the relative size of the substituents, is expected to play a major role in the stereochemical control of the catalytic reactions: as shown in Fig. 9, the chiral environment of the metal in monodentate phosphetane complexes is mainly defined by the relative size of the phosphorus substituent and the α-carbon substituents (note that one of the α substituents R′ being faraway from the metal, its sterical hindrance is negligible).
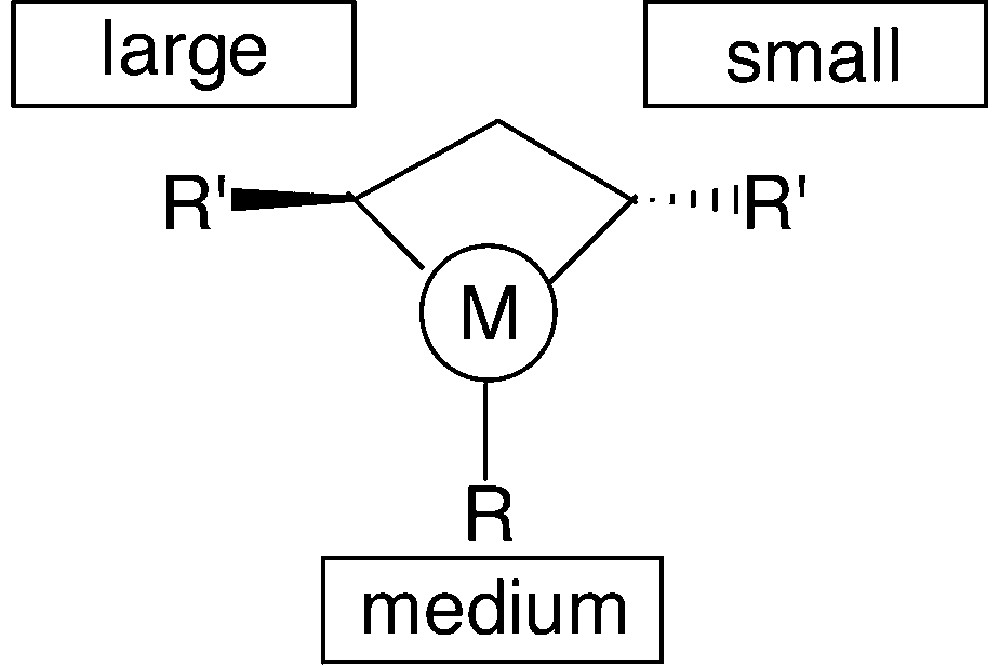
Consequently, an appropriate choice of R and R′ allows optimisation of the catalytic properties of these ligands. This has been shown through studies on a model reaction: the rhodium promoted hydrogenation of α-acetamidocinnamic acid [17] (Fig. 10).
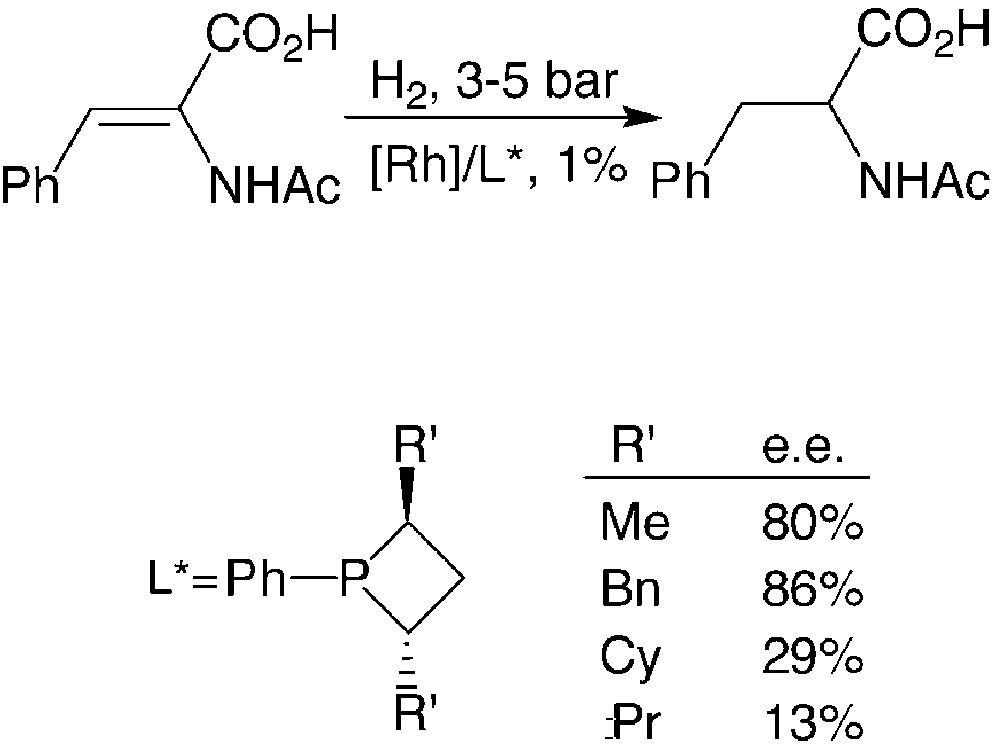
The enantiomeric excesses afforded by P-phenyl-substitutes phosphetanes are strongly dependent on the nature of the R′ substituent: they vary from 13%, for R′ = iPr, to 86%, for R′ = PhCH2. Full rational for the observed trend is not available to date.
The best e.e.s values obtained in these reactions compare favourably with those attained by other monodentate phosphines [24] and confirm the promising potential of these phosphetanes in asymmetric catalysis. Monodentate phosphetanes are not expected to successfully compete with bidentate species in the rhodium-promoted asymmetric hydrogenations above, however they should find specific use in reactions that require monodentate ligands, such as palladium-promoted olefin hydrosilylations [25] and nickel-promoted hydrovinylations [26].
Concerning rhodium, as well as ruthenium promoted enantioselective hydrogenations of unsaturated substrates, the choice ligands are, usually, C2-symmetric bidentate phosphines. This has been observed in the phosphetane series as well: the bidentate, C2-symmetric phosphetanes 20, 21 and 22 have been prepared and successfully used in these hydrogenation reactions (Fig. 11).
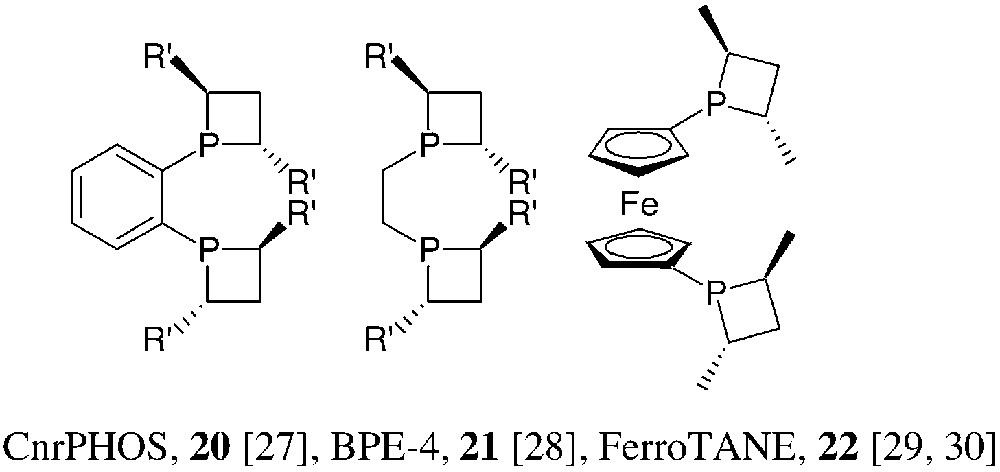
The synthetic method used for the preparation of 20–22 follows the general strategy of Fig. 6, starting from 1,2-bis(phosphino)benzene, 1,2-bis(phosphino)ethane and 1,1′-bis(phosphino)ferrocene, respectively. Yields are moderate (30–60%). CnrPHOS and BPE-4 bearing small R′ substituents are rather air-sensitive. Consequently, they are more easily isolated as their mono- or bis-borane complexes. The mono-borane complex 23 of (S,S)-iPr-CnrPHOS has been characterised by X-ray crystallography [31] (Fig. 12).
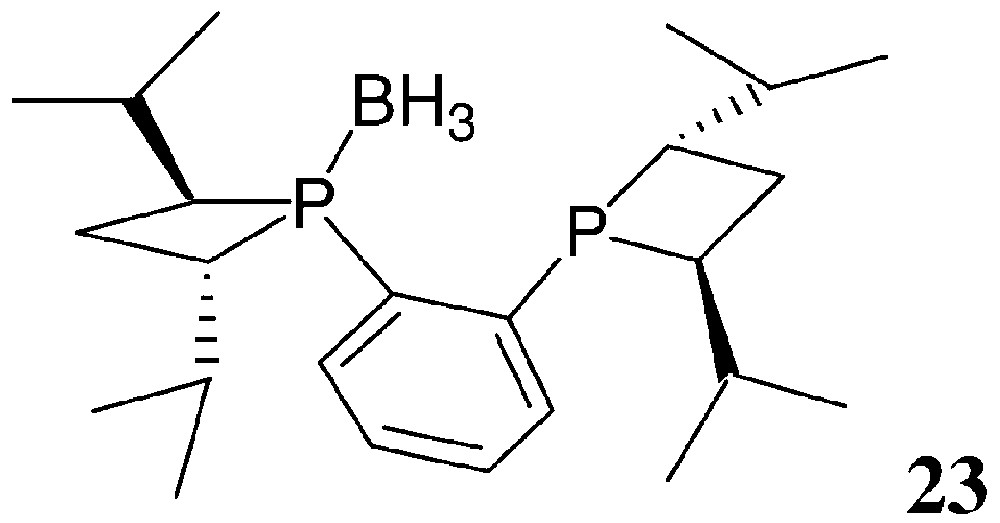
FerroTANEs and sterically hindered CnrPHOS and BPE-4 are, however, air-stable, easy to handle compounds.
All these diphosphines behave as chelating ligands toward transition metals and give stable rhodium, ruthenium and palladium complexes (Fig. 13). Some of them have been structurally characterised.
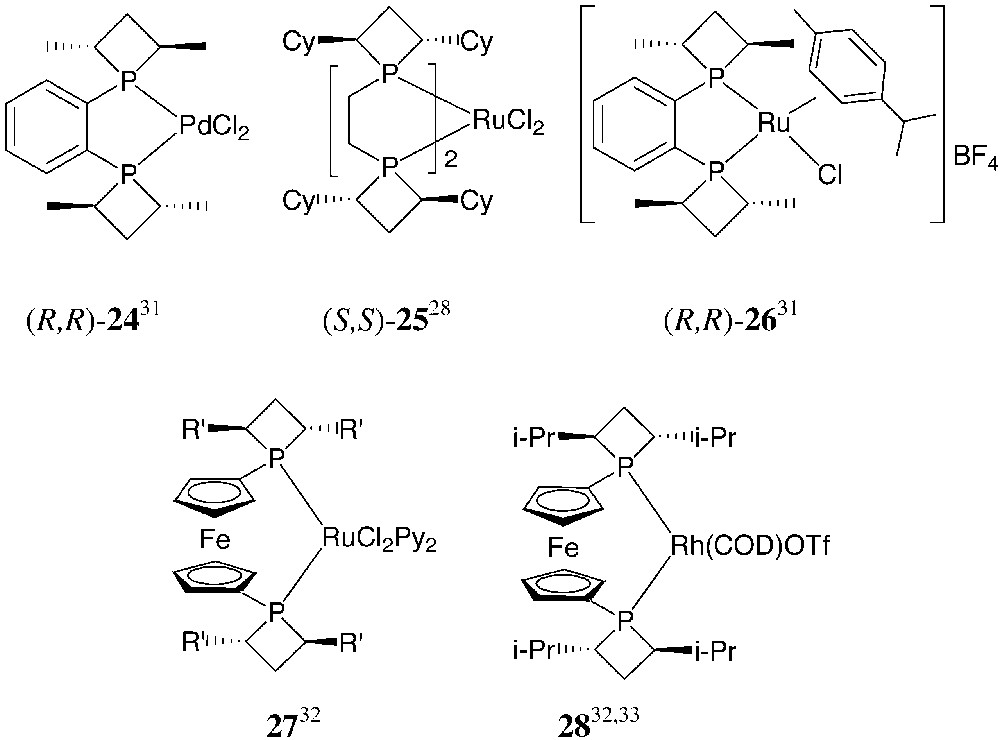
From [28, 31–33].
Ruthenium complexes of phosphetanes 20–22 have been used as catalysts in the enantioselective hydrogenation of functionalised carbonyls [27, 28, 31, 32]. β-Ketoesters and 2,4-pentanedione have been considered as model substrates. Selected results are summarised in Table 1.
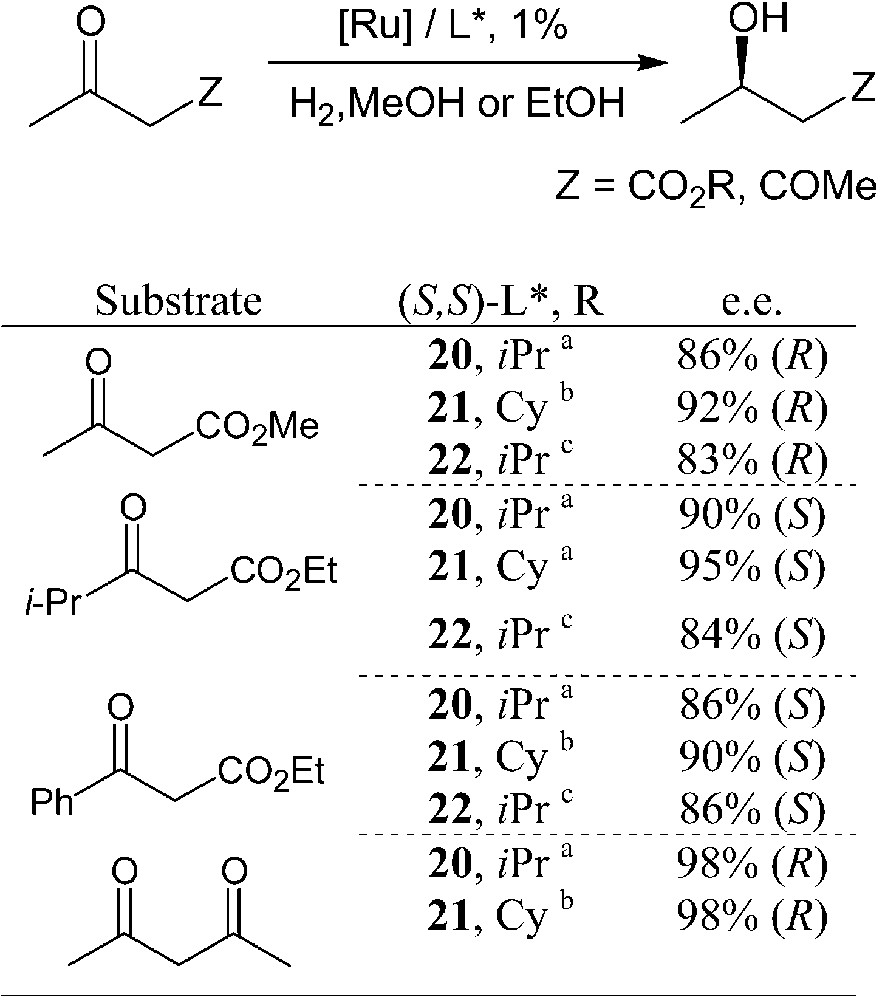
Enantioselective hydrogenations of carbonyl derivatives promoted by ruthenium phosphetane complexes.
Catalyst precursors: (a) (COD)Ru(2-Me-allyl)2 + HBr; (b) [(C6H6]RuCl2]2; (c) L*RuCl2Py2 + HBr.
Bidentate phosphetanes 20–22 display moderate-to-high enantioselectivities in these hydrogenation reactions. Within each series, the highest efficiency is attained by using sterically hindered ligands, that is for R′ = isopropyl or cyclohexyl. According to these preliminary catalytic tests, Cy-BPE-4 seems to be the most suitable ligand for these hydrogenations. It display, however, lower catalytic activity – high temperatures and/or high H2 pressure are required – and a somewhat lower enantioselectivity than the analogous phospholane-based BPE ligands [34].
From catalytic tests on rhodium-promoted hydrogenations of functionalised olefins, FerroTANEs 22 emerged as the most suitable ligands, within the bis-phosphetane series. Selected results are summarised in Table 2 [29, 30, 32, 35]. FerroTANEs displayed especially high catalytic efficiency and enantioselectivities in the hydrogenations of unsaturated α- and β-aminoacids and itaconate derivatives (entries 12, 13, 4).
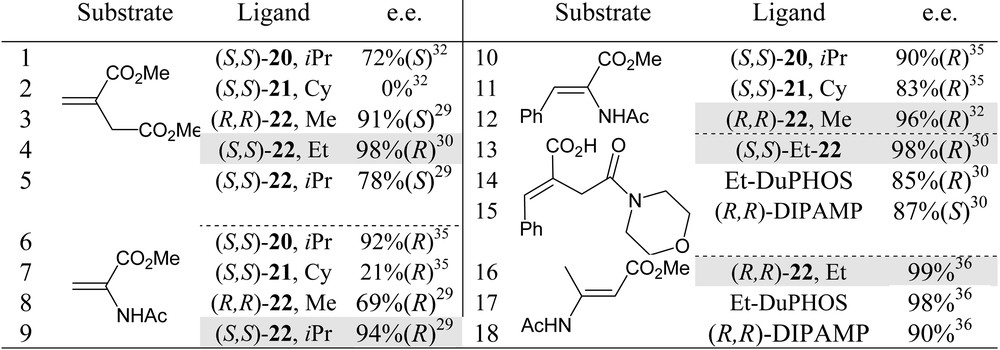
Hydrogenation of functionalised olefins promoted by rhodium complexes of phosphetane 20, 21 and 22. Comparative results.
Either preformed diphosphine–rhodium complexes or catalysts generated in situ from (COD)2Rh+X– were used in these studies. Catalyst amount: from 1 to 0.1%.
For each single substrate, fine tuning of the phosphetane α-substituents is still required to attain very high enantioselectivities: as shown in entries 3–5 and 8,9 respectively: Et-FerroTANE is the best ligand for the hydrogenation of dimethyl itaconate, while iPr-FerroTANE performs better in the hydrogenation of α-acetamidoacrylate.
Results in Table 2 also suggest that FerroTANEs rank among the best chiral phosphines for the asymmetric hydrogenation of (E)-methyl β-acetamido-2-butenoate (entries 13–15) [36] and of monoamido itaconate (entries 16-18) [30].
Finally, in these rhodium promoted hydrogenations, FerroTANEs were found to be far superior to the structurally analogous 1,1′-bis(phospholanyl)ferrocenes [29, 33, 37]: in the hydrogenation of dimethyl itaconate, for instance, 1,1′-bis(2,5-dialkylphospholanyl) ferrocenes give a maximum enantiomeric excess of 70%, to be compared with the 98% e.e. attained with phosphetanes (entry 4) [30]. The reasons for such an increase in selectivity upon moving from five- to four-membered heterocycles is unclear at present.
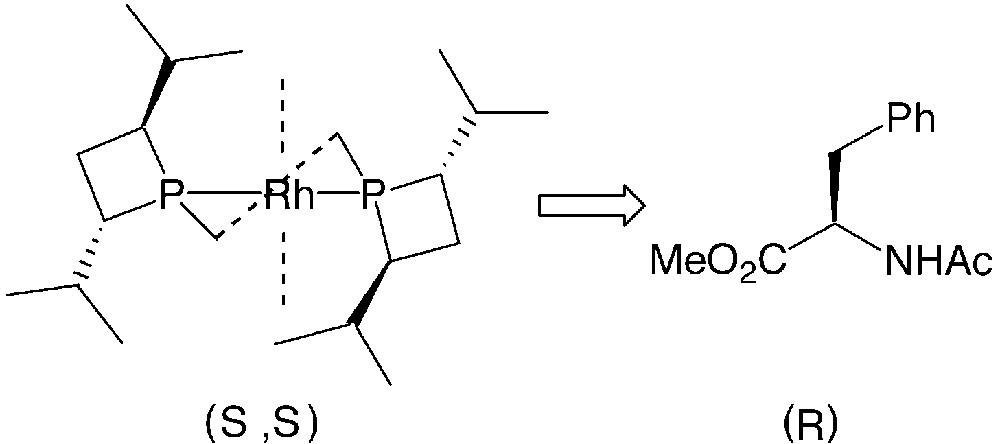
Concerning the stereochemical course of the hydrogenations of dehydroaminoacid derivatives (entries 6–12), FerroTANEs 22 behave like other electron-rich phosphines [38]: the expected (R)-configured product is obtained from the (S,S)-configured iPr-FerroTANE (Fig. 14).
CnrPHOS and BPE-4 display a more complex behaviour. The enantioselectivity is strongly affected by variations of the reaction conditions and especially by changes in the hydrogen pressure. In some cases, the stereochemical issue of these hydrogenations may even be reversed by increasing the hydrogen pressure [34]. This interesting behaviour and its mechanistic implications need to be analysed in more depth.
4 Conclusions
During the past 10 years, there has been considerable progress in the design of phosphetane-based chiral ligands. Requirements for high efficiency and high enantioselectivity in catalytic reactions have been fulfilled through the synthesis of C2-symmetric bidentate derivatives. Within this series, 1,1′-bis(phosphetanyl)ferrocenes emerged recently as particularly useful ligands, thus further developments in their catalytic applications are reasonably expected.
In addition, monodentate phosphetanes, which are easily accessible through a very flexible approach, displayed a promising potential on model catalytic reactions. They should find applications in specially targeted reactions, where the use of monodentate ligands is required.